
Abstract
Cardiomyopathies are a heterogeneous group of cardiac muscle disorders that result in dilated, hypertrophic, or restrictive pathophysiological entities. Dilated cardiomyopathy (DCM) is the most common form in sub-Saharan Africa (SSA). However, population-specific research studies reporting the actual burden of DCM in this region are still lacking. Also, little is known about the genetic basis of DCM in this population, and genetic testing is still not readily accessible. This review describes the common pathogenic genes implicated in DCM globally and discusses the evidence-based management of patients with DCM. We also present a summary of studies describing genes implicated or associated with DCM in patients residing in SSA.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Dilated cardiomyopathy (DCM) is Africa’s second most common cause of heart failure, preceded only by hypertensive heart diseases [1]. Although DCM is prevalent in Africa, little is known about the genetic aetiology or mutations responsible for monogenic forms of this type of cardiomyopathy. In clinical practice, most patients are given a working diagnosis of idiopathic dilated cardiomyopathy (IDCM) without any formal genetic testing. This is partly because of limited access to genetic testing and the lack of cardiomyopathy gene panel tests based on data on genetic mutations found in African patients with DCM.
Population-based data on the burden of IDCM in Africa are lacking. In a post-mortem analysis of 90 subjects with underlying cardiovascular disease in South Africa, 17% had idiopathic DCM [2]. In a prospective registry of heart failure cases in a tertiary-level hospital in Johannesburg involving 5328 patients, 9.4% had a primary diagnosis of IDCM [3]. Although DCM is common, patients in low- and middle-income countries still present with advanced heart failure symptoms at a younger age than those in high-income countries [4, 5]. In this review, we discuss the mutational profile of DCM genetics and provide clinical guidance to clinicians managing DCM patients.
Definitions
Cardiomyopathies are a heterogeneous group of cardiac muscle disorders associated with cardiac dysfunction. Therefore, the terminology used to define dilated cardiomyopathies varies and may overlap (Table 1).
Clinical manifestation
The onset of heart failure in sub-Saharan African patients generally occurs at a younger age, thus affecting the economically active group [4]. In the early stages, some patients may remain asymptomatic, whilst in others, the nonspecific symptoms of heart failure may easily be misinterpreted as related to exhaustion or pulmonary diseases. Individuals with DCM may present with acute or gradual onset of heart failure symptoms. In some instances, individuals with DCM may also present with conduction abnormalities and sudden cardiac death.
Genetic determinants and mechanisms of genetic dilated cardiomyopathy
Globally, more than 50 genes that play a role in DCM have been identified, with some included in commercially available gene testing panels [9]. Of these genes, twelve have a definitive or strong relationship with DCM, seven had moderate evidence, and 25 had a limited role in DCM. In addition, seven have been disputed or assigned as having no known disease relationship due to the lack of evidence in human studies [9].
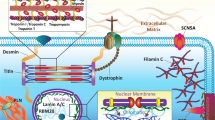
Most identified genes responsible for the DCM phenotype encode proteins within the myocytes. These are the sarcomere components, mitochondria, cytoskeleton, and desmosomal proteins. Genetic mutations implicated in DCM are predominantly composed of rare variants, making it challenging to diagnose a truly monogenic form of DCM [10]. A variant is considered pathogenic or likely pathogenic if there is a ≥ 90% likelihood of causing DCM [11]. The variable penetrance implies that although individuals may carry the disease-causing genotype, the DCM phenotype varies in clinical manifestation and severity. The DCM phenotype may only be evident in some candidates if the affected individual is further exposed to additional environmental or toxic insults such as alcohol. An international panel of clinical and scientific experts in DCM genetics has reviewed the available evidence on DCM genetics and identified twelve implicated genes from 8 gene ontologies as having a strong or definitive association with DCM [9]. Figure 1 depicts these “monogenic” genes classified according to their location in cardiac myocytes.

Genes with a definitive or strong relationship with dilated cardiomyopathy grouped according to their location in cardiac myocytes. DES desmin, FLNC filamin C, LMNA lamin A/C, MYH7 myosin heavy chain 7, PLN phospholamban, TTN titin, TNNC1 troponin C, TNNT2 troponin T2, RBM20 RNA-binding motif protein 20, SCN5A sodium voltage-gated channel, α subunit 5
Mechanism of cardiomyopathy based on causative genes
In up to 50% of cases, DCM is inherited in an autosomal dominant pattern [12]. Titin (TTN) has been reported as the primary causative gene [12]. Titin encodes the largest protein in the heart and functions as a spring that ties myosin to the Z-band in sarcomeres [13]. The second common gene implicated in the pathogenesis of DCM is the LMNA gene, which encodes protein lamins A and C. Mutations in this gene account for 5–10% of DCM and are responsible for the disruption of the chromatin organisation in dividing cells and signal transduction in non-dividing cells. Furthermore, LMNA DCM is often associated with atrioventricular blocks, atrial fibrillation, and ventricular arrhythmias. Patients with LMNA gene mutations may require primary prevention therapy, including a cardiac defibrillator device implantation, due to a 46% risk of sudden cardiac death [14, 15].
