
Abstract
The growing fight against infections caused by bacteria poses new challenges for development of materials and medical devices with antimicrobial properties. Silver is a well known antimicrobial agent and has recently started to be used in nanoparticulate form, with the advantage of a high specific surface area and a continuous release of enough concentration of silver ions/radicals. The synthesis of MgO–Ag nanocomposite coatings by in situ deposition of silver nanoparticles during plasma electrolytic oxidation of a magnesium substrate is presented in this study. The process was performed in an electrolyte containing Ag nanoparticles under different oxidation conditions (i.e., current density, oxidizing time, silver nanoparticles concentration in the electrolyte). Surface morphology, phase composition and elemental composition (on the surface and across the thickness of MgO–Ag nanocomposite coatings) were assessed by scanning electron microscopy, X-ray diffraction, energy X-ray dispersive spectrometry and radio frequency glow discharge optical emission spectroscopy, respectively. The coatings were found to be porous, around 7 μm thick, consisting of a crystalline oxide matrix embedded with silver nanoparticles. The findings suggest that plasma electrolytic oxidation process has potential for the synthesis of MgO–Ag nanocomposite coatings.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
In order to enhance the corrosion resistance and the wear properties of magnesium alloys, different coatings can be produced, such as porous oxide layers grown by plasma electrolytic oxidation (PEO) process. By the PEO process, the magnesium substrate is made anode in an electrolytic cell and its surface is converted into the corresponding oxide under the electrical field. The oxide layer consists of crystalline phases, with a highly porous surface and with components derived from both, the electrolyte and the substrate. The electrolytes used include multi-component water based solutions with silicates and phosphates as possible constituents. PEO coatings can offer very good wear, corrosion and heat resistance, low electrical conductivity and aesthetic/decorative properties [1, 2], but also potential biofunctionality.
Magnesium alloys have regained their potential as metallic biomaterials for medical devices due to their low Young’s modulus relative to titanium and cobalt–chromium alloys, the physiological role of the Mg2+ in the human body, alloys biodegradability and the technological advances in material processing [3]. However, no Mg alloys specifically designed for biomedical applications are yet available. PEO porous coatings could provide extended biofunctionalization to the magnesium substrate through controlled biodegradation rate and modulation of the events at the tissue-implant interface.
The growing fight against infections caused by bacteria poses new challenges for development of materials and medical devices with antimicrobial properties. Metallic silver and silver salts have been extensively used as antimicrobial agents in the form of impregnated dressings for burn injuries [4, 5], polymers bearing silver salts [6], silver coatings deposited by magnetron co-sputtering [7, 8], electrophoretic deposition [9, 10] or galvanic deposition [11]. The silver antimicrobial effectiveness, low toxicity and the growth-inhibitory capacity against microorganisms it is well recognized [11–14]. Feng et al. [15] showed that the release of silver ions from AgNO3 would penetrate the cell wall, will subsequently turn the DNA into a condensed form and at the same time will react with proteins. All these phenomena will damage and eventually kill the microorganisms. Kim et al. [16] reported some limitations in using metallic silver and silver salts as antimicrobial agents, such as the interfering effects of salts and the possibility to continuously release enough concentration of silver ions from the metal form. An alternative to overcome these limitations is the use of silver nanoparticles.
Several studies have been reported on the effectiveness of the antimicrobial effect of silver nanoparticles although the mechanism is not yet fully understood [16–19]. Kim et al. [16] studied the antimicrobial mechanism of Ag nanoparticles and reported the formation of Ag free radicals, from the surface of nanoparticles, and the subsequent free radical-induced damage of the cellular membrane leading to bacteria death. Sondi and Salopek-Sondi [17] showed that the antimicrobial activity of Ag nanoparticles is associated with the formation of “pits” in the cell wall of bacteria that leads to an accumulation of Ag in the bacterial membrane resulting in cell death. Amro et al. [20] also reported the formation of pits in the outer cell membrane and the change of membrane permeability caused by the release of the membrane proteins and lipopolysaccharide molecules. In addition, release of silver ions, in the range of 70–100 ppm, from nanocrystalline silver with a particle size of less than 20 nm was previously reported [21].
Possible antimicrobial applications of silver coated magnesium alloys include various areas that require special attention to hygiene such as: healthcare facilities, educational institutions, retirement facilities, public transport, etc. and also for implantable medical devices to fight the implant associated infections.
