
Abstract
Finding the most stable tautomer or a set of low-energy tautomers of molecules is critical in many aspects of molecular modelling or virtual screening experiments. Enumeration of low-energy tautomers of neutral molecules in the gas-phase or typical solvents can be performed by applying available organic chemistry knowledge. This kind of enumeration is implemented in a number of software packages and it is relatively reliable. However, in esoteric cases such as charged molecules in uncommon, non-aqueous solvents there is simply not enough available knowledge to make reliable predictions of low energy tautomers. Over the last few years we have been developing an approach to address the latter problem and we successfully applied it to discover the most stable anionic tautomers of nucleic acid bases that might be involved in the process of DNA damage by low-energy electrons and in charge transfer through DNA. The approach involves three steps: (1) combinatorial generation of a library of tautomers, (2) energy-based screening of the library using electronic structure methods, and (3) analysis of the information generated in step (2). In steps 1–3 we employ combinatorial, computational and chemoinformatics techniques, respectively. Therefore, this hybrid approach is named “Combinatorial*Computational*Chemoinformatics”, or just abbreviated as C3 (or C-cube) approach. This article summarizes our developments and most interesting methodological aspects of the C3 approach. It can serve as an example how to identify the most stable tautomers of molecular systems for which common chemical knowledge had not been sufficient to make definite predictions.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Finding the most stable tautomer or a set of low-energy tautomers is critical in many aspects of molecular modelling or virtual screening experiments. The first selection of potentially important (low-energy) tautomers is typically done based on common organic chemistry knowledge. This knowledge implies that tautomers are formed by migration of hydrogen atoms accompanied by migration of one or more double bonds in a molecule (prototropic tautomers) [1]. There are software tools available for generation of tautomers [2–4]. They first identify proton donor and acceptor sites typically involved in tautomerisation such as electronegative atoms: N or O. Next, a library of compounds is generated with various tautomers resulting from proton transfers between these sites accompanied by readjustments of double bond pattern.
Recently we have been studying anionic tautomers of nucleic acid bases (NABs), which are expected to be important in radiation induced mutagenesis (These anionic tautomers discussed throughout this article are in fact radical anions but for simplicity will be referred to as anions). Contrary to earlier experimental and computational predictions, we demonstrated that the most stable valence anions of pyrimidine bases, such as 1-methylcytosine [5], uracil [6], and thymine [7] do not result from a proton transfer between electronegative atoms, N or O. Instead they result from enamine-imine transformations, i.e., a proton is transferred between a NH site and a carbon site. Moreover, some of the most stable tautomers are not prototropic tautomers sensu stricto. That is, they violate the definition of (prototropic) tautomerization, which states that the inter-conversion of tautomers is accompanied by migration of one or more double bonds. In contrast, the neutral structures corresponding to these stable anions result from migration of one electron (i.e. they are diradicals, see Fig. 1). Some of these valence anions proved to be adiabatically bound with respect to the most stable tautomers of neutral NABs (as well as neutral canonical tautomers). It was an important finding because so far it was believed that the only adiabatically bound anions of NABs have a dipole-bound character [8]. The importance of valence anions results from the fact that dipole-bound anions are strongly perturbed by other atoms or molecules and their relevance in condensed phase environments is questionable. Our discovery of adiabatically bound valence anions of pyrimidine NABs was facilitated by a series of studies on proton transfer reactions in anionic complexes of NABs with various proton donors [9–13]. For the clarity of further text, note that we define adiabatic electron affinity (AEA) with respect to the neutral canonical tautomer [14]. Under such definition, the AEA’s of different tautomers reflect their relative stability and adiabatically bound anions, which have been in focus of our research due to their importance in radiation induced processes, are also the most stable anionic tautomers.

Tautmers of uracil (only one of possible resonant structures shown). Structures correspond to: a neutral canonical tautomer, b second-most stable neutral tautomer, c most stable anionic tautomer (radical anion), d third most-stable anionic tautomer (radical anion). For neutral species singlets are considered whereas doublets for anions
Our initial studies were focused on pyrimidine NABs (uracil, thymine, cytosine), because the number of potentially relevant tautomers was managable—a few tens of structures [5–7]. In addition, we had some insights from earlier studies which proton donor and proton acceptor sites should be considered [9–13]. The number of analogous anionic tautomers for purine NABs (guanine and adenine), for which we wanted to perform pre-screening using the density functional level of theory (DFT), was as large as 500–700, as there were no additional suggestions on relevant proton donor and acceptor sites. This was problematic not so much because of the computer time but rather the human time required to prepare, run, and analyze the calculations, which became prohibitive. To overcome these limitations, we developed a hybrid approach involving both combinatorial and accurate quantum chemical methods [15]. The procedure involved: (1) combinatorial generation of a library of tautomers; (2) pre-screening based on the results of geometry optimization/energy minimization of initial structures performed at the DFT level of theory. We call this step an “energy-based virtual screening” because, in contrast to the “structure-based virtual screening”, the most stable tautomers are the target of this screening; and (3) the final refinement at higher levels of theory of geometry and stability for the top hits determined at stage (2). The library of initial structures of various tautomers is generated with TauTGen [16], a tautomer generator program developed by us. TauTGen provides great flexibility in defining constraints used to enumerate tautomers. It can, for example, generate tautomers outside of typically considered prototropic tautomers. TauTGen can easily generate as many as 1,000 tautomers for molecules of the size of purine bases.
