
Abstract
A range of anionic and cationic polycarboxylate ether (PCE) plasticizers with different molecular architectures (molecular weights, side chain lengths, and ratios of side chain density to backbone charge) are synthesized and tested to determine their effects on the rheological properties of fresh alkali-activated slag (AAS) pastes. A higher density of long side chains in the lower molecular weight polymers can provide a noticeable yield stress reduction, indicating a mild increase in workability compared to that of an unmodified AAS paste. It is hypothesized that side chains may have two important roles, i.e., providing steric hindrance to disperse particles after PCE adsorption on a particle surface, and also providing partial protection of the backbone charges against attachment of one PCE molecule to two or more slag particles, which is called bridging. This enhances the likelihood of adsorption on single particles, and thus increases the plasticizing action. A very similar plasticizing mechanism is observed for PCEs with similar structures but differing charge signs (cationic/anionic), which indicates that both anionic and cationic adsorption sites are available on AAS particle surfaces. The measured flow curves of all pastes are well described by the Herschel–Bulkley model with shear thinning behavior.
Similar content being viewed by others

Introduction
There have been a large number of experimental studies over the past decades aimed at finding suitable alternative materials which can be used in place of ordinary Portland cement (OPC) in specific (or general concreting) applications, with a particular focus, in recent years, on low CO2 emissions materials. Any cement replacement material must provide satisfactory mechanical properties in the hardened state, good durability, fluidity, and rheological properties in the fresh state, which are at least similar to those of Portland cement, to enable processing and placement using established techniques and skills which already exist in the construction industry [1, 2]. One such option is alkali-activated slag (AAS) which has shown comparable performance and major reductions in CO2 emissions compared to OPC [3]; slag is a by-product of the production of iron in a blast furnace, which is granulated and ground to form a fine powder which can react in an alkaline environment to form cement-like binders based on calcium aluminosilicate gels [4]. However, AAS does provide some processing challenges regarding its rheological properties. Generally, at the same water to binder ratio (w/b), the fluidity of OPC is considerably greater than that of AAS, meaning that the workability of the AAS is less. This difficulty needs to be overcome to produce AAS-based concretes displaying comparable rheological properties to those of concretes based on OPC.
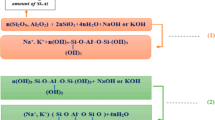
To circumvent fluidity problems in OPC-based concretes, and to enable the development of specialist products such as self-compacting concretes, a vast number of chemical admixtures that can be used to reduce the required w/b for a given workability have been developed. Such admixtures, known as plasticizers or water reducers, typically act by controlling dispersion and flocculation of particles in cement paste, and depend on the surface chemistry of the particles to enable them to function effectively [5]. Some highly effective plasticizing agents are classified as “superplasticizers,” and one of the most widely used families of modern superplasticizers is based on the chemistry of polycarboxylate ethers (PCEs). A PCE typically contains a charged backbone chain with polyethylene glycol (PEG) side chains. The backbone charges adhere to charged sites on cement particle surfaces, and the side chains provide steric hindrance to maintain particle–particle distances by limiting the attractive forces which would otherwise cause the particles to agglomerate [6, 7]. A schematic diagram showing the plasticizing action of a PCE is displayed in Fig. 1.