This study is assessing the potential of the PEO process for the synthesis of MgO–Ag nanocomposite coatings by in situ deposition of Ag nanoparticles during the oxidation of a magnesium substrate. The approach enables to enhance the mechanical properties, the corrosion resistance of the Mg alloy and to possibly add an extra functionality, i.e. antimicrobial activity, using a single step process. Structural and morphological characterization of the resultant coatings by X-ray diffraction (XRD), scanning electron microscopy/energy X-ray dispersive spectrometry (SEM/EDX), and radio frequency glow discharge optical emission spectroscopy (RF-GDOES) analyses is presented in this study.
2 Materials and methods
2.1 Zeta potential and size distribution of Ag particles
A Zetasizer Nano ZS equipped with MPT-2 Titrator (Malvern Instruments, UK) was used to determine the zeta potential (derived from electrophoretic mobility) and size distribution of Ag nanoparticles in the electrolyte used for anodic oxidation. Therefore, a dispersion of 1.0 g/l of Ag particles (Sigma-Aldrich) in the PEO electrolyte was prepared. The zeta potential measurements were carried out at 25°C, and between pH values 2 and 12.6 while the size measurements were performed at 25°C and pH of the electrolyte (i.e. pH = 12.6). For reproducibility at least 3 measurements were conducted for each pH value.
2.2 Plasma electrolytic oxidation of Mg substrate
The substrate used was a Mg–Zn–Zr–RE based alloy with 1.55wt.% Zn and 0.51wt.% Zr. Extruded Mg rods were machined into small cylindrical disks having a thickness of 8 mm and a diameter of 20 mm. The disks were manually ground with successive SiC paper grades from 1,200 to 2,400 (Struers, Denmark) using 100% ethanol as lubricating fluid. After grinding, the surface area of the substrates was calculated as 0.113 dm2. The samples were ultrasonically cleaned in ethanol and dried in a stream of compressed air prior to oxidation process. PEO was carried out in a double-wall glass electrolytic cell with a volume of 800 ml. The electrolyte used was Keronite (Keronite Limited, UK), a non hazardous, low concentrated alkaline solution (98% demineralized water, chrome and ammonia free), bearing 1.0, 3.0 and 5.0 g/l colloidal Ag particles. Cooling of the electrolyte was performed by water circulation through the electrolytic cell jacket. The temperature of the electrolyte was maintained in the range of 22–35°C. Magnesium disks were screwed to an insulated metallic rod and suspended in the centre of the electrolytic cell as anode, surrounded by a cylindrical steel cathode. The agitation of the electrolyte was maintained at a speed of 250 rpm using a magnetic stirrer (Ika, NL). PEO was performed under galvanostatic conditions using two different current densities, i.e. 3.0 and 5.0 A/dm2. The latter was selected as an extreme condition to possibly determine burning. The current was applied using an AC power supply type ACS 1500 (ET Power Systems Ltd, UK). Oxidation time was 1 to 10 min. The current and voltage transients were recorded during PEO at intervals of 1 s by a computer interfaced with the power supply through a National Instruments SCXI data acquisition system. After oxidation, the samples were thoroughly cleaned with deionised water, dried with blowing air and stored in desiccator until further testing.
2.3 Phase composition of MgO–Ag nanocomposite coatings
The XRD measurements were performed on a Bruker-AXS D5005 diffractometer, equipped with a Huber incident-beam CuKα1 monochromator and a Braun metal wire position sensitive detector (PSD). The 2θ range was 20–100° with a step size of 0.0426° and a counting time per step of 1 s.
2.4 Surface morphology of MgO–Ag nanocomposite coatings
The surface morphology of the magnesium oxide layers was investigated by SEM with a microscope JEOL JSM-6500F combined with an EDX probe, using an electron beam energy of 10 to 20 keV and a beam current of 40 nA. Prior to investigation, the oxidized magnesium samples were coated with a uniform carbon layer for good electrical conductivity. EDX analyses were performed to identify the elemental composition at specific locations on the surface of the oxides.
2.5 Chemical composition across the thickness of MgO–Ag nanocomposite coatings
The depth profile of the elemental composition of the composite coatings was qualitatively determined using a Leco GDS-750A rf GDOES operating at a true rf power emission of 14 W.
3 Results and discussion
3.1 Stability of Ag nanoparticles in the PEO electrolyte
The average size of Ag particles dispersed in the Keronite electrolyte, as determined by dynamic light scattering (DLS) method, was found to be 39 nm (Fig. 1). SEM examination of particles indicated a spherical shape (Fig. 2).