A good measure of success of our approach was our finding about valence anions of guanine. This base, which was believed to have the smallest electron affinity among nucleobases [17], supports at least 13 anionic tautomers, which are adiabatically bound with respect to the neutral canonical tautomer [18]. The most stable anion of guanine is adiabatically bound by as much as 8.5 kcal/mol. Using the same approach, we found at least one anion of adenine that is adiabatically bound with respect to the neutral canonical tautomer, the adiabatic electron affinity is 0.9 kcal/mol [19]. There findings were also confirmed experimentally [19–21]. The approach applied to cytosine demonstrated that it does not support an adiabatically bound valence anion [15]. In cases of all anionic NABs, some of the identified most stable tautomers do not correspond to prototropic tautomers and therefore would not be discovered using standard approaches. Finally, we have performed a preliminary screening of tautomers of cationic uracil to explore the posibility of formation of unusual tautomers [15]. Although we did not find those, we demonstrated that the relative energy differences between the most stable tautomers are much smaller for the cationic than for the neutral species.
Although the application of the hybrid combinatorial–computational (quantum chemical) approach proved to be successful in the identification of the most stable tautomers, at the same time it brought new challenges. For example, how to analyze tens of structures characterized at the high level of theory in the computational step of our approach? The natural path forward was to use chemoinformatics techniques, which have been developed to deal with large quantities of chemical data. This, however, brought another challenge: how to process quantum chemical (QC) data using existing chemoinformatics tools? We have taken steps to meet these challenges [22]. We have developed a number of approaches that allow us to combine data from QC calculations (e.g., orbitals, electron density and molecular geometries) with the chemoinformatics analysis methods (e.g., similarity calculations, clustering). For example, we proposed new, simple vector representations of 3D grid data such as an electron density distribution or an orbital bonding/antibonding character distribution. These vectors (called holograms) were combined with well established distance measures to perform similarity comparisons, clustering and to gain more insight from results of quantum chemical characterizations. Moreover, we proposed to use the results of our combinatorial–computational searches to divide the library of tautomers into two subset of: (1) the most stable tautomers and (2) less stable tautomers. The library supplemented with the stability information could be re-analyzed using substructure analysis techniques and clustering to identify the set of structural features determining the stability as well as to demonstrate that the most stable tautomers form “islands of stability” in the tautomeric space. The latter steps correspond to “knowledge extraction”, which may be used in the future to conduct more efficient searches for most stable tautomers.
In this article we summarize our developments of methodology, tools and approaches to find and analyze low-energy tautomers. Our contributions, discussed in the following sections, include: (1) a combinatorial–computational exploration of tautomeric spaces in order to identify the most stable tautomers of a molecule; (2) chemoinformatics approaches to analyze vast quantities of data harvested in quantum chemical calculations of (1). Step (2) also includes approaches to improve efficiency of the combinatorial–computational searches by using partial information on the studied chemical space. Both steps involve combinatorial, computational and chemoinformatics techniques. Therefore, we call our approach “Combinatorial*Computational*Chemoinformatics”, or just abbreviated as C3 (or C-cube) approach. When discussing our approach we will briefly illustrate its applications using an example of anionic guanine, which is a NAB that we have studied most extensively [15, 18, 20, 22, 23]. However, as our goal is to present the C3 approach, which is generally applicable, we will limit the discussion of results to absolute minimum, providing references to original, extensive studies.
Combinatorial–computational identification of the most stable tautomers
Overview
The combinatorial–computational identification of low energy tautomers of a molecule consists of three steps: (1) combinatorial generation of a library of tautomers with TauTGen, a tautomer generator program, (2) screening based on the results of geometry optimization/energy minimization of initial structures performed at the density functional level of theory, and (3) the final refinement of geometry for the top hits at the second order Møller-Plesset level of theory (MP2) followed by single-point energy calculations at the coupled cluster level of theory with single, double, and perturbative triple excitations (CCSD(T)) [24]. The details of steps 1–3 will be discussed in the following three sections.
Combinatorial generation of libraries of tautomers
An important part of the C3 approach is the generation of a diverse library of molecular tautomers. As the C3 approach is targeted at “difficult” molecular cases (charged species with or without a solvent), we can expect that the relevant tautomers might result from some uncommon transformations of the canonical tautomer on the neutral form, i.e., a proton could be transferred between N and C atoms. We developed a program for generation of tautomers, TauTGen [15, 16]. This program builds 3D structures of all possible tautomers from a molecular frame built of heavy atoms (the core) and a specified number of hydrogen atoms (Fig. 2). The hydrogens are attached to the sites specified by a user and a library of tautomers is combinatorially generated within a user-defined list of constraints. As the program does not have the embedded chemical knowledge, it provides great flexibility in enumeration of tautomers. For example, it can generate structures highly unstable as neutral species such as tautomers discussed in this article.

TauTGen uses a fixed frame of heavy atoms and a given number of hydrogen atoms to create tautomers of the resulting molecular system (here guanine is used as example)
The user has to provide an initial geometry of the molecular frame and to specify the minimum and maximum number of hydrogen atoms connected to each heavy atom. Sites for placement of hydrogen atoms are also defined by the user. To define a site, the user has to provide the following information:
-
Name—a string of characters used to build up a (file) name for each tautomer
-
A point where the hydrogen atom is to be placed. The point is defined relative to the fixed molecular frame
-
Information which heavy atom is the holder of this site (connectivity information)
-
The required total number of hydrogen atoms assigned to the heavy atom, which would make the specific site available for occupation (a site constraint)
-
Stereoconfiguration information, which tells the program if occupying a particular site will lead to the R or S configuration of the connected heavy atom.
Special care is taken to precisely name the sites. These names are used to create the names of tautomers that are later used as the filenames. For example, sites A and B (Fig. 3a) are named “N2cis” and “N2trans” to distinguish possible rotamers resulting from rotation of the N2H imino group. The connectivity information is used to count the number of hydrogen atoms at each heavy atom, N s . The number of available sites for hydrogen might be 2 even when N s = 1. Each site has a defined constraint, which tells for which values of N s the site becomes available for occupation. This is what we mean by the site constraint. This option is used to build proper hybridizations of heavy atoms. For example, the C and E sites (Fig. 3 b) are occupied only when N s = 2 for C8. Then C8 attains the sp3 hybridization. On the other hand, the D site is occupied only when N s = 1 for C8—the sp2 hybridization is then assigned to C8.