Schematic diagram of PCE adsorption on slag particles, and the plasticizing mechanism at high pH
The ability to tailor the molecular architecture of a PCE is of strong interest in the field of rheology control of cements, because this design flexibility allows control of the interactions of the PCE with solid particles and dissolved species, to achieve desirable performance. Effective design of the molecular architecture will increase the adsorption efficiency of PCEs on particles, and also optimizes the steric hindrance; both of these factors are essential in achieving good plasticizing performance. There are a variety of structural parameters that can be optimized in the design of a PCE for use in controlling cement paste rheology, including the length of the PEG side chains, molecular weight, and the ratio of backbone charge to side chain density. There exists a great deal of (sometimes contradictory) information in the literature regarding the role of these parameters on the performance of a PCE in Portland cement [6–15], and this is now also beginning to be understood for hybrid (OPC-containing) alkali-activated binders [16]. In addition, the nature of the functional groups responsible for the backbone charge will influence the adsorption performance via differences in electrostatic forces, depending on the charge environments present on the particle surfaces [12, 13, 17].
The majority of the PCE structures that are used highly effectively in OPC-based binder systems appear to have little or no beneficial effect in AAS binders. Research on some PCE structures in alkali-activated binders has shown a number of factors that could contribute to this lack of performance, and it is proposed that the most important reasons for this lack of performance are the degradation of polymer chains under the very alkaline environments present in this system [18, 19], and/or binding of polymers by the high concentration of dissolved ions in the fluid component of the paste (i.e., a chelating effect) [20].
Despite the major differences in chemistry and reaction processes between OPC and AAS binders, the PCE molecular architectures that have been used in past studies of alkali-activated systems have been mostly the same polymer structures which display good performance in OPC-based systems and generally have negatively charged backbones [8]. Slag has silanol groups that deprotonate in alkaline solutions, which may result in a negatively charged slag surface [21]—but this will also depend on the details of the aqueous electrolyte solution environment within the fresh paste, because some positively charged ions such as Ca2+, K+, and Na+ may adsorb onto the silanol groups, and then the surface may also carry some positive charges [20]. Therefore, both positively and negatively charged PCEs are potentially able to adsorb on slag particles and plasticize an AAS paste. In addition, the side chain length and density, and molecular weight, may play important roles in determining the plasticizing effects via manipulation of the steric hindrance effect.
Therefore, in this study, a broad range of PCE molecular architectures, with either cationic or anionic backbone charges, different lengths and densities of PEG side chains, and different molecular weights (M n ), are designed and synthesized. These new PCEs are examined in AAS systems to determine, for the first time, the effects of PCE molecular architecture on the rheological properties of these pastes.
Experimental section
Materials
Ground granulated blast furnace slag, with chemical composition as displayed in Table 1, and anhydrous sodium metasilicate powder (Na2O 50.9 wt% and SiO2 49.1 wt%) as activator were provided by Zeobond Pty Ltd, Australia. The AAS samples were formulated by mixing slag, sodium metasilicate (8 wt% by mass of slag), and the synthesized PCEs (0.5 wt% by mass of slag, on a dry polymer basis) with Milli-Q grade purified water. The water to binder ratio was kept at 0.45 for all pastes. After hand mixing to combine the components, the paste was mechanically mixed for 10 min at 500 rpm. The rheology of the homogenous paste was then measured immediately after mixing.
The PCE plasticizers were synthesized in three steps. In the first step, PEG macromonomers were synthesized by reaction of acryloyl or methacryloyl chloride and PEG with different chain lengths: 8, 22, and 43 ethylene oxide (EO) repeating units. These were used to form the side chains of the final copolymers. In the second step, the prepolymers were prepared by controlled free radical polymerization of the polyethylene glycol acrylate or methacrylates prepared in the previous step, with dimethylaminoethyl methacrylate (for cationic polymers) or tert-butyl acrylate (for anionic polymers), using the reversible addition–fragmentation chain transfer (RAFT) technique due to its advantages of well-controlled molecular weights and narrow molecular weight distributions [22]. The final step was the ionization of the prepolymers, which included deprotection of the tert-butyl group in trifluoroacetic acid/dichloromethane for anionic polymers, and quaternization of tertiary amines by iodomethane in tetrahydrofuran (THF) for cationic polymers. A linear polymer was synthesized by catalytic reaction of PEG methyl ether (M n = 5000 Da) and succinic anhydride.
The compositional ratios of the two comonomers in the final prepolymers were controlled by their feed ratios and determined by 1H NMR spectroscopic analysis on a Varian Unity Plus 400 MHz spectrometer using a deuterated solvent (CDCl3 or D2O) as reference; this information is shown in Table 2 as the PA:PEG ratio. The molecular weights of the polymers were determined by gel permeation chromatography (GPC) using HPLC-grade N,N-dimethylformamide (DMF) as eluent on a Shimadzu liquid chromatography system. Astra software was used to process the data to determine the molecular weights, based on the assumption of 100 % mass recovery of the polymers. The polydispersity index (PDI) which is the ratio of weight average molar mass (M w) to number average molar mass (M n) for each polymer is shown in Table 2.
Rheological tests