Size distribution of Ag nanoparticles dispersed in the PEO electrolyte

Morphology of Ag nanoparticles revealed by SEM
In order to have a homogeneous distribution of particles in the oxide coating, a stable dispersion of particles (i.e., no settling) in the PEO electrolyte is required. Factors that influence dispersions stability include: particle size, particle surface charge, solution concentration (ionic strength) and solution pH. Zeta potential (i.e., potential at the slipping plane of a particle) is used to evaluate the surface charge of particles and their dispersion stability in a certain solution. The variation of zeta potential of Ag particles with pH in the Keronite electrolyte is shown in Fig. 3. At pH 12.6 used for PEO, the zeta potential of Ag particles was −26.8 mV indicating a good dispersion stability due to the strong repulsive forces between the nanosized particles. With pH decrease, the zeta potential decreased (absolute value) and reached the zero value at pH 2.85 (isoelectric point, IEP) when all particles are settled.

Zeta potential of Ag nanoparticles in Keronite electrolyte as a function of pH (IEP = isoelectric point)
3.2 Synthesis of nanocomposite coatings
The magnesium substrates have been oxidized using two different current densities, i.e. 3.0 and 5.0 A/dm2 and three different concentrations of Ag nanoparticles in the PEO electrolyte, i.e. 1.0, 3.0 and 5.0 g/l. Experiments with no Ag nanoparticles in the electrolyte were also performed. The colour of the coated specimens changed with particles concentration in the electrolyte. Thus, PEO performed without particles in the electrolyte revealed a white-grey oxide film whereas upon addition of 1.0 g/l Ag nanoparticles in the electrolyte, the resulting oxide films were shiny light yellow. The colour of the oxide films became darker with the increase of Ag nanoparticles concentration. Thus, a concentration of 3.0 g/l Ag produced a dark yellow coating while the specimen oxidized with 5.0 g/l Ag formed a matt dark brown composite film.
The voltage-time evolution for the samples oxidized at different concentrations of Ag nanoparticles in the PEO electrolyte (i.e. 0, 1.0, 3.0 and 5.0 g/l) using a current density of 3.0 A/dm2 is shown in Fig. 4. The voltage-time transients revealed two characteristic stages during the formation of porous oxide composite coatings: (i) a rapid increase of the voltage up to the sparking voltage followed by (ii) a relatively slow voltage rise until the burning stage of the intensive sparking is reached. At a current density of 3.0 A/dm2, in the first stage, the voltage increased at a rate of 2.66 V/s, regardless of the Ag concentration in the electrolyte. The sparking voltage (when visible sparking commenced) was identified at 160 V after about 1 min of anodizing. The sparking voltage was reached faster with an increase in current density (e.g., approx. after 40 s at 5.0 A/dm2). The voltage-time transients (Fig. 4) revealed the highest value of the anodic forming voltage, i.e. 280 V for the substrates oxidized in the electrolyte without particles or with 1 g/l Ag. The anodic forming voltage decreased with increasing of the Ag nanoparticles concentration in the electrolyte. For the samples oxidized at 3.0 g/l and 5.0 g/l Ag, the highest anodic forming voltage was 220 V and 170 V, respectively. By the end of the second stage, the voltage fluctuations became pronounced being accompanied by relatively less but more powerful sparks, which caused local destructive effects of the oxide layer. An oxidizing time of 3 min at a current density of 3.0 A/dm2 was considered optimum to limit the damaging effect of the burning voltage and partial detachment of the oxide film at all Ag concentrations.

Voltage transients at different Ag concentrations in the electrolyte (3.0 A/dm2)
3.3 Composition and morphology of the nanocomposite coatings
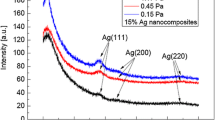
XRD patterns of the nanocomposite coatings produced by PEO are shown in Fig. 5. Apart from a variation of Ag peaks intensity there were no substantial differences in the layer phase compositions. Peaks attributable to Mg (substrate), crystalline MgO periclase (cubic structure) and Ag were identified. The presence of the Ag peaks suggests the deposition of Ag particles in the coating.