Information needed to define sites for hydrogen attachment. The sites are marked with capital letters
If one wants to generate stereoisomers, then two sites have to be used for each asymmetric atom in order to describe the R and S configurations. In the case of planar or nearly planar NABs the sites F and G that are “below” and “above” the molecular plane might be distinct (Fig. 3c). Each of these sites bears additional information describing the configuration, e.g. 1 or 2 for the “above” or “below” configuration, respectively.
As soon as the framework, available sites, the total number of hydrogen atoms Nhydrogens, and all constraints are defined, TauTGen generates all possible distributions of Nhydrogens hydrogens among Nsites sites. For each distribution TauTGen checks whether all applied constraints are respected. The constraints are checked in the following order:
-
constraints on the maximum and minimum number of hydrogens connected to each heavy atom
-
site constraints; check if the sites are used consistently with the actual values of N s
-
stereoconfiguration; check whether other enantiomer has already been generated (this check is not done by default).
Each new distribution of hydrogen atoms needs to pass all these checks to become an entry in the library of tautomers.
The stereoconfiguration check is done by a separate routine that detects enantiomers of a given distribution. If an enantiomer of the previously generated stereoisomer has been built, the distribution is rejected so the final set of stereoisomers consists of diasteroisomers only. The following steps are parts of the stereoconfiguration check:
-
A stereoconfiguration fingerprint is assigned to each new distribution. The fingerprint contains information if hydrogens occupying stereosensitive sites are above or below the molecular plane. In other words, we keep track whether the involved heavy atoms are R or S.
-
An inverse stereoconfiguration fingerprint is created for the distribution. It is then compared against the stereoconfiguration fingerprints of all previously generated stereoisomers of the same tautomer.
If there is no match between the fingerprints, the current distribution is a diastereoisomer of the previously generated stereoisomers and it is accepted to the library. If there is a match then the current distribution is an enantiomer and hence it is rejected.
Finally, TauTGen generates filenames and saves atomic coordinates of each member of the library to a separate file, which makes it easy to process the structures in the following steps of the C3 approach. The TauTGen program was written in the C programming language and the source code, documentation, and example input files are available online [16].
Example
We demonstrate an application of the TauTGen program by generation of anionic tautomers of guanine. A set of constraints used for guanine is presented in Table 1. We defined 23 sites available for hydrogen attachment. 17 sites were available for heavy atoms with N s = 1. Within these 17 sites, 4 sites were available to build rotamers of the N2 imino and O4 hydroxy groups and 2 sites were available for each of the C2, C4, C5 and C6 atoms to build stereoisomers with different positions of hydrogens in relation to the molecular plane. Additional 6 sites were available to build tautomers with two hydrogen atoms at N2, O4 and C8. The final molecular frames and sites are displayed in Fig. 4.

Molecular framework of guanine with all sites for hydrogen attachment. The total number of sites differ from the number of sites in Table 1 because some sites overlap
Within these constraints TauTGen generated 499 unique structures for guanine. In the course of generation of tautomers of guanine the TauTGen program generated initially 33,649 distributions, which were later reduced to 9,768, 907 and finally to 499 in the series of constraint checks.
Energy-based screening of the combinatorially generated library of tautomers
The energy-based screening corresponds to a selection of top structures from a list of tautomers sorted by descending order of stability. Stability of each tautomer is estimated by its energy at the optimal geometry at the employed level of theory. The major challenge in practical implementation of energy-based screening comes from the number of tautomers to be characterized. Although performing a geometry optimization using standard methods implemented in quantum chemical packages is a straightforward procedure, performing of such for hundreds of structures in an automatic, unsupervised manner is not so anymore. The common problems are related to convergence of both the self-consistent field (SCF) and geometry optimization procedures as well as computer system-related failures. We developed algorithms and tools that allow handling these problems automatically and therefore allow running a large number of electronic structure geometry optimizations in a hassle-free manner. We briefly summarize our developments in the following paragraphs.
The molecular structures generated by the TauTGen program were expressed in Cartesian coordinates and stored in typical.xyz files. They are used to build input files to the Gaussian03 [25] and NWChem [26, 27] programs. Initial screening was performed at the DFT level of theory with a B3LYP exchange–correlation functional [28–30] and 6-31++G** basis set [25]. A tendency of B3LYP to overestimate the excess electron binding energy helps to avoid false negatives when screening for adiabatically bound anions. The 6-31 ++G** basis set has an advantage that the time required to perform geometry optimization for a NAB is acceptable. This choice of the method and the basis sets was also supported by our earlier experience with calculations of adiabatic electron affinities (AEAs) for some pyrimidine NABs [5–7].
It is known that “buckling” of the ring of a NAB might increase the electronic stability of the anion, because the excess electron occupies a π* orbital [5–7]. For this reason, all initial structures of anions were built from buckled molecular frames. In the case of about 15% of generated structures, the initial SCF procedure failed to converge. In these cases we applied one, or a combination of up to four approaches: (a) start the calculation from orbitals generated with a smaller basis set (3-21G or 6-31G*), (b) start the calculation from orbitals generated in water solution simulated with the IEF-PCM method and the cavity built up using the United Atom (UA0) model [31] (c) try to converge the SCF procedure using a quadratically converging algorithm, (d) start the calculation from a slightly distorted geometry (the distortion was introduced by performing 2 optimization steps, but for the neutral molecule). In consequence, we recorded only a few cases when the SCF procedure failed to converge for the initial structure of the anion. The screening calculations of some guanine tautomers were performed using Gaussian03, others using NWChem. The B3LYP geometry optimizations were followed by single point calculations for neutral systems at the optimal anionic geometries to determine tautomers’ electron vertical detachment energies (VDEs). A negative value of VDEs suggest an unbound anion which is not correctly described at the employed DFT level. Energies for such anions were marked as unreliable in the final results table.