Genetic studies in the African context
We conducted a systematic literature search in PubMed, Scopus, and Web of Science to identify original clinical research studies conducted in Africa that report genes and variants associated with dilated cardiomyopathy. The search string was used “Dilated cardiomyopathy AND genetics AND Africa”. We excluded studies reporting on genetic findings in peripartum cardiomyopathy. The Preferred Reporting Items For Systematic Reviews and Meta-Analyses (PRISMA) flow chart showing the selection of studies is available as a supplementary file.
The literature search yielded twelve studies (Table 2), with very few studies (n = 2) on familial or monogenic cases of DCM in Africa. Eight studies (67%) were conducted in South Africa, and the rest in Tunisia (n = 2), Morocco (n = 1), and Egypt (n = 1).
A comprehensive approach to diagnosing dilated cardiomyopathy
History taking and clinical examination
The first step in diagnosing DCM entails obtaining a comprehensive clinical history, physical examination, laboratory investigations, and imaging. History taking should include a three-generation family history and a pedigree [10]. Figure 2 demonstrates an example of a four-generation pedigree of a proband with IDCM. Clinical details related to the cohort, including the proband depicted in the pedigree, are described elsewhere [28].

A four-generation family pedigree of a 62-year-old male (black arrow indicates proband) diagnosed with idiopathic dilated cardiomyopathy with a left ventricular ejection fraction (LVEF) of 30% at the age of 43. Squares shaded in black indicate male relatives with DCM. Females are represented with circles. Crossed-out circles and squares denote demised relatives
Clinicians should enquire about a family history of sudden cardiac death, unexplained deaths before age 50, heart transplantation, and pacemaker insertion before age 55. Furthermore, a history of death due to unnatural causes or drownings should be elicited. If a family member died due to unnatural causes, post-mortem reports should be reviewed to exclude or confirm primary or secondary causes of death [10]. History taking should be followed by a thorough clinical examination that includes a neurological examination focused on identifying neuromuscular diseases such as muscular dystrophy, which may manifest as muscle wasting, contractures of the elbows, spine, and Achilles tendons [29].
Electrocardiogram
Various electrocardiographic abnormalities may be found in patients with DCM, including a prolonged PR interval, evidence for ventricular hypertrophy, pathological Q waves, or bundle branch block [30,31,32]. In addition, poor prognostic factors on ECG include atrial fibrillation and a left bundle branch block [32].
Echocardiogram
An echocardiogram is mandatory to confirm ventricular dilatation and calculate the LVEF, which may guide the selection of appropriate heart failure therapy. In addition, features of cardiac remodelling, which include increased left atrial size, functional mitral insufficiency, alteration of diastolic function, and involvement of other chambers, should be elicited as these features are also associated with an unfavourable prognosis [32, 33]. Speckle-tracking echocardiography may also help to identify asymptomatic patients with left ventricular systolic dysfunction before they manifest with an overt DCM phenotype [34].
Laboratory tests
Laboratory biochemical tests are an integral part in the clinical workup of patients suspected to have IDCM. These may identify DCM’s endocrine, infectious, and haematological causes. Detecting micronutrient deficiencies such as selenium or thiamine may suggest malnutrition, which is strongly associated with alcohol misuse (Fig. 3). Abnormally high serial cardiac troponin and creatine kinase serum levels may indicate an acute myocyte injury due to myocarditis. Thyroid stimulating hormone and thyroxine levels should be assessed to exclude hypothyroidism or hyperthyroidism. Low calcium levels may indicate underlying chronic hypocalcaemia, while elevated iron levels may suggest underlying iron overload. When clinically suspected, bacterial and fungal infections should be excluded by performing blood cultures. Furthermore, a significantly elevated brain natriuretic peptide level has been reported to suggest a poor prognosis in patients with DCM [35,36,37].

Causes of dilated cardiomyopathy that should be considered after excluding coronary artery disease
Coronary angiography
Coronary angiography is currently the gold standard for evaluating coronary epicardial vessels’ atherosclerotic disease. Therefore, all patients with DCM should ideally be referred for a diagnostic coronary angiogram to exclude coronary artery disease. Furthermore, this test is mandatory since the therapeutic approach varies in patients with ischaemic and non-ischaemic cardiomyopathy.
Cardiovascular magnetic resonance imaging
Magnetic resonance imaging is crucial for excluding infiltrative conditions such as sarcoidosis and amyloidosis. In addition, the visualisation of gadolinium enhancement on late images may indicate underlying fibrosis. Several research studies have reported an association between the visualisation and burden of late gadolinium enhancement (LGE) and all-cause mortality in DCM [38,39,40,41].