XRD patterns of anodic oxide layers produced at 3.0 A/dm2, 3 min oxidation
The surface morphology of MgO–Ag nanocomposite coatings was investigated by SEM. Figure 6 shows the SEM micrographs of the Mg specimens oxidized at 3.0 A/dm2 and 3 min duration with no particles in the electrolyte and with 1.0, 3.0 and 5.0 g/l Ag nanoparticles addition, respectively. For the Ag-free electrolyte, smooth and highly porous coatings were revealed. Pores of 0.1–1 μm were observed in the anodic oxide films regardless the presence of Ag nanoparticles. The specimens oxidized with 3.0 g/l and 5.0 g/l Ag nanoparticles in the electrolyte (Fig. 6c, d) revealed a relatively rough surface appearance possibly due to in situ Ag particles deposition that may affect the distribution and uniformity of the sparks which lead to a non-uniform growth of the oxide film. Microcracks could be observed on the surface of the oxide films and they seem to become more numerous with increasing the Ag nanoparticles concentration in the electrolyte. When using 5.0 g/l Ag nanoparticles in the electrolyte, damaged areas with partial detachment of the oxide film were detected. The thickness of the oxide films was estimated at about 7 μm by cross-section SEM analysis.

Morphology of anodic oxide layers produced at 3.0 A/dm2 and 3 min duration with different Ag nanoparticles concentrations in the electrolyte
Figure 7 shows the EDX analyses of the nanocomposite coatings (surface analysis) produced by PEO at a constant applied current density of 3.0 A/dm2 and 3 min oxidation, in the electrolyte containing 1.0, 3.0 and 5.0 g/l Ag nanoparticles. Different concentrations of Ag were detected in the coating being associated with the presence of particles. An increase of Ag nanoparticles concentration in the electrolyte resulted in an increase of the amount of Ag nanoparticles present in the composite coating. The EDX analysis of the coatings also indicated the presence of silicon and phosphorus in the coatings originating from the Si and P species in the PEO electrolyte. Important deposits of agglomerated Ag nanoparticles (with local Ag concentrations up to 69.8 wt.%) were present in the areas where partial detachment of the oxide occurred (Fig. 7d).

SEM micrographs and EDX patterns of anodic oxide layers produced at 3.0 A/dm2, 3 min duration and (a) 1.0 g/l Ag, (b) 3.0 g/l Ag, (c) 5.0 g/l Ag, (d) 5.0 g/l Ag (damaged area)
Further compositional analyses were performed to indicate whether the Ag nanoparticles were also present across oxide layers thickness. Therefore, the elemental composition across the oxide layer thickness was determined by rf GDOES analysis. Figure 8 reveals the varying intensity of the main elements versus sputtering time for the specimens oxidized at 3.0 A/dm2 for 3 min in the electrolyte bearing 1.0 and 3.0 g/l Ag nanoparticles. Silver was present across the thickness of the oxide layers. However, its distribution was not uniform, with relatively larger intensities towards the alloy/oxide interface. According to rf GDOES analysis, phosphorus and silicon species derived from the PEO electrolyte are distributed uniformly throughout the coating thickness.

GDOES depth profile of the nanocomposite coatings produced at 3.0 A/dm2 for 3 min duration with (a) 1.0 g/l Ag and (b) 3.0 g/l Ag in the electrolyte
The XRD, SEM/EDX and rf GDOES analyses suggested the in situ deposition of Ag nanoparticles during oxidation of the magnesium substrate by the PEO process. Further research is required to investigate the deposition mechanism and to control the concentration and the distribution of Ag nanoparticles in the oxide layers. Also, the release kinetics of silver from the coatings, the antimicrobial activity and the biodegradation rate of coated magnesium samples will be assessed.
4 Conclusions
Plasma electrolytic oxidation of a magnesium substrate in an electrolyte containing Ag nanoparticles has been performed under different process parameters in an attempt to evaluate the technical feasibility of the process for the synthesis of MgO–Ag nanocomposite coatings.
A zeta potential of −26.8 mV of Ag nanoparticles dispersed in the PEO electrolyte, at pH 12.6, prior to the PEO process, was found to induce sufficient strong repulsive forces between the particles to maintain system stability during the oxidation process.
The anodic forming voltage was higher in the electrolyte without any particle incorporation and decreased with increasing the silver nanoparticles concentration in the electrolyte.
The MgO–Ag nanocomposite coatings were found to be porous, around 7 μm thick, consisting of a crystalline oxide phase with embedded silver nanoparticles as confirmed by XRD, EDX and rf GDOES analyses. The successful deposition of Ag nanoparticles during the PEO process suggests that the process can be used for the synthesis of MgO–Ag nanocomposite coatings. Further research is required to assess the properties of such coatings for biomedical applications.