We used UNIX scripting tools to automate the screening procedure. We developed Gaussian Output Tools (GOT) scripts [32] to analyze output files from Gaussian03. The GOT scripts are written in the Practical Extraction and Report (Perl) language and can extract final energies, geometries and forces from the Gaussian03 output files. Analogous scripts were developed for NWChem output files. Other shell scripts were used to prepare and submit initial calculations as well as identify and restart the calculations of tautomers for which the SCF or geometry optimizations failed to converge. The final B3LYP energies for the neutral and anionic species were used to calculate the relative energies as well as AEAs and VDEs (or adiabatic and vertical ionization potentials in the case of cationic species [15]). We used graph isomorphism algorithm to check if the initial structure of a tautomer is the same as the optimized one. This allowed us to automatically mark tautomers, which decompose or convert to another tautomer. These tautomers, although they have an energy assigned, were considered unstable and excluded from further characterization. The molecular structures were sorted according to their relative energies and tabularized. In some cases we found that two, or more initial structures converged to the same energy. We have analyzed these cases to find out whether the same energy resulted from the same converged structures or from an accidental degeneracy. Additionally, all of the most stable structures selected for further investigation were visualized and analyzed.
Example
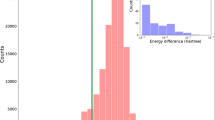
The 499 structures of guanine were optimized at the B3LYP/6-31++G** level. With the goal being the determination of adiabatically bound anions, we compared the final B3LYP energies for anions with the B3LYP energy of the neutral canonical tautomer at its optimal geometry. The histograms are presented in Fig. 5, which illustrate the relative stability in terms of AEA for all considered structures. It might be seen that the values of AEA smoothly decrease for about 90% of the structures. A sudden decrease of AEA for the remaining 10% of the structures is related to the fact that some of these structures decompose in the course of geometry optimization. The strucutres of anions based on two most stable neutral tautomers (G, GN) and fourteen most stable anionic structures G1–G14 are presented in Fig. 6.

Results of the energy-based screening of a library of anionic tautomers of guanine. Stability of anions is defined in respect of the neutral canonical structure. It is equivalent to AEA without corrections for zero-point vibrational energy. The values for the 50 most stable tautomers are presented on a larger plot whereas a smaller plot covers 499 tautomers. The tautomers are ordered according to decreasing stability

Important tautomers of anionic guanine
Refinement of the energies of the selected tautomers
The energy differences between structurally different tautomers might be as small as a fraction of a kcal/mol. Thus a meaningful study of the relative stability of tautomers typically requires employing the most accurate ab initio methods. For example, the relative energies of tautomers of cytosine in the gas phase predicted at the second order Möller-Plesset (MP2) level of theory differ qualitatively from the relative energies predicted at the CCSD(T) level of theory, suggesting that only the latter method might be accurate enough for determining the relative stability of close-lying tautomers [33].
Our approach therefore refines the energy and structure of the most stable tautomers identified at the energy based screening stage. The B3LYP geometries were further optimized at the MP2/AVDZ level of theory [34]. The final single-point calculations were performed at the coupled cluster level of theory with single, double, and non-iterative triple excitations (CCSD(T)/AVDZ) [34] at the optimal MP2 geometries. The obtained energies were corrected for the energies of zero-point vibrations. The MP2 geometry optimizations and frequency calculations were performed with Gaussian03 and the CCSD(T) calculations with the MOLPRO package [35].
We found the selected level of theory to be accurate enough to refine results of the DFT prescreening. Further refinement of our results is possible but much more computationally expensive. Please refer to the studies of Bachorz and coworkers [36] for an example. They used the state-of-the-art methodology to estimate stability of the valence anion of uracil. They used explicitly-correlated second-order Møller–Plesset perturbation theory (RI-MP2-R12) in conjunction with the conventional coupled cluster method with single, double, and perturbative triple excitations (CCSD(T)) supplemented with basis set extrapolation techniques. The final energies were corrected for zero-point vibration energies, determined in harmonic approximation at the UHF-RI-MP2/aug-cc-pVTZ level of theory. Their best estimate of the VDE is 0.60 eV while 0.51 eV is obtained at our accurate level. A discrepancy of 0.09 eV is not negligible, but not critical for the purpose of our projects.
Another question raised after the discovery of the new tautomers was regarding the stability of these species in condensed phases. Initially the relative stability of hydrated anionic tautomers was estimated at the DFT level with solvent effects approximated through continuum models [31]. These studies exposed the need for approaches that accurately predict the relative stability of important anionic tautomers of NABs in water solution. We addressed this by improving approaches for prediction of free energies of solvation. They are based on the microscopic solvent model and quantum mechanical/molecular mechanics (QM/MM) simulations. Please refer to Refs. [37–39] for further details of these approaches.
Example
In case of guanine and the B3LYP/6-31++G** level of theory, we found 14 anionic tautomers which were more stable than the canonical neutral, i.e., candidates for adiabatically bound anions. All of them were further studied at the MP2 and CCSD(T) levels with the AVDZ basis set. These calculations revealed that only 13 of 14 tautomers support adiabatically bound anions. The most stable anion is characterized by an AEA of 8.5 kcal/mol. A detailed discussion of the energetic and biological relevance of the new tautomers, suggested possible formation pathways, discussion of kinetics of formation as well as the experimental confirmation of our findings (a photoelectron spectroscopy data) were discussed in Ref. [20].
Chemoinformatics analysis of results of multiple quantum mechanical calculations
Overview
The development of the combinatorial–computational approach presented in the previous section provided an automated way to characterize hundreds of tautomers in the process of identification of the most stable species. At the same time it triggered development of new approaches to facilitate the analysis of large quantities of data coming from these studies. These approaches will be discussed in the following paragraphs.