Endomyocardial biopsy
An endomyocardial biopsy provides a definite histological, immunohistochemistry, and molecular evaluation of myocardial tissue [11]. It is indicated in patients suspected to have acute myocarditis or chronic inflammatory cardiomyopathy. The evaluation of endomyocardial biopsy samples of DCM patients may show nonspecific histopathological signs such as hypertrophy and vacuolar changes of myocytes and fibrosis [42]. Although invasive, the risk of complications associated with an endocardial biopsy is low, with 11% of patients experiencing atrioventricular block [42].
Endomyocardial biopsies should be considered if the test will likely alter the therapeutic management of patients, mainly if conditions such as sarcoidosis, giant cell myocarditis, eosinophilic myocarditis, or hemochromatosis are considered differential diagnoses. Strategies that could improve access to myocardial biopsy, particularly in low- and middle-income countries (LMIC), include the implementation of referral pathway protocols that prioritise the performance of biopsies in these patients, ensuring that personnel are adequately trained, as well as increasing the number of available catheterisation laboratories.
Genetic testing
Pre-test genetic counselling should be provided to DCM patients and their families. A detailed family history should be taken, and the possible findings and implications of genetic results should be explored during the counselling session. Defining terminology such as “pathogenic mutations, variants of uncertain significance and benign genetic variants” should be explained [8, 13]. Post-test counselling should focus on interpreting results, discussing reproductive risk, and the need for cascade family testing [13, 28].
Cardiomyopathy gene testing is still not widely available in most LMICs. However, diagnostic testing should be performed in carefully phenotyped patients with evidence of disease. In contrast, predictive testing is recommended in asymptomatic individuals (usually family members of an individual with a known DCM mutation) to predict the future risk of disease [43]. Currently available genetic tests involve sequencing a single gene or an individual variant, cardiomyopathy gene panel sequencing, whole-exome sequencing, and whole-genome sequencing [44].
Sequencing a single gene or individual variant should be considered a confirmatory test in a family member with a proband carrying a pathogenic/likely pathogenic variant detected through other techniques. Although this approach is cost-effective, it is not appropriate for diagnostic testing in a proband from another family with DCM where the causal mutation has not yet been identified [44, 45]. Cardiomyopathy gene panel sequencing involves sequencing the coding regions of several cardiomyopathy genes simultaneously in a single experiment. The diagnostic yield is higher. However, this test may not capture non-coding variants [13].
Whole-exome sequencing involves the analysis of a sequence of the entire coding region of the human genome. Limited non-coding regions may be included, but much of the non-coding DNA is not analysed. Whole-exome sequencing is not limited to genes previously linked with disease, thus enabling the potential to identify novel variants in new genes of interest. However, there is also a higher likelihood of identifying a variant of uncertain significance with whole-exome sequencing, and these usually need further investigation or careful explanation to patients as to their unknown implications [46]. In contrast, whole-genome sequencing captures both coding and non-coding variants, including deep intronic variants. Although more expensive, whole-genome sequencing will capture all variants and can be used to calculate polygenic risk scores for multifactorial causality of DCM and to report pharmacogenetic variants [47]. If a causal genetic mutation for monogenic DCM is identified, cascade screening of family members should be implemented, where a specific mutation rather than a gene panel is evaluated.
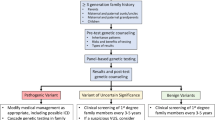
Genetic testing within the African context is challenging, partly due to the diverse genetic makeup of populations within the continent, making the possibility of identifying a variant of uncertain significance higher [48]. Furthermore, currently available DCM gene panels could be limiting in the African context as the diagnostic yield may be lower since African-specific DCM-causing genes (should they be present) have not yet been identified. In addition, the current gene panels are based on DCM patients who are primarily not of African descent. Figure 4 summarises an approach to genetic testing in a proband with DCM.

An approach to genetic testing in a proband with dilated cardiomyopathy. Diagram modified from Tayal et al. [10]. DCM dilated cardiomyopathy, ECG electrocardiogram, ICD implantable cardioverter defibrillator, LP likely pathogenic, LV left ventricular, MRI magnetic resonance imaging, P pathogenic, VUS variant of uncertain significance
Differential diagnosis
Diagnosing DCM may be challenging in most LMICs and other remote regions without access to diagnostic modalities such as coronary angiography or cardiovascular MRI. In such scenarios, the diagnosis is generally made on clinical grounds. The higher prevalence of infectious diseases in sub-Saharan Africa makes viral myocarditis highly likely. As such, clinicians faced with patients presenting acutely with the clinical syndrome of heart failure should routinely evaluate and monitor serum troponins, creatine kinase-MB levels, and viral antibody titres to exclude viral myocarditis. The viral antibody titre tests may include coronavirus, enterovirus, HIV, cytomegalovirus, Epstein-Barr virus, adenovirus, human herpes virus 6, parvovirus B19, hepatitis, and influenza virus antibodies [49].