The rheological behavior of each AAS sample was measured using a Haake VT550 rheometer with a four-blade vane with length of 50.1 mm and diameter of 15.3 mm, in a sample volume sufficient to simulate an infinite medium [23]. Different types and strengths of forces between particles exist in concentrated suspensions such as AAS paste, including short range repulsion forces between particles, normal and shear stresses, particle pressure, diffusion or migration, inertial and hydrodynamic effects, and sedimentation [24]. The balance of these forces between particles in a suspension determines the response to applied external forces. The yield stress (σ y, Pa), which is the minimum applied stress necessary to overcome the interparticle forces and cause deformation, was selected as the key mechanical property to be used to determine the influence of each PCE on paste properties, along with consideration of the apparent viscosity at higher shear strain. A direct yield stress measurement at a low rotational rate (0.2 rpm) was used because of the accuracy and reproducibility of the measured data [25], and the yield stress was calculated from the applied torque based on the geometry of the vane and the rotation rate according to [25]. The percentage yield stress reduction achieved through the addition of a PCE was then calculated for each sample by Eq. 1:
The flow curve (relationship between shear stress and shear strain) was also determined for each sample by increasing the rotation rate in seven consecutive steps, to study the effect of the PCEs on AAS paste flow behavior at higher shear rates.
Zeta potential measurement
A ZetaProbe analyzer from Colloidal Dynamics was used to measure the zeta potential of slag particles in MilliQ-grade purified water (3 wt% suspension). This method measures the electrokinetic sonic amplitude as a result of an applied alternating electrical field; the movement of colloidal particles due to the applied alternating electrical field depends on the particle size and zeta potential [26]. Na2SiO3 solution (2 g/L) was used for titration of the sample to monitor surface charge changes after addition of the activator. At each step, 2.7 g Na2SiO3 per g slag was added from an alkaline solution to the slag dispersion, and after equilibration the zeta potential was measured based on the initial sample volume and the added volume of the titrant. Titration continued up to 10.8 wt% of the activator to slag. All related calculations were performed by the ZetaProbe Polar software (v.2.14).
PCE adsorption measurement
Pastes were mixed as described in “Rheological tests” section, then centrifuged for 10 min at 4500 rpm. The supernatant was filtered (0.45-μm filter) and washed with 500 mL of 0.1 N HCl to dilute to a concentration less than 100 mg carbon/L, and to prevent carbonation of the alkalis present. This solution was analyzed in a total organic carbon analyzer (TOC; Shimadzu) to measure the mass of organic carbon per volume of solution. This value is used to calculate the mass of the polymer in the supernatant, and thus the extent of adsorption via a mass balance calculation.
Results and discussion
The effects on AAS paste rheology of factors, including PCE molecular weight, side chain length, and charge density, are all interrelated. Additionally, the complex chemistry of the reacting binder system increases the complexity of the problem, as the nature and amount of solids present, and the ionic strength and pH in the paste liquid, are continually varying. This induces different interactions of inorganic components with plasticizers depending on the stage and extent of the reaction, and the polymeric molecules may also be degraded over time in the alkaline environment. However, this study focuses on the rheological properties in a very short period of time after mixing, to provide consistency of the results between samples and to limit the possible influences due to degradation, retarding, or accelerating effects of different PCEs.
Cationic and anionic polymers with different PEG side chain lengths (8, 22, and 43 EO repeating units), different ratios of density of backbone charges to side chains (5:1, 1:1, and 1:5), and various molecular weights, were synthesized. The polymer analytical data and structures are shown in Table 2. Each PCE is named according to its structural characteristics: anionic or cationic backbone (A or C), M n in kDa, side chain length, and backbone charge:side chain ratio—so, for example, the anionic PCE with M n = 27 kDa, 8-unit side chains, and 4 backbone charges per side chain, is named A27(8)-4:1. According to the conformation diagram of Gay and Raphael [27], all comb polymers in this study are categorized as the flexible backbone worm (FBW) type, because the number of monomers in the side chain (P) is higher than number of monomers in the backbone (N), and also far below the N3 which would be required to form a star-shape polymer.
Effect of PCE backbone length on paste yield stress
The A27(8)-4:1 polymer has a relatively long backbone chain with numerous negative charges, and a few short side chains that facilitate the adsorption of positively charged particles/ions in the paste. A yield stress increase of about 90 % is observed in the presence of this polymer, as shown in Fig. 2. This may be explained by the potential for a high degree of bridging to take place in the presence of this polymer, as the charged backbone can become adsorbed to multiple slag particles (Fig. 3). According to this mechanism, more attractive interparticle forces are present as a result of this effect, increasing the agglomeration of particles and causing an increase in yield stress. If this is the case, restricting each polymer to adsorption on a single particle, by introducing steric hindrance via longer side chains and by shortening the backbone chain, should limit this effect. As shown in Fig. 2, for A9(43)-5:1 in comparison with A27(8)-4:1, this is in fact the case: longer side chains and shortening of the backbone chain are likely to limit the extent of bridging. However, this change does not actually give any improvement in regard to yield stress reduction. The reasons for this will be discussed in detail below.