Visual comparison of tautomers
Typically computational chemists using electronic structure methods deal with a small number of molecules. Therefore they can heavily rely on visualization to gain insights into the studied systems. Geometrical parameters are typically measured using functionalities implemented in graphical user interfaces (GUIs) of visualization programs. Similarly, the values of electron density in a molecular fragment and the bonding/antibonding character of an orbital are investigated since they contribute to chemical properties of this fragment. Their plots obtained for different systems are visually compared by placing them next of each other. At this point it is worth to mention one difficulty related with visual comparison of electron density distribution and the related properties that we have identified and addressed [40]. Orbitals and electron densities are typically visualized as finite volumes limited by a boundary defined by a preselected contour value (CV). When comparing molecular orbitals or electron densities of different systems one usually prepares plots using consistent CVs. This approach works fine when the charge distributions do not differ much in their spatial extension. We found, however, the same approach misleading when the studied charge distributions span a broad range of extension. The problem becomes particularly relevant when dealing with orbitals, which are characterized by very different orbital energies, and therefore different electron binding energies. This is often the case for a singly occupied molecular orbital (SOMO) in anionic species. Please refer to Ref. [40] for an extensive discussion and graphical examples. We suggested that an unbiased way to visualize orbitals or electron densities that differ much in the extension of charge distributions would be to assure that a consistent and preselected fraction of the total charge is reproduced in each plot. We have presented an algorithm how to identify a CV value for a preselected fraction of the total charge (Fe). We have implemented it in the OpenCubMan package [41], which also provides the following functionality: (1) identification of a CV that corresponds to a preselected value of Fe, (2) determination of Fe associated with a given CV, (3) selection of a particular part of the grid limited by a pre-defined plane. This selection is made by zeroing the to-be-discarded part of the grid. The last functionality will be used for some tasks described in the following paragraphs.
The visual comparison of molecular data, whether structures or 3D data such as molecular orbitals or electron densities, becomes impractical when the number of systems becomes significant. In the following paragraphs we present approaches that do not involve visualization and can be used to compare a large number of structures characterized in the course of electronic structure calculations.
Analysis of charge distributions
Although, the analysis of the excess charge distribution presented in this and the following sections is based on the Hartree–Fock singly occupied molecular orbitals obtained at the optimal MP2/APVDZ geometries in the final step of the combinatorial–computational approach, the analysis scheme is general and can be adopted to other levels of theory.
The major differences in the distribution of the excess electron could be, in principle, identified by comparing the SOMO plots. However, to get the quantitative information we developed a novel approach, in which the electron density contribution coming from the SOMO is assigned to heavy atoms using Bader’s analysis [42, 43]. Bader’s analysis defines a unique way of dividing molecules into atoms. The definition of an atom is based purely on the electronic charge density. The atoms are divided by so-called zero flux surfaces, which are 2-D surfaces on which the charge density is a minimum perpendicular to the surface. Having defined the atom limiting surfaces, the charge density is integrated over the volumes occupied by particular atoms. Then we define an orbital density hologram (in short an orbital hologram) as a vector the components of which hold information about population of the excess electron on each heavy atom. We calculate dissimilarity between two orbitals by calculating the Euclidian distance \( D_{orb}^{AB} \) between orbital holograms:
where xiA and xiB are the i-th components of the orbital holograms for the tautomers A and B. An obvious advantage of the orbital hologram representation is its small size comparing to large 3D grid data of the original orbital. The smaller size allows for easier handling and analysis, especially when the number of orbitals/tautomers becomes large.
Having defined a dissimilarity measure between orbitals, pairwise dissimilarities can be calculated and then clustering can be performed to group the most similar orbitals. In our work [22] we have used hierarchical clustering, which can be presented in an easy to analyze dendrogram, which reveals similarities between orbitals. The information that is available from orbital clustering contributes to our understanding of the binding modes of the excess electron. A similar shape of the electron density distribution corresponding to the SOMO orbital suggests a similar nature of the corresponding electronic state of the molecule. For specific examples and a summary of alternative approaches refer to Ref. [22].
Analysis of bonding/antibonding effects of singly occupied molecular orbitals
The π* orbitals occupied by the excess electron in the anionic NABs tautomers have partly a bonding and partly an antibonding character. The major differences in the distribution of bonding and antibonding areas of SOMO orbital could be identified by visual inspection although it becomes impractical for a larger number of tautomers. To verify if the bonding and antibonding character of the π* orbital correlates with the stability of a particular tautomer, we developed an approach that can quantitatively measure the bonding or antibonding character. This is done by summing the contributions over the chemical bonds present in the molecular framework built from heavy atoms.
In our approach, the determination of bonding/antibonding character has been designed in the spirit of the Hückel model of π-electron systems [44]. In this model, π orbitals are expressed as linear combination of pz atomic orbitals (AO) of atoms forming the π-system. It is a minimal basis set for π electrons. Moreover, it is assumed that the AO’s are orthonormal and only first immediate neighbors couple through the Hamiltonian. The way to estimate the bonding/antibonding character between two atoms is to look at bond orders resulting from a given orbital. For a given orbital, a contribution to the bond order between atoms X and Y is given by cX*cY, where the c’s are the LCAO coefficients of the contributing pz functions of atoms X and Y, respectively. Furthermore, the contribution from a given orbital to electronic charge localized on atoms X and Y are c 2X and c 2Y , respectively.
In the spirit of the Hückel method, we introduce a minimal basis set for π electrons. This hypothetical basis does not contain conventional pz atomic orbitals but rather effective atom-centered basis functions that reproduce an accurate occupied molecular orbital that we want to analyze. This molecular orbital has been obtained with a conventional extended basis set, e.g., AVDZ. We assume that all Hückel model assumptions apply to the new, hypothetical, minimal basis set. Moreover, we assume that bond orders and charges on atoms are calculated in the analogous way. The question remains how to find the LCAO coefficients cx and cY that accompany the hypothetical basis functions centered on the X and Y atom, respectively. For the molecular orbital of interest we determine Bader’s charges and we monitor the sign of the orbital in the neighborhood of each heavy atom X. This information is sufficient to determine the cx coefficients. The details of this procedure will be described below. We demonstrated in Ref. [22] that for a test case—the benzene molecule—this approach gives practically the same results as the Hückel model.