A genetic aetiology should be considered in females with idiopathic cardiomyopathy in the peripartum phase [50,51,52,53]. A discussion on the genetic basis of peripartum cardiomyopathy is beyond the scope of this manuscript, and the reader is referred to literature published elsewhere [54].
Management of patients with genetic causes of dilated cardiomyopathy
The pharmacological management of a proband with a genetic cause for DCM is not different from that of any individual with non-genetic causes of DCM. It is guided by the presence of heart failure symptoms, the baseline LVEF, and the presence or risk for potentially lethal arrhythmias. Pharmacological therapy for DCM is mainly based on neurohumoral blockade [55]. Diuretics and inotropes may be considered in the acute phase in patients with congestive heart failure complicated by cardiogenic shock. Agents such as beta-blockers, angiotensin-converting enzyme inhibitors (ACE-I) or angiotensin receptor blockers in ACE-I intolerant individuals, angiotensin receptor neprilysin inhibitors (ARNI), mineralocorticoid antagonists (MRA), and more recently the sodium-glucose cotransporter-2 (SGLT2) inhibitors are currently considered essential foundational therapies in the management of heart failure with reduced ejection fraction [37].
Mutations in the LMNA gene have been linked to atrioventricular block and atrial or ventricular arrhythmias [56, 57]. To prevent lethal ventricular arrhythmias, implantable cardioverter defibrillators should be considered, particularly in patients who have survived a malignant ventricular tachyarrhythmia or those presenting with symptomatic ventricular tachycardia [15]. Unfortunately, device therapy is still not widely available in most LMICs due to its high cost.
Targeted therapies
Several strategies that address genetic abnormalities in patients with DCM are available. These methods include targeting specific single mutations, exon skipping, and gene replacement targeting all mutations at once by gene transfer of the full-length complementary DNA [55]. Also, newer molecules such as EMD 57,033 bind in the same region of myosin, increasing the rate of ATP binding, hydrolysis, and actin interactions. Furthermore, CK-1827452, a molecule that accelerates the transition of the actin-myosin complex from weakly to strong bound, is being investigated for potential clinical use [58]. Also, there is an ongoing randomised, double-blind placebo-controlled trial evaluating the efficacy of ARRY-371797, an inhibitor of the p38α MAPK pathway in symptomatic patients with DCM due to a mutation of the gene encoding the lamin A/C protein [59].
Future directions and recommendations
There is an urgent need to establish registries for IDCM/DCM patients and their families in Africa to facilitate the investigation of a genetic basis for IDCM in patients of African ancestry [60]. This should be coupled with the creation of African genome banks to enable the ease of determining the pathogenicity of VUS against a suitable reference population. Establishing African genomic consortia such as the Human Heredity and Health in Africa (H3Africa) Consortium promises to make these goals a reality [61].
Conclusion
IDCM remains a common disease in many African regions and continues to cause significant morbidity and mortality in young patients. Given that Africa has remarkable genetic diversity, a study of the genetic aetiology of this condition in an African setting is likely to yield novel insights and could be of clinical relevance in diagnosis and treatment. This highlights the need for improved access to IDCM genetic testing. Once widely implemented, the term “idiopathic dilated cardiomyopathy” should only be reserved for patients with a comprehensive clinical workup that includes genetic testing that fails to identify a plausible cause for DCM.
Availability of data and materials
Not applicable.