Yield stress reduction of two anionic PCEs with high density of backbone charges, and different M n and side chain lengths (negative values mean yield stress increase)

Schematic diagram of bridging effect of a PCE; the polymer backbone links two particles
Cationic or anionic PCE: the effect of charge sign on yield stress reduction
Considering the yield stress reduction results for both anionic and cationic PCEs with similar molecular structure (Fig. 4), it appears that regardless of the sign of the polymer backbone charge, the response of rheology to other chemical structural factors such as side chain density, length, and molecular weight follows a similar trend. However, cationic polymers with charges protected through steric hindrance by dense, long side chains such as C14(43)-1:5 or C8(43)-1:1 show a slightly higher plasticizing efficiency compared to their anionic counterparts. Thus, before putting forward a discussion on the performance of PCEs based on their chemical structure, some further discussion of the chemical environments in activated slag is necessary.

Effect of the ratio of backbone charges to side chain density for PCEs with long side chains and lower molecular weights on yield stress reduction (negative values mean that yield stress increases)
The sodium silicate-activated slag paste investigated in this study shows a pH of around 13 (as measured using a glass electrode), which is almost constant during the first hour after mixing. At this pH, silanol groups deprotonate and give a net slightly negatively charged surface, but it is also possible that cationic species (Ca2+ and Na+) in the fluid component of the paste can adsorb on these silanol groups and provide some sites with positive charges [28]. Zeta potential measurement of a dilute dispersion of slag in water (3 wt%) shows a slightly negative surface charge (Fig. 5), which is consistent with the presence of both cationic and anionic sites on the slag surface, but with a higher concentration of anionic charges. Upon addition of sodium silicate solution to the system, as shown in Fig. 5, the negative charge on the surface increases rapidly. At an Na2SiO3/slag ratio of 8 wt%, which is the same as the ratio of sodium silicate to slag in the rheological measurements reported here, the system shows a more negative charge. Addition of sodium silicate raises the pH, and so in the highly alkaline environment of the slag dispersion, as stated by Song et al. [29], the concentrations of Al and Si anions increase in the paste, but those of Ca2+ and Mg2+ cations will decrease correspondingly. Also, for the sodium silicate activator, the addition of deprotonated silicate species to the system, and their bonding to the surface (possibly via adsorbed cationic species such as Ca2+ on the surface) can contribute to the net negative charge of the slag surface, although positive sites are also still available. This could provide more scope for adsorption of cationic polymers. However, analysis of the extent of adsorption for cationic and anionic PCEs [C14(43)-1:5 and A11(43)-1:5] according to the method described in “PCE adsorption measurement” section shows that both polymers have very high (and similar) adsorption extents, measured at 97 % in both cases. The high ionic concentration of the liquid component in AAS may provide some complications for PCEs containing many charged sites, as will be discussed below.

Zeta potential of dispersion of slag in water (3 wt%), with the addition of different amounts of sodium silicate
Effects of side chain density of PCE at different M n on paste yield stress
Three ratios of backbone charges to side chains, 5:1, 1:1, and 1:5, were investigated in both anionic and cationic polymers, with side chain lengths of 43 EO units and M n around 10 kDa. The yield stress reduction data for these polymers are presented in Fig. 4. By increasing the side chain density compared to the number of backbone charges, the effect on yield stress is enhanced for both cationic and anionic polymers, following almost the same trend. This shows that limiting the number of the PCE charged sites results in better plasticizing performance. The aim here is that each PCE molecule should adsorb on only one slag particle, so that they are then able to disperse the particles by greater steric hindrance, as a larger number of side chains are able to reduce the yield stress. For C14(43)-1:5, the yield stress reduction was more than 40 %, which is the greatest among the polymers tested. This PCE has an average of one charge per molecule, and therefore bridging is almost impossible. Coupled with this, the high density of long side chains increases the steric hindrance and thus improves the performance of the polymer as a plasticizer after adsorption onto a charged particle surface.
At higher molecular weight (>50 kDa; Fig. 6), the effect of side chain density was reversed, and with increasing side chain density, less yield stress reduction was observed in the paste with C51(43)-1:1 than with C66(43)-6:1, although the difference was relatively small. The molecular weight of the backbone charge unit is very low compared to that of the PEG side chains with 43 EO repeating units, and therefore increasing M n in a polymer with more backbone charges than side chains results in a long backbone chain with quite sparse side chains. This may suggest some degree of inefficiency, considering the possibility of a large extent of bridging in AAS paste rather than efficient plasticizing, but another contributing factor at higher molecular weight is chain folding. A long backbone chain in a polymer with high M n (>50 kDa) is likely to fold on itself through conformational effects and thus, by creating extra steric hindrance, shield some of the backbone charges and protect them from side interactions, especially bridging. This limited access to charged sites in C66(43)-6:1 is thus identified as the main reason for the more effective yield stress reduction achieved with this polymer compared to C9(43)-5:1, which has roughly the same side chain length and density (Fig. 6). However, it appears that the chain folding is not sufficient to effectively reduce the effects of bridging, as the amount of yield stress reduction achieved with C66(43)-6:1 is less than half of that which is given by C14(43)-1:5.

Effect of backbone charge to side chain density ratio on yield stress reduction for PCEs with different molecular weights (negative values mean that yield stress increases)
Figure 7 shows the effect of molecular weight on yield stress reduction for PCEs with higher side chain density and a moderate side chain length, 22 EO units. This is in agreement with the results of Zingg et al. [15], who used a PCE with higher M n (~70 kDa) and long side chains (about 100 EO units), in a Portland cement system. They showed that a polymer with a high density of long side chains and high M n does not have the ability to reduce the viscosity of a cement paste as effectively when compared to a polymer with same side chain length but lower in graft density [15]. This is due to the very high extent of steric hindrance that exists for the polymer structure with a high graft density. The steric hindrance observed for polymers with an increased number of longer side chains is so high that the synthesis of a polymer with the same structure as C14(43)-1:5, but with higher M n was found in this investigation to be impractical.