The details of employed procedure [22] to calculate a contribution from a π orbital to the bond order between neighboring atoms X and Y is as follows. The valence anions of NABs typically do not have Cs symmetry and one needs to define an approximate molecular plane. This plane is selected in a way to minimize the distance of heavy atoms to the plane and it is defined by eigenvectors of the inertia tensor. The molecular plane is consistent for all tautomers as they were superimposed before calculating of inertia tensor. This plane can be used to select electron density on either side of the plane by the algorithms implemented in OpenCubMan program. Next, we integrate the electron density associated with the π* orbital over the spaces associated with atoms X and Y (where the atomic spaces result from Bader’s analysis discussed in the previous section), and the resulting atomic charges are denoted δX and δY, respectively. In addition, we focus attention on one side of the approximate molecular plane and we monitor which sign, plus or minus, dominates in the space associated with X and Y. These signs are labeled sign(X) and sign(Y), respectively (Fig. 7). In the case of tautomers of NABs there was no ambiguity in determining the signs. Finally, the cX and cY coefficients are determined as:
and a contribution to the bond order between X and Y is given by cXcY. The positive and negative sign of cXcY determines whether the interaction is of bonding or antibonding character, respectively. The result does not depend which side of the molecular plane is used to determine sign(X) and sign(Y).

Determining the sign of SOMO orbital for the purpose of calculating bonding and antibonding effect on a chemical bond between X and Y
Having defined a method to measure the bonding/antibonding character of the SOMO orbital for each bond, we can define a vector, the components of which hold this information for all bonds present in the molecule. We will refer to these vectors as bonding character holograms. Similarly to the orbital holograms defined in the previous section, the similarity between bonding character holograms can be calculated using the Euclidean or Manhattan distance (or other distance measure), which give qualitatively the same results in this case [22]. The bonding character holograms can be clustered using, for example, the hierarchical clustering methods and the corresponding dendrogram can be generated to facilitate analysis. Refer to Ref. [22] for an example and further discussion.
The total bonding and antibonding character of the SOMO orbital can be calculated as a sum of, respectively, bonding and antibonding contributions over all the components of a bonding character hologram. These summed values indicate to which extent the SOMO is dominated by bonding and antibonding interactions.
Analysis of geometrical parameters
Apart from comparing excess electron distributions, we were comparing structural parameters of most stable tautomers. An important structural feature of anionic tautomers of NABs is buckling of the molecule. The geometrical parameters related to buckling are the dihedral angles defined among the atoms of molecular frame of non-hydrogen atoms. One can compute the dissimilarity between the buckling modes (\( D_{BM}^{AB} \)) of different tautomers using, for example, the Euclidean distance:
where γiA and γiB are the i-th dihedral angle related to the buckling of tautomer A and B, respectively. For a given optical isomer of A this optical isomer of B is selected, which provides a smaller value of \( D_{BM}^{AB} \). Again, having defined a similarity measure between buckling modes, all pairwise similarities can be calculated, and clustering then performed to group the most similarly buckled tautomers. In our studies we have used hierarchical clustering [22].
Further analysis of the tautomeric space
We also applied off-the-shelf chemoinformatics methods to study the tautomeric space, rather than limiting ourselves to particular chemical structures. In this context we used the results of our combinatorial–computational searches to divide the library of tautomers into two sets: (1) tautomers corresponding to adiabatically bound anions (most stable tautomers), and (2) less stable tautomers. Subdivision led to an interesting tautomer structure-stability relationship (SSR) analysis summarized here. The obtained two sets were compared to identify the set of structural features determining the stability.
In case of guanine, the SSR analysis was carried out on a reduced set of 165 tautomers (because of software limitations, multiple stereoisomers and rotamers were removed from the set of 499 tautomers as they would become redundant in the 2D fingerprint representation presented in this section, which does not take into account the spatial orientation of the bonded atoms). There are 10 adiabatically bound anions in the reduced set. We coded 2D substructure features present in each of 165 tautomers into Boolean vectors (called fingerprints). The generation of substructure dictionary as well as fingerprints themselves were performed using the BCI fingerprint package available from Digital Chemistry [45]. The substructure dictionary derived from our set of 165 tautomers contained 1,492 fragments (meaning that 1492-bit fingerprints were generated).
Then weighted and modal fingerprints were generated to represent groups of adiabatically bound (most stable tautomers) and unbound anions (less stable tautomers). Substructural analysis was carried out based on the occurrence of particular substructure features represented in the groups’ fingerprints. The analysis suggested that the characteristic features of the set of adiabatically bound anions are the absence of hydrogen atoms at C4, C5 and C6 carbons (Fig. 6). This finding nicely correlates with the excess electron distribution analysis which suggested a bonding character of π* orbital in the C4, C5 and C6 regions. Please refer to Ref. [22] for figures and further discussion of these results.
In addition to the SSR analysis presented above that identified structural features that distinguish the most stable anions from least stable tautomers, we tried to answer a related question: are the most stable anionic tautomers also similar to each other? In other words, do they form an island of stability in the tautomeric space? In order to answer this question we performed clustering of the set of 165 anionic tautomers. We used again the hierarchical clustering and the Tanimoto similarity measure. Indeed, we observed that most of the adiabatically bound anions cluster together, suggesting the existence of an “island of stability” in the chemical space of guanine tautomers. For example, we were able to find a cluster containing 24 elements including 7 out of the top 10 most stable tautomers. These 7 tautomers correspond to the 5 most stable tautomers (equal to 3% of the total tautomers) and to two further adiabatically bound anions from the second part of top 10 list. When compared with canonical guanine, the characteristic substructural features of tautomers in this cluster are additional hydrogen atoms at C8 and/or C2 atoms. For more details please refer to Ref. [22].