Abbreviations
- ACE:
-
Angiotensin-converting enzyme
- AR:
-
Adrenoreceptor
- DCM:
-
Dilated cardiomyopathy
- DE S :
-
Desmin
- DNA:
-
Deoxyribonucleic acid
- ECG:
-
Electrocardiogram
- FLNC :
-
Filamin C
- HIV:
-
Human immunodeficiency virus
- HLA :
-
Human leucocyte antigen
- HO-1 :
-
Heme oxygenase 1
- IDCM:
-
Idiopathic dilated cardiomyopathy
- IQR:
-
Interquartile range
- LMNA :
-
Lamin A/C
- LGE:
-
Late gadolinium enhancement
- LV:
-
Left ventricle
- LVEF:
-
Left ventricular ejection fraction
- MHC:
-
Major histocompatibility complex
- MRI:
-
Magnetic resonance imaging
- MYH7 :
-
Myosin heavy chain 7
- OR:
-
Odds ratio
- PLN :
-
Phospholamban
- RBM20 :
-
Ribonucleic acid binding motif protein 20
- RNA:
-
Ribonucleic acid
- SCN5A :
-
Sodium voltage-gated channel α subunit 5
- TTN :
-
Titin
- TNNC1 :
-
Troponin C
- TNNT2 :
-
Troponin T2
- VUS:
-
Variant of uncertain significance
References
Agbor VN, Essouma M, Ntusi NaB, Nyaga UF, Bigna JJ, Noubiap JJ (2018) Heart failure in sub-Saharan Africa: a contemporaneous systematic review and meta-analysis. Int J Cardiol 257. https://doi.org/10.1016/j.ijcard.2017.12.048
Steenekamp JH, Simson IW, Theron W (1992) Cardiovascular causes of death at Tshepong Hospital in 1 year, 1989–1990. A necropsy study. S Afr Med J 81(3)
Stewart S, Carrington M, Pretorius S, Methusi P, Sliwa K (2011) Standing at the crossroads between new and historically prevalent heart disease: effects of migration and socio-economic factors in the Heart of Soweto cohort study. Eur Heart J 32(4). https://doi.org/10.1093/eurheartj/ehq439
Dokainish H, Teo K, Zhu J, Roy A, Alhabib KF, Elsayed A et al (2013) Heart failure in Africa, Asia, the Middle East and South America: the INTER-CHF study. Int J Cardiol 204. https://doi.org/10.1016/j.ijcard.2015.11.183
Callender T, Woodward M, Roth G, Farzadfar F, Lemarie JC, Gicquel S et al (2014) Heart failure care in low- and middle-income countries: a systematic review and meta-analysis. PLoS Med 11(8). https://doi.org/10.1371/journal.pmed.1001699
Elliott P, Andersson B, Arbustini E, Bilinska Z, Cecchi F, Charron P et al (2008) Classification of the cardiomyopathies: a position statement from the European Society Of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J 29(2). https://doi.org/10.1093/eurheartj/ehm342
Fatkin D, Johnson R, Mcgaughran J, Weintraub RG, Atherton JJ, Group CGCW (2017) Position statement on the diagnosis and management of familial dilated cardiomyopathy. Heart Lung Circ 26(11). https://doi.org/10.1016/j.hlc.2017.04.021
Rosenbaum AN, Agre KE, Pereira NL (2020) Genetics of dilated cardiomyopathy: practical implications for heart failure management. Nat Rev Cardiol 17(5). https://doi.org/10.1038/s41569-019-0284-0
Jordan E, Peterson L, Ai T, Asatryan B, Bronicki L, Brown E et al (2021) Evidence-based assessment of genes in dilated cardiomyopathy. Circulation 144(1). https://doi.org/10.1161/CIRCULATIONAHA.120.053033
Tayal U, Ware JS, Lakdawala NK, Heymans S, Prasad SK (2021) Understanding the genetics of adult-onset dilated cardiomyopathy: what a clinician needs to know. Eur Heart J 42(24). https://doi.org/10.1093/eurheartj/ehab286
Schultheiss HP, Fairweather D, Caforio ALP, Escher F, Hershberger RE, Lipshultz SE et al (2019) Dilated cardiomyopathy. Nat Rev Dis Primers 5(1). https://doi.org/10.1038/s41572-019-0084-1
Shaboodien G, Spracklen TF, Kamuli S, Ndibangwi P, Van Niekerk C, Ntusi NaB (2020) Genetics of inherited cardiomyopathies in Africa. Cardiovascular diagnosis and therapy 10(2). https://doi.org/10.21037/cdt.2019.10.03
Mcnally EM, Mestroni L. Dilated cardiomyopathy: genetic determinants and mechanisms. Circ Res. 2017;121(7). https://doi.org/10.1161/CIRCRESAHA.116.309396
Van Berlo JH, De Voogt WG, Van Der Kooi AJ, Van Tintelen JP, Bonne G, Yaou RB et al (2005) Meta-analysis of clinical characteristics of 299 carriers of LMNA gene mutations: do lamin A/C mutations portend a high risk of sudden death? J Mol Med (Berl) 83(1). https://doi.org/10.1007/s00109-004-0589-1
Ciarambino T, Menna G, Sansone G, Giordano M (2021) Cardiomyopathies: an overview. Int J Mol Sci 22(14). https://doi.org/10.3390/ijms22147722
Adadi N, Radi FZ, Lahrouchi N, Hara L, Ratbi I, Elalaoui SC et al (2018) Inherited dilated cardiomyopathy in a large Moroccan family caused by LMNA mutation. Anatol J Cardiol 20(1). https://doi.org/10.14744/AnatolJCardiol.2018.69639