Effect of molecular weight on yield stress reduction for PCEs with high side chain density, and a moderate side chain length (22 EO units)
For fresh AAS binders, where the ionic strength of the liquid component of the paste is very high because of the dissolved ions present in the activator, the molecular architecture of the polymer must be tailor designed to avoid PCE consumption in a way that does not help in dispersing particles and plasticizing. For the case of a polymer with higher density and availability of backbone charges compared to side chains, this could facilitate undesired bridging. According to this mechanism, particles are linked together rather than being dispersed.
Effects of different side chain lengths on yield stress reduction
Another important aspect in the design of PCE structures is the length of the side chains. Longer side chains are likely to provide a greater plasticizing effect according to the mechanism described in Fig. 1, assuming that adsorption onto the particle surfaces is effective and then longer side chains provide increased steric hindrance. However, there are contradictory results between studies, where both short and long side chains are able to give effective plasticizing in OPC systems, and/or where the differences between molecules with different side chain lengths sometimes seem insignificant [14, 15, 30]. In the case of AAS, as shown in Fig. 8, increasing the side chain length from 22 to 43 EO repeating units, while maintaining constant charge density and number of side chains per polymer, resulted in a more marked reduction in yield stress, demonstrating a considerable difference between long and moderate side chain lengths. As was discussed above, when a PCE molecule adsorbs on a particle surface, longer side chains provide more steric hindrance for particle dispersion, allowing lubrication of the particle surfaces by water, which results in a reduction in the paste yield stress. However, the steric hindrance of longer side chains can also reduce the ability of the PCE to adsorb onto the particle surface. Also, interaction between free ions and a polymer in the paste liquid, with no contribution from the particles, could reduce the adsorption by reducing the availability of charged PCE sites.

Yield stress reduction of PCEs with long (43 EO units) and moderate (22 EO units) side chain lengths, at low molecular weight and with 1:5 ratio of backbone charges to side chains
Effects of PCE steric structure on yield stress reduction
The general structure of PCEs investigated in this study was a comb-like copolymer, where PEG side chains were grafted to the charged backbone of the polycarboxylate. Considering this structure, the C14(43)-1:5 for cationic and A11(43)-1:5 for anionic polymers provided the best performance as plasticizers for the AAS system studied. The molecular architecture of these two polymers, regarding their M n and side chain density, comprised a backbone with an average of five side chains and one charged site per molecule. This is formally described as a comb polymer, but with a very short backbone chain. To provide a comparison between this structure and a simple linear structure with one charged site per molecule and a similar molecular weight embodied in a long tail of PEG (about 110 EO repeating units), the additional polymer A5(110)-L was synthesized. It was expected that the head charge would adsorb on particles and the long tail would provide steric hindrance to disperse particles from each other. However, no notable change in yield stress was observed for the linear polymer (Fig. 9), whereas the anionic polymer with the comb-like structure showed a yield stress reduction of around 35 %. This shows that a comb-like structure, even with a very short backbone, causes greater steric repulsion in dispersing the particles than a linear structure.

Yield stress reduction of linear and comb-like anionic polymers in AAS
AAS paste flow behavior with and without PCE
The rheological behavior of concentrated suspensions such as cement and AAS pastes is non-Newtonian [30, 31]. In this study, AAS pastes both with and without added PCEs showed a yield pseudo-plastic type of behavior. When increasing the shear strain applied to a fresh AAS paste, the point at which the yield stress is reached is accompanied by a transition from a stationary state with many attractive interparticle forces, to a partially deflocculated system in which some of these bonding forces are irreversibly altered by shear forces. The intensity of the deflocculation increases with higher shear strains, and a more dispersed AAS suspension shows lower apparent viscosity. For OPC pastes, both shear-thinning and shear-thickening types of behavior have been reported in the literature depending on the particle size, applied shear strain, and the type of supplementary materials and admixtures such as superplasticizers used in cement pastes [32–34]. However, the addition of slag to OPC results in a lower apparent viscosity and a consistent shear-thinning behavior [35]. Addition of superplasticizers results in viscosity reduction due to adsorption on particle surfaces, which overcomes some of the attractive interparticle forces and reduces flocculation of the suspension due to steric repulsion.
Shear stresses measured at a range of shear strains for AAS paste without PCEs, and with A11(43)-1:5 and C14(43)-1:1, are represented in Fig. 10. The positions of data points in this figure (and also data for other pastes, not shown, containing PCEs that decreased the yield stress compared to the reference paste) suggest that the flow behavior can be described according to the Herschel–Bulkley (H–B) model (Eq. 2), where τ is the shear stress (Pa), τ 0 is the yield stress (Pa), \( \dot{\gamma } \) is the shear strain (s−1), k is the consistency factor (Pa/sn), and n is the flow index. The flow index represents the type of flow behavior; for n < 1 the material is shear thinning, while materials with n > 1 are shear thickening. If n = 1, Eq. 2 reduces to the Bingham model.