Accelerated searches of most stable tautomers
The existence of an “island of stability” in the tautomeric space may be exploited in the future to develop faster methods for the identification of the most stable tautomers. An example of such method, briefed below, was suggested in Ref. [22].
One could reduce the number of calculations required to screen the tautomeric space to find the most stable species. Such a reduction could be achieved in the following steps:
-
Generate t tautomers and the corresponding fingerprints.
-
Perform hierarchical agglomerative clustering, stopping at one cluster.
-
Choose a level of P clusters.
-
Select p molecules, one molecule from each of P clusters.
-
Run quantum chemical calculations for the p molecules to obtain their relative energy.
-
Perform energy-based screening of the p molecules to get the m most stable molecules representing M clusters.
-
Analyze the dendrogram representing the clustering. Identify S clusters at the level of F clusters (F < P) that contain M clusters.
-
Run quantum chemical calculations for all molecules (s) contained in the S clusters.
-
Perform energy-based screening of the s molecules to get the most stable tautomers.
The efficiency of such a procedure was estimated using the data collected from the clustering of anionic guanine tautomers. For example, p = 80 clusters are selected and the energy based screening is performed for 80 representative molecules. In the worst case, we would find only two adiabatically bound anions (as only two clusters have 100% concentration of adiabatically bound anions). We select only one (m = 1), the most stable molecule in the set of 80, and trace it in the dendrogram up to the level of 15 clusters (F = 15). In this case only one cluster (S = 1) is selected. The cluster has 24 elements and we need to characterize 23 of them at the quantum chemical (QC) level (one is already characterized). This procedure would require us to perform QC calculations for 103 tautomers instead of 165, giving 37.6% of CPU time saving while retrieving all the five most stable tautomers (and seven adiabatically bound anions total)! If the safer option of m = 3 is selected, we would end up with 126 calculations (80 at first stage and remaining elements of S = 3 clusters)—23.6% of CPU time saving and eight adiabatically bound anions retrieved. Our work on anions of adenine and cytosine suggests that such optimized search procedures would successfully identify the most stable tautomers of these molecules: the general application of such procedure would, however, need further investigation. Such a procedure might be valuable for an initial rough exploration of tautomeric spaces of large molecules, or molecules for which little is known about their chemistry (either due to the nature of the molecule or the environment in which it is placed).
Summary
We have summarized our developments of an approach that can be used to identify most stable tautomers for molecular systems for which common chemical knowledge had not been sufficient to make such predictions. The approach involves three steps: (1) combinatorial generation of libraries of tautomers, and (2) energy-based screening of the library using electronic structure methods; (3) analysis of generated data. The steps 1–3 correspond to combinatorial, computational and chemoinformatics techniques, respectively. Therefore, this hybrid approach is named “Combinatorial*Computational*Chemoinformatics”, or just abbreviated as the C3 (or C-cube) approach. This approach was successfully applied to discover the most stable anionic tautomers of nucleic acid bases that might be involved in the process of DNA damage by low-energy electrons and in charge transfer through DNA. These species are new phenomena that require further characterization. Although we have investigated their thermodynamic stability, electron binding energies and kinetics of formation, other important issues like their reactivity in the gas and condensed phases need to be addressed in the future.
Our C3 approach is not limited to identification of the most stable anionic tautomers of NABs. On the contrary, it can serve as a template, which can be easily modified and adopted to identify most stable tautomers of molecules in esoteric oxidation states and chemical environments. Also components of the C3 approach can be used as framework to compare many systems studied at a quantum chemical level of theory. For example, orbital density holograms developed to compare distribution of the excess electron in a set of molecules, can be easily adopted to compare distribution of any other property among atoms in a set of molecules. One can imagine a hologram with Fukui indices—a vector representation of a molecule storing information about atomic Fukui indices (also of interest in the context of nucleic acid bases [46]). Such representations could be compared and clustered in exactly the same manner as the presented orbital density holograms. As our future direction of study, we plan to compare different hologram representations to find a set of complementary approaches, which would carry unique information about a molecule.