Fish M, Shaboodien G, Kraus S, Sliwa K, Seidman CE, Burke MA et al (2016) Mutation analysis of the phospholamban gene in 315 South Africans with dilated, hypertrophic, peripartum and arrhythmogenic right ventricular cardiomyopathies. Sci Rep 6. https://doi.org/10.1038/srep22235
Sayed S, Idriss NK, Blann A, Sayyed HG, Raafat DM, Fouad D et al (2015) The number of GT(n) repeats in the hemeoxygenase-1 gene promoter is increased in pediatric heart failure but is unrelated to renal, antioxidant and anti-inflammatory markers. Pediatric cardiology 36(6). https://doi.org/10.1007/s00246-015-1146-0
Mahjoub S, Mehri S, Ghazouani E, Ouarda F, Boussada R, Zaroui A et al (2010) HLA class II polymorphisms in Tunisian patients with dilated cardiomyopathy. Tissue Antigens 75(6). https://doi.org/10.1111/j.1399-0039.2009.01432.x
Mahjoub S, Mehri S, Ourda F, Boussaada R, Zouari B, Ben Arab S (2011) [Epidemiological study of the idiopathic dilated cardiomyopathy in Tunisia]. Ann Cardiol Angeiol (Paris) 60(4). https://doi.org/10.1016/j.ancard.2011.04.006
Shaboodien G, Engel ME, Syed FF, Poulton J, Badri M, Mayosi BM (2009) The mitochondrial DNA T16189C polymorphism and HIV-associated cardiomyopathy: a genotype-phenotype association study. BMC Med Genet 10. https://doi.org/10.1186/1471-2350-10-37
Du Preez J, Matolweni LO, Greenberg J, Mntla P, Adeyemo AA, Mayosi BM (2018) The α2CDel322–325 adrenergic receptor polymorphism is not associated with heart failure due to idiopathic dilated cardiomyopathy in black Africans
Woodiwiss AJ, Badenhorst D, Sliwa K, Brooksbank R, Essop R, Sareli P et al (2008) β1- and α2c-adrenoreceptor variants as predictors of clinical aspects of dilated cardiomyopathy in people of African ancestry
Badenhorst D, Norton GR, Sliwa K, Brooksbank R, Essop R, Sareli P et al (2007) Impact of beta2-adrenoreceptor gene variants on cardiac cavity size and systolic function in idiopathic dilated cardiomyopathy. Pharmacogenomics J 7(5). https://doi.org/10.1038/sj.tpj.6500426
Khogali SS, Mayosi BM, Beattie JM, Mckenna WJ, Watkins H, Poulton J (2001) A common mitochondrial DNA variant associated with susceptibility to dilated cardiomyopathy in two different populations. Lancet (London, England) 357(9264). https://doi.org/10.1016/S0140-6736(00)04422-6
Candy GP, Skudicky D, Mueller UK, Woodiwiss AJ, Sliwa K, Luker F et al (1999) Association of left ventricular systolic performance and cavity size with angiotensin-converting enzyme genotype in idiopathic dilated cardiomyopathy. The American journal of cardiology 83(5). https://doi.org/10.1016/s0002-9149(98)00981-3
Tiago AD, Badenhorst D, Skudicky D, Woodiwiss AJ, Candy GP, Brooksbank R et al (2002) An aldosterone synthase gene variant is associated with improvement in left ventricular ejection fraction in dilated cardiomyopathy. Cardiovasc Res 54(3). https://doi.org/10.1016/s0008-6363(02)00281-x
Bailly C, Henriques S, Tsabedze N, Krause A (2019) Role of family history and clinical screening in the identification of families with idiopathic dilated cardiomyopathy in Johannesburg, South Africa. S Afr Med J 109(9). https://doi.org/10.7196/SAMJ.2019.v109i9.13936
Brown CA, Lanning RW, Mckinney KQ, Salvino AR, Cherniske E, Crowe CA et al (2001) Novel and recurrent mutations in lamin A/C in patients with Emery-Dreifuss muscular dystrophy. Am J Med Genet 102(4). https://doi.org/10.1002/ajmg.1463
Dec GW, Fuster V (1994) Idiopathic dilated cardiomyopathy. New England Journal of Medicine 331(23). https://doi.org/10.1056/NEJM199412083312307
Lakdawala NK, Winterfield JR, Funke BH (2013) Dilated cardiomyopathy. Circ Arrhythm Electrophysiol 6(1). https://doi.org/10.1161/CIRCEP.111.962050
Merlo M, Cannata A, Gobbo M, Stolfo D, Elliott PM, Sinagra G (2018) Evolving concepts in dilated cardiomyopathy. Eur J Heart Fail 20(2). https://doi.org/10.1002/ejhf.1103
Japp AG, Gulati A, Cook SA, Cowie MR, Prasad SK (2016) The diagnosis and evaluation of dilated cardiomyopathy. J Am Coll Cardiol 67(25). https://doi.org/10.1016/j.jacc.2016.03.590
Murtaza G, Virk HUH, Khalid M, Rahman Z, Sitwala P, Schoondyke J et al (2017) Role of speckle tracking echocardiography in dilated cardiomyopathy: a review. Cureus 9(6). https://doi.org/10.7759/cureus.1372