Flow curves for AAS paste without PCE, and with A11(43)-1:1, and C14(43)-1:5 (dashed lines are the Herschel–Bulkley model fit for each paste)
Flow curve data were compared with the H–B model for AAS paste without PCE, and for the samples containing PCEs with effective yield stress reduction. The H–B model was fitted to the data, with n values between 0.54 and 0.65 determined for each of the samples, as shown by the dashed lines in Fig. 10.
For all PCEs with effective yield stress reduction, n was lower than 1.0, showing shear-thinning behavior, although the value of n was different in each case. Also, the value of n was consistently lower than in AAS without the polymer additives, showing a greater degree of shear-thinning behavior with the addition of effective PCEs to AAS paste. The “flow index reduction” was calculated according to Eq. 3 for each sample containing a PCE, where n P and n R are the flow indexes for the AAS paste with and without PCE, respectively.
The higher degree of shear-thinning behavior of AAS pastes in the presence of those PCE additives which were able to reduce yield stress shows a mutual benefit of these plasticizers. It is also observed that differences in the molecular architecture of PCEs also affect the magnitude of the flow index reduction.
Effect of different PCE structures on AAS shear-thinning behavior
Flow index reduction is one possible tool to enable comparison of the magnitude of shear-thinning behavior between AAS pastes with and without PCE additives. According to the discussion presented in “Effects of side chain density of PCE at different M n on paste yield stress” section, the use of a PCE with low molecular weight and more side chains than backbone charges resulted in a greater extent of yield stress reduction. The same trend was observed in the flow index reduction data, as shown in Fig. 11 for PCEs with different molecular weights but the same (moderate) side chains with higher density, and in Fig. 12 for PCEs with different side chain densities but the same (long) side chain length and low molecular weights.

Effect of PCE molecular weight on flow index reduction for polymers with high side chain density and moderate side chain length

Effect of backbone charge:side chain density ratio on flow index reduction, for PCEs with long side chains and low molecular weight
The effect of side chain length on flow index is opposite to what was observed for the yield stress reduction in “Effects of different side chain lengths on yield stress reduction” section, and as is shown in Fig. 13, a lower flow index was observed at higher side chain length. However, this does not mean that at higher shear strain a PCE with shorter chains is more dominant in viscosity reduction. For a better comparison at higher shear strain, the relative viscosity (the ratio of apparent viscosity of the samples with PCE to apparent viscosity of the reference AAS paste) of samples with PCEs of different side chain lengths, as a function of shear strain, is presented in Fig. 14. This demonstrates that PCEs with longer side chains still have consistently lower apparent viscosity, although at higher shear rate, the difference compared to PCEs with shorter side chains decreases significantly. A longer side chain is important in the stationary or very near-stationary state (when yield stress is measured), to give effective steric hindrance to disperse particles, but at higher shear strains, while the particle collisions will increase due to the particle motion, longer side chains do not cause a considerable viscosity difference compared to moderate side chain length PCEs. For this reason, the apparent viscosity at higher shear strains in Fig. 14 for the polymer with moderate side chain length becomes closer to the relative viscosities for PCEs with longer side chains, showing that the side chain length is less important at higher shear strains. This may also explain the higher flow index reduction for polymers with moderate side chain length.

Flow index reduction of PCEs with long (43 EO units) and moderate (22 EO units) side chain lengths, with low molecular weight and 1:5 ratio of backbone charges to side chains

Relative viscosity as a function of shear strain, for PCEs with different side chain lengths
Effect of silicate activator and high pH on the performance of PCEs
Sodium silicate-activated slag has a pH of around 13. At this pH, there are some concerns about hydrolysis of ester bonds in PCE molecules leading to detachment of side chains and loss of workability. Although this degradation phenomenon has not been fully experimentally proven, it has been suggested as a reason for the loss of performance of some PCEs in alkali-activated systems [16, 17]. However, Marchon et al. [16] showed that this was not the case in their hybrid alkali-activated binder systems, and that the effectiveness of PCEs depends instead on the competitiveness of adsorption between hydroxyls and PCEs. For that reason, the performance of C14(43)-1:5 polymer (with the highest yield stress reduction) was studied here in activated and nonactivated slag pastes, and the results are shown in Fig. 15. The performance of this polymer is notably better in the activated-slag system than in the nonactivated paste. It is hard to find an explanation for this observation of better performance in the activated system, but it does clearly show that there is no loss of performance of this PCE at high pH. It could also be concluded that silicate species do not directly reduce PCE performance through competitive effects.