References
Raczynska ED, Kosinska W, Osmialowski B, Gawinecki R (2005) Chem Rev 105:3561–3612
Sayle R, Delany J (1999) Canonicalization and enumeration of tautomers, In: Innovative computational applications. Institute for International Research, Sir Francis Drake Hotel, San Francisco, 25–27 Oct 1999
TAUTOMER, developed and distributed by Molecular networks GmbH, Erlangen, Germany
Pospisil P, Ballmer P, Scapozza L, Folkers G (2003) J Recept Signal Transduct 23:361–371
Haranczyk M, Rak J, Gutowski M (2005) J Phys Chem A 109:11495–11503
Bachorz RA, Rak J, Gutowski M (2005) Phys Chem Chem Phys 7:2116–2125
Mazurkiewicz K, Bachorz RA, Gutowski M, Rak J (2006) J Phys Chem B 110:24696–24707
Hendricks JH, Lyapustina SA, de Clercq HL, Snodgrass TJ, Bowen KH (1996) J Chem Phys 104:7788–7791
Gutowski M, Dąbkowska I, Rak J, Xu S, Nilles JM, Radisic D, Bowen KH Jr (2002) Eur Phys J D 20:431–439
Haranczyk M, Bachorz RA, Rak J, Gutowski M, Radisic D, Stokes ST, Nilles JM, Bowen KH (2003) J Phys Chem B 107:7889–7895
Haranczyk M, Rak J, Gutowski M, Radisic D, Stokes ST, Nilles JM, Bowen KH Jr (2004) Israel J Chem 44:157–170
Haranczyk M, Dabkowska I, Rak J, Gutowski M, Nilles M, Stokes S, Radisic D, Bowen KH Jr (2004) J Phys Chem B 108:6919–6921
Haranczyk M, Rak J, Gutowski M, Radisic D, Stokes ST, Bowen KH Jr (2005) J Phys Chem B 109:13383–13391
In case on neutral NABs, canonical tautomer is either the most stable tautomer or among few most stable tautomers
Haranczyk M, Gutowski M (2007) J Chem Inf Model 47:686–694
Freely available at http://tautgen.sourceforge.net Accessed 25 Jan 2010
Li X, Cai Z, Sevilla MD (2002) J Phys Chem A 106:1596–1603
Haranczyk M, Gutowski M (2005) Angewandte Chemie Int Ed 44:6585–6588
Haranczyk M, Gutowski M, Li X, Bowen KH Jr (2007) Proc Natl Acad Sci (PNAS) 104:4804–4807
Haranczyk M, Gutowski M, Li X, Bowen KH (2007) J Phys Chem B 111:14073–14076
Li X, Bowen KH, Haranczyk M, Bachorz RA, Mazurkiewicz K, Rak J, Gutowski M (2007) J Chem Phys 127:174309
Haranczyk M, Holliday J, Willett P, Gutowski M (2008) J Comput Chem 29:1277–1291
Haranczyk M, Gutowski M (2005) J Am Chem Soc (JACS) 127:699–706
Taylor PR (1994) In: BO Roos (ed) Lecture notes in quantum chemistry II, Springer, Berlin
Gaussian 03, Revision C.02, Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JrJA, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, and Pople JA, Gaussian Inc., Wallingford CT 2004
Straatsma TP, Aprà E, Windus TL, Bylaska EJ, de Jong W, Hirata S, Valiev M, Hackler M, Pollack L, Harrison R, Dupuis M, Smith DMA, Nieplocha J, Tipparaju V, Krishnan M, Auer AA, Brown E, Cisneros G, Fann G, Früchtl H, Garza J, Hirao K, Kendall R, Nichols J, Tsemekhman K, Wolinski K, Anchell J, Bernholdt D, Borowski P, Clark T, Clerc D, Dachsel H, Deegan M, Dyall K, Elwood D, Glendening E, Gutowski M, Hess A, Jaffe J, Johnson B, Ju J, Kobayashi R, Kutteh R, Lin Z, Littlefield R, Long X, Meng B, Nakajima T, Niu S, Rosing M, Sandrone G, Stave M, Taylor H, Thomas G, van Lenthe J, Wong A, Zhang Z, NWChem (2004) A computational chemistry package for parallel computers, Version 4.6, Pacific Northwest National Laboratory, Richland, Washington 99352-0999, USA
Kendall RA, Aprà E, Bernholdt DE, Bylaska EJ, Dupuis M, Fann GI, Harrison RJ, Ju J, Nichols JA, Nieplocha J, Straatsma TP, Windus TL, Wong A (2000) Computer Phys Comm 128:260–283
Becke AD (1988) Phys Rev A 38:3098–3100
Becke AD (1993) J Chem Phys 98:5648–5652
Lee C, Yang W, Paar RG (1988) Phys Rev B 37:785–789
Tomasi J, Persico M (1994) Chem Rev 94:2027–2094
GOT: Gaussian output tools available at http://gaussot.sf.net Accessed 25 Jan 2010
Fogarasi G (2002) J Phys Chem A 106:1381–1390 and references cited therein
Kendall RA, Dunning TH Jr, Harrison RJ (1992) J Chem Phys 96:6796–6806
Amos RD, Bernhardsson A, Berning A, Celani P, Cooper DL, Deegan MJO, Dobbyn AJ, Eckert F, Hampel C, Hetzer G, Knowles PJ, Korona T, Lindh R, Lloyd AW, McNicholas SJ, Manby FR, Meyer W, Mura ME, Nicklass A, Palmieri P, Pitzer R, Rauhut G, Schütz M, Schumann U, Stoll H, Stone AJ, Tarroni R, Thorsteinsson T, Werner H-J, MOLPRO, a package of ab initio programs designed by H-J Werner and PJ Knowles, version 2002.1
Bachorz RA, Klopper W, Gutowski M (2007) J Chem Phys 126:085101
Rosta E, Haranczyk M, Chu ZT, Warshel A (2008) J Phys Chem B 112:5680–5692
Haranczyk M, Gutowski M, Warshel A (2008) Phys Chem Chem Phys 10:4442–4448
Kamerlin S, Haranczyk M, Warshel A (2009) J Phys Chem B 113:1253–1272
Haranczyk M, Gutowski M (2008) J Chem Theory Comput 4:689–693
Open-source Cubefile Manipulator Program (OpenCubMan) is available free of charge at SourceForge archive: http://opencubman.sourceforge.net Accessed 15 Jan 2010
Henkelman G, Arnaldsson A, Jónsson H (2006) Comput Mater Sci 36:254–360
Sanville E, Kenny SD, Smith R, Henkelman G (2007) J Comput Chem 28:899–908
Atkins P, De Paula J (2006) Atkins’ physical chemistry, 8th edn. Oxford University Press, Oxford, p 387
http://www.digitalchemistry.co.uk Accessed 25 Jan 2010
Mineva T, Russo N (2010) J Mol Struct Theochem 943:71–76
Acknowledgments
Authors thank John Holliday and Peter Willett for stimulating discussions that contributed to the development of the C3 approach. M.H. is a 2008 Seaborg Fellow at Lawrence Berkeley National Laboratory. This research was supported in part (to M.H.) by the U. S. Department of Energy under contract DE-AC02-05CH11231.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Haranczyk, M., Gutowski, M. Combinatorial–computational–chemoinformatics (C3) approach to finding and analyzing low-energy tautomers. J Comput Aided Mol Des 24, 627–638 (2010). https://doi.org/10.1007/s10822-010-9344-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10822-010-9344-6