Gardner RS, Ozalp F, Murday AJ, Robb SD, Mcdonagh TA (2003) N-terminal pro-brain natriuretic peptide. A new gold standard in predicting mortality in patients with advanced heart failure. Eur Heart J 24(19). https://doi.org/10.1016/j.ehj.2003.07.005
Kim H, Cho YK, Jun DH, Nam CW, Han SW, Hur SH et al (2008) Prognostic implications of the NT-ProBNP level and left atrial size in non-ischemic dilated cardiomyopathy. Circ J 72(10). https://doi.org/10.1253/circj.cj-07-1087
Mcdonagh TA, Metra M, Adamo M, Gardner RS, Baumbach A, Bohm M et al (2021) 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur Heart J 42(36). https://doi.org/10.1093/eurheartj/ehab368
Becker MaJ, Cornel JH, Van De Ven PM, Van Rossum AC, Allaart CP, Germans T (2018) The prognostic value of late gadolinium-enhanced cardiac magnetic resonance imaging in nonischemic dilated cardiomyopathy: a review and meta-analysis. JACC Cardiovasc Imaging 11(9). https://doi.org/10.1016/j.jcmg.2018.03.006
Di Marco A, Anguera I, Schmitt M, Klem I, Neilan TG, White JA et al (2017) Late Gadolinium enhancement and the risk for ventricular arrhythmias or sudden death in dilated cardiomyopathy: systematic review and meta-analysis. JACC Heart Fail 5(1). https://doi.org/10.1016/j.jchf.2016.09.017
Pi SH, Kim SM, Choi JO, Kim EK, Chang SA, Choe YH et al (2018) Prognostic value of myocardial strain and late gadolinium enhancement on cardiovascular magnetic resonance imaging in patients with idiopathic dilated cardiomyopathy with moderate to severely reduced ejection fraction. J Cardiovasc Magn Reson 20(1). https://doi.org/10.1186/s12968-018-0466-7
Zhang K, Wang W, Zhao S, Katz SD, Iervasi G, Gerdes AM et al (2018) Long-term prognostic value of combined free triiodothyronine and late gadolinium enhancement in nonischemic dilated cardiomyopathy. Clin Cardiol 41(1). https://doi.org/10.1002/clc.22858
Seferovic PM, Tsutsui H, Mcnamara DM, Ristic AD, Basso C, Bozkurt B et al (2021) Heart Failure Association, Heart Failure Society of America, and Japanese Heart Failure Society position statement on endomyocardial biopsy. J Card Fail 27(7). https://doi.org/10.1016/j.cardfail.2021.04.010
Evans JP, Skrzynia C, Burke W (2001) The complexities of predictive genetic testing. BMJ 322(7293). https://doi.org/10.1136/bmj.322.7293.1052
Hershberger RE, Givertz MM, Ho CY, Judge DP, Kantor PF, Mcbride KL et al (2018) Genetic evaluation of cardiomyopathy: a clinical practice resource of the American College of Medical Genetics and Genomics (ACMG). Genet Med 20(9). https://doi.org/10.1038/s41436-018-0039-z
Bozkurt B, Colvin M, Cook J, Cooper LT, Deswal A, Fonarow GC et al (2016) Current diagnostic and treatment strategies for specific dilated cardiomyopathies: a scientific statement from the American Heart Association. Circulation. 134(23). https://doi.org/10.1161/CIR.0000000000000455
Ramchand J, Wallis M, Macciocca I, Lynch E, Farouque O, Martyn M et al (2020) Prospective evaluation of the utility of whole exome sequencing in dilated cardiomyopathy. J Am Heart Assoc 9(2). https://doi.org/10.1161/JAHA.119.013346
Minoche AE, Horvat C, Johnson R, Gayevskiy V, Morton SU, Drew AP et al (2019) Genome sequencing as a first-line genetic test in familial dilated cardiomyopathy. Genet Med 21(3). https://doi.org/10.1038/s41436-018-0084-7
Krause A (2019) New genetic testing technologies: advantages and limitations. S Afr Med J 109(4). https://doi.org/10.7196/SAMJ.2019.v109i4.13990
Tschope C, Ammirati E, Bozkurt B, Caforio ALP, Cooper LT, Felix SB et al (2021) Myocarditis and inflammatory cardiomyopathy: current evidence and future directions. Nat Rev Cardiol 18(3). https://doi.org/10.1038/s41569-020-00435-x
Horne BD, Rasmusson KD, Alharethi R, Budge D, Brunisholz KD, Metz T et al (2011) Genome-wide significance and replication of the chromosome 12p11.22 locus near the PTHLH gene for peripartum cardiomyopathy. Circ Cardiovasc Genet 4(4). https://doi.org/10.1161/CIRCGENETICS.110.959205
Morales A, Painter T, Li R, Siegfried JD, Li D, Norton N et al (2010) Rare variant mutations in pregnancy-associated or peripartum cardiomyopathy. Circulation 121(20). https://doi.org/10.1161/CIRCULATIONAHA.109.931220
Sheppard R, Hsich E, Damp J, Elkayam U, Kealey A, Ramani G et al (2016) GNB3 C825T Polymorphism and myocardial recovery in peripartum cardiomyopathy: results of the multicenter investigations of pregnancy-associated cardiomyopathy study. Circ Heart Fail 9(3). https://doi.org/10.1161/CIRCHEARTFAILURE.115.002683