Performance of C14(43)-1:5 performance in sodium metasilicate-activated slag, and in nonactivated slag (slag + water)
The adsorption extent of the polymers on slag is a key measure by which to characterize PCE performance. As noted above, the best cationic and anionic PCEs [C14(43)-1:5 and A11(43)-1:5] show very high adsorption extents, around 97 %. These polymers have an average of approximately one charge per molecule, which is the lowest among the polymers tested, but adsorb extensively on the slag particles. So, adsorption of these polymers in alkali-activated slag appears not to be a limiting factor, and other factors which were discussed earlier in “Effect of PCE backbone length on paste yield stress, Effects of side chain density of PCE at different M n on paste yield stress, Effects of different side chain lengths on yield stress reduction, Effects of PCE steric structure on yield stress reduction” sections have greater importance. However, it is anticipated that this extent of adsorption would vary in different alkali-activated systems, depending on the type of precursors, activator, and structure of PCEs used.
Conclusions
In the high ionic strength environment prevailing in AAS pastes, the number of charged sites in the backbone of a PCE plays an important role in determining its effectiveness in reducing yield stress. The presence of more available, unprotected charges in a polymer backbone can result in more side interactions such as bridging, resulting in consumption of plasticizer in a way that does not help in dispersion and deflocculation of the particles to achieve the aim of yield stress and viscosity reduction. This loss of workability properties can be avoided by controlling molecular weight, side chain length, and density, which suppresses the availability of PCE charges for side interactions by providing steric hindrance around the charges. However, careful control of this suppression is important to ensure that adsorption of each PCE onto a single particle can occur. The optimal polymer geometry, among those tested, was a very short charged backbone chain with a cationic charge and around five long PEG side chains (43 EO units) per PCE. Linear PEGs with a charged head and long tail could not plasticize the AAS paste.
AAS pastes with or without PCE addition showed non-Newtonian, shear-thinning behavior which can be described according to the Herschel–Bulkley model. Addition of PCEs which gave yield stress reduction also resulted in reduction of the Herschel–Bulkley flow index, compared to AAS pastes without plasticizer addition. The effect of side chain length appears less important in viscosity reduction at higher shear strains than in yield stress reduction.
This work, therefore, provides a pathway toward a better understanding of the effects of molecular architecture on plasticizing performance of PCEs in alkali-activated binders and provides new knowledge which is important in the development of a highly effective plasticizer for AAS or other geopolymer binders.
References
Schneider M, Romer M, Tschudin M, Bolio H (2011) Sustainable cement production–present and future. Cem Concr Res 41:642–650
Provis JL, van Deventer JSJ (2014) Alkali-activated materials: state of the art report, RILEM TC 224-AAM. Springer, Dordrecht
van Deventer JSJ, Provis JL, Duxson P (2012) Technical and commercial progress in the adoption of geopolymer cement. Miner Eng 29:89–104
Bernal SA, Mejía de Gutierrez R, Pedraza AL, Provis JL, Rodríguez ED, Delvasto S (2011) Effect of binder content on the performance of alkali-activated slag concretes. Cem Concr Res 41:1–8
Kwan AKH, Fung WWS (2012) Roles of water film thickness and SP dosage in rheology and cohesiveness of mortar. Cem Concr Compos 34:121–130
Yamada K, Hanehara S (2003) Working mechanism of polycarboxylate superplasticizer considering the chemical structure and cement characteristics. In: 11th international congress on the chemistry of cement, Durban (CD-ROM Proceedings)
Ferrari L, Kaufmann J, Winnefeld F, Plank J (2010) Interaction of cement model systems with superplasticizers investigated by atomic force microscopy, zeta potential, and adsorption measurements. J Colloid Interface Sci 347:15–24
Hanehara S, Yamada K (1999) Interaction between cement and chemical admixture from the point of cement hydration, absorption behaviour of admixture, and paste rheology. Cem Concr Res 29:1159–1165
Sakai E, Yamada K, Ota A (2003) Molecular structure and dispersion-adsorption mechanisms of comb-type superplasticizers used in Japan. J Adv Concr Technol 1:16–25
Yamada K (2011) Basics of analytical methods used for the investigation of interaction mechanism between cements and superplasticizers. Cem Concr Res 41:793–798
Plank J, Pollmann K, Zouaoui N, Andres PR, Schaefer C (2008) Synthesis and performance of methacrylic ester based polycarboxylate superplasticizers possessing hydroxy terminated poly(ethylene glycol) side chains. Cem Concr Res 38:1210–1216