References
G.E. Thompson, F. Monfort, E. Matykina, A. Berkani, P. Skeldon, Corros. Rev 25, 631 (2007)
C. Blawert, W. Dietzel, E. Ghali, G. Song, Adv. Eng. Mater. 6, 511 (2006). doi:10.1002/adem.200500257
M.P. Staiger, A.M. Pietak, J. Huadmai, G. Dias, Biomaterials 27, 1728 (2006). doi:10.1016/j.biomaterials.2005.10.003
B.S. Atiyeh, M. Costagliola, S.N. Hayek, S.A. Dibo, Burns 33, 139 (2007). doi:10.1016/j.burns.2006.06.010
K. Dunn, V. Edwards-Jones, Burns 30, 772 (2004). doi:10.1016/S0305-4179(04)90000-9
R. Kumar, H. Munstedt, Biomaterials 26, 2081 (2005)
W. Chen, Y. Liu, H.S. Courtney, M. Bettenga, C.M. Agrawal, J.D. Bumgardner et al., Biomaterials 27, 5512 (2006). doi:10.1016/j.biomaterials.2006.07.003
V. Zaporojtchenko, R. Podschun, U. Schurmann, A. Kulkarni, F. Faupel, Nanotechnology 17, 4904 (2006). doi:10.1088/0957-4484/17/19/020
G. Zhang, X. Chen, J. Zhao, Y. Chai, W. Zhuang, L. Wang, Mater. Lett. 60, 2889 (2006). doi:10.1016/j.matlet.2006.02.009
M. Zarbov, D. Brandon, L. Gal-Or, N. Cohen, Key Eng. Mater. 314, 95–96 (2006)
J. Hardes, H. Ahrens, C. Gebert, A. Streitbuerger, H. Buerger, M. Erren et al., Biomaterials 28, 2869 (2007). doi:10.1016/j.biomaterials.2007.02.033
T.N. Kim, Q.L. Feng, J.O. Kim, J. Wu, H. Wang, G.C. Chen et al., J. Mater. Sci. Mater. Med. 9, 129 (1998). doi:10.1023/A:1008811501734
J. Pratten, S.N. Nazhat, J.J. Blaker, A.R. Boccaccini, J. Biomater. Appl. 19, 47 (2004). doi:10.1177/0885328204043200
G.J. Chi, S.W. Yao, J. Fan, W.G. Zhang, H.Z. Wang, Surf. Coat. Tech. 157, 162 (2002). doi:10.1016/S0257-8972(02)00150-0
Q.L. Feng, J. Wu, G.Q. Chen, F.Z. Cui, T.N. Kim, J.O. Kim, Biomed. Mater. Res 52, 662 (2000). doi :10.1002/1097-4636(20001215)52:4<662::AID-JBM10>3.0.CO;2-3
J.S. Kim, E. Kuk, K.N. Yu, J.H. Kim, S.J. Park, H.J. Lee et al., Nanomed. Nanotechnol. Bio. Med. 3, 95 (2007). doi:10.1016/j.nano.2006.12.001
I. Sondi, B. Salopek-Sondi, J. Colloid Interface Sci. 275, 177 (2004)
A.R. Shahverdi, A. Fakhimi, H.R. Shahverdi, S. Minaian, Nanomed. Nanotechnol. Bio. Med. 3, 168 (2007)
V. Alt, T. Bechert, P. Steinrucke, M. Wagener, P. Seidel, E. Dingeldein et al., Biomaterials 25, 4383 (2004). doi:10.1016/j.biomaterials.2003.10.078
N.A. Amro, L.P. Kotra, K. Wadu-Mesthrige, A. Bulychev, S. Mobashery, G. Liu, Langmuir 16, 2789 (2000). doi:10.1021/la991013x
A. Melaiye, W.J. Youngs, Expert Opin. Ther. Pat. 15, 125 (2005). doi:10.1517/13543776.15.2.125
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Necula, B.S., Fratila-Apachitei, L.E., Berkani, A. et al. Enrichment of anodic MgO layers with Ag nanoparticles for biomedical applications. J Mater Sci: Mater Med 20, 339–345 (2009). https://doi.org/10.1007/s10856-008-3589-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10856-008-3589-9