Ware JS, Li J, Mazaika E, Yasso CM, Desouza T, Cappola TP et al (2016) Shared genetic predisposition in peripartum and dilated cardiomyopathies. N Engl J Med 374(3). https://doi.org/10.1056/NEJMoa1505517
Goli R, Li J, Brandimarto J, Levine LD, Riis V, Mcafee Q et al (2021) Genetic and phenotypic landscape of peripartum cardiomyopathy. Circulation 143(19). https://doi.org/10.1161/CIRCULATIONAHA.120.052395
Verdonschot JaJ, Hazebroek MR, Ware JS, Prasad SK, Heymans SRB (2019) Role of targeted therapy in dilated cardiomyopathy: the challenging road toward a personalized approach. J Am Heart Assoc 8(11). https://doi.org/10.1161/JAHA.119.012514
Crasto S, My I, Di Pasquale E (2020) The Broad Spectrum of LMNA Cardiac diseases: from molecular mechanisms to clinical phenotype. Front Physiol 11. https://doi.org/10.3389/fphys.2020.00761
Ollila L, Nikus K, Holmstrom M, Jalanko M, Jurkko R, Kaartinen M et al (2017) Clinical disease presentation and ECG characteristics of LMNA mutation carriers. Open Heart 4(1). https://doi.org/10.1136/openhrt-2016-000474
Repetti GG, Toepfer CN, Seidman JG, Seidman CE (2019) Novel therapies for prevention and early treatment of cardiomyopathies. Circ Res 124(11). https://doi.org/10.1161/CIRCRESAHA.119.313569
Pfizer (2021) A study of ARRY-371797 (PF-07265803) in patients with symptomatic dilated cardiomyopathy due to a lamin A/C gene mutation. Available from: https://www.pfizerclinicaltrials.com/find-a-trial/nct03439514.
Kraus SM, Shaboodien G, Francis V, Laing N, Cirota J, Chin A et al (2021) Rationale and design of the African Cardiomyopathy and Myocarditis Registry Program: the IMHOTEP study. Int J Cardiol 333. https://doi.org/10.1016/j.ijcard.2021.02.026
Peprah E, Wiley K, Sampson U, Narula J (2017) A New Age for African-Driven Genomics Research: Human Heredity and Health in Africa (H3Africa). Glob Heart 12(2). https://doi.org/10.1016/j.gheart.2017.05.003
Acknowledgements
The authors would like to thank Dr Claude Bailly, from the Division of Human Genetics, who assisted with constructing the pedigree included in this manuscript. All figures in this manuscript were created with Biorender software.
Funding
Open access funding provided by University of the Witwatersrand. The Genetics of idiopathic dilated cardiomyopathy in Johannesburg Study is funded by the Carnegie Corporation of New York Grant No. B8749.RO1, Discovery Foundation Academic Fellowship Award, South African Heart Association Research Scholarship Award, and the South African Medical Association PhD Supplementary Scholarship. None of the funders played a role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Author information
Authors and Affiliations
Contributions
NT was involved in the conceptualisation of the review paper, data collection, writing and review of the first draft of the manuscript, and funding acquisition. DM was involved in the data collection and review and editing of the manuscript. MR, AK, QW, and PM reviewed and edited the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethical approval
Ethical approval waived since the article is a narrative review that does not contain identifiable patient data. However, the data depicted in Fig. 2 was collected as part of “The Genetics of idiopathic dilated cardiomyopathy in Johannesburg Study”. The study complied with the Declaration of Helsinki, and informed consent was obtained from the study participants. Approval to conduct the study was granted by the University of the Witwatersrand Human Research Ethics Committee (Ethics Clearance certificate number: M150467).
Competing interests
NT is a cardiologist and has received consultation fees from Acino Health Care Group, AstraZeneca, Boston Scientific, Novartis Pharmaceuticals, Novo Nordisk, Pfizer, Sanofi, Phillips, Servier, Takeda, and Merck. He has also received educational and travel grants from Medtronic, Biotronik, Boston Scientific, and Vertice Health Care Group. None of the other authors has any relevant financial or professional disclosures.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tsabedze, N., Ramsay, M., Krause, A. et al. The genetic basis for adult-onset idiopathic dilated cardiomyopathy in people of African descent. Heart Fail Rev 28, 879–892 (2023). https://doi.org/10.1007/s10741-023-10302-9
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10741-023-10302-9