Lesti M, Ng S, Plank J (2010) Ca2+-mediated interaction between microsilica and polycarboxylate comb polymers in a model cement pore solution. J Am Ceram Soc 93:3493–3498
Qiu XQ, Peng XY, Yi CH, Deng YH (2011) Effect of side chains and sulfonic groups on the performance of polycarboxylate-type superplasticizers in concentrated cement suspensions. J Dispers Sci Technol 32:203–212
Ran QP, Somasundaran P, Miao CW, Liu JP, Wu SS, Shen J (2009) Effect of the length of the side chains of comb-like copolymer dispersants on dispersion and rheological properties of concentrated cement suspensions. J Colloid Interface Sci 336:624–633
Zingg A, Winnefeld F, Holzer L, Pakusch J, Becker S, Gauckler L (2008) Adsorption of polyelectrolytes and its influence on the rheology, zeta potential, and microstructure of various cement and hydrate phases. J Colloid Interface Sci 323:301–312
Marchon D, Sulser U, Eberhardt A, Flatt RJ (2013) Molecular design of comb-shaped polycarboxylate dispersants for environmentally friendly concrete. Soft Matter 9:10719–10728
Plank J, Sachsenhauser B (2006) Impact of molecular structure on zeta potential and adsorbed conformation of α-allyl-ω-methoxypolyethylene glycol–maleic anhydride superplasticizers. J Adv Concr Technol 4:233–239
Palacios M, Puertas F, Bowen P, Houst YF (2009) Effect of PCs superplasticizers on the rheological properties and hydration process of slag-blended cement pastes. J Mater Sci 44:2714–2723. doi:10.1007/s10853-009-3356-4
Criado M, Palomo A, Fernandez-Jimenez A, Banfill PFG (2009) Alkali activated fly ash: effect of admixtures on paste rheology. Rheol Acta 48:447–455
Habbaba A, Plank J (2010) Interaction between polycarboxylate superplasticizers and amorphous ground granulated blast furnace slag. J Am Ceram Soc 93:2857–2863
Plank J, Schrofl C, Gruber M, Lesti M, Sieber R (2009) Effectiveness of polycarboxylate superplasticizers in ultra-high strength concrete: the importance of PCE compatibility with silica fume. J Adv Concr Technol 7:5–12
Xu J, Tao L, Boyer C, Lowe AB, Davis TP (2010) Combining thio–bromo “click” chemistry and RAFT polymerization: a powerful tool for preparing functionalized multiblock and hyperbranched polymers. Macromolecules 43:20–24
Nguyen QD, Boger DV (1992) Measuring the flow properties of yield stress fluids. Annu Rev Fluid Mech 24:47–88
Johnson SB, Franks GV, Scales PJ, Boger DV, Healy TW (2000) Surface chemistry–rheology relationships in concentrated mineral suspensions. Int J Miner Proc 58:267–304
Dzuy NQ, Boger DV (1985) Direct yield stress measurement with the vane method. J Rheol 29:335–347
O’Brien RW, Jones A, Rowlands WN (2003) A new formula for the dynamic mobility in a concentrated colloid. Colloids Surf A 218:89–101
Gay C, Raphaël E (2001) Comb-like polymers inside nanoscale pores. Adv Colloid Interface Sci 94:229–236
Labbez C, Jonsson B, Pochard I, Nonat A, Cabane B (2006) Surface charge density and electrokinetic potential of highly charged minerals: experiments and Monte Carlo simulations on calcium silicate hydrate. J Phys Chem B 110:9219–9230
Song S, Jennings HM (1999) Pore solution chemistry of alkali-activated ground granulated blast-furnace slag. Cem Concr Res 29:159–170
Hanehara S, Yamada K (2008) Rheology and early age properties of cement systems. Cem Concr Res 38:175–195
Palacios M, Banfill PFG, Puertas F (2008) Rheology and setting of alkali-activated slag pastes and mortars: effect of organic admixture. ACI Mater J 105:140–148
Cyr M, Legrand C, Mouret M (2000) Study of the shear thickening effect of superplasticizers on the rheological behaviour of cement pastes containing or not mineral additives. Cem Concr Res 30:1477–1483
Jayasree C, Krishnan JM, Gettu R (2011) Influence of superplasticizer on the non-Newtonian characteristics of cement paste. Mater Struct 44:929–942
Feys D, Verhoeven R, De Schutter G (2009) Why is fresh self-compacting concrete shear thickening? Cem Concr Res 39:510–523
Grzeszczyk S, Janowska-Renkas E (2012) The influence of small particle on the fluidity of blast furnace slag cement paste containing superplasticizers. Constr Build Mater 26:411–415
Acknowledgements
This work has been funded through an Australian Research Council Linkage Project grant, cofunded by Zeobond Pty. Ltd., and also benefited from support through the Particulate Fluids Processing Centre, a Special Research Centre of the Australian Research Council.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kashani, A., Provis, J.L., Xu, J. et al. Effect of molecular architecture of polycarboxylate ethers on plasticizing performance in alkali-activated slag paste. J Mater Sci 49, 2761–2772 (2014). https://doi.org/10.1007/s10853-013-7979-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-013-7979-0