
Abstract
The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) that causes coronavirus disease 2019 (COVID-19) has infected more than 42.5 million people globally resulting in the death of over 1.15 million subjects. It has inflicted severe public health and economic hardships across the world. In addition to acute respiratory distress syndrome, respiratory failure, sepsis, and acute kidney injury, COVID-19 also causes heart failure (HF). COVID-19-induced HF is manifested via different mechanisms, including, but not limited to, (1) virus-induced infiltration of inflammatory cells, which could impair the function of the heart; (2) pro-inflammatory cytokines (monocyte chemoattractant protein-1, interleukin-1β; interleukin-6; tumor necrosis factor-α) that could cause necrosis and death of the myocardium; (3) endothelial injury coupled with micro-thrombosis which could damage the endocardium; and (4) acute respiratory distress syndrome and respiratory failure that could lead to heart failure due to severe hypoxia. It is concluded that the etiology of COVID-19-induced HF is multifactorial and mitigation of the development of HF in patients with COVID-19 will require different approaches such as social distancing, drug therapy, and the urgent development of a vaccine to eradicate the disease.
Similar content being viewed by others

Introduction
Coronavirus Disease 2019 (COVID-19) caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) was first reported in Wuhan, China, in December 2019 [1]. Since then COVID-19 has spread across the globe and has caused a serious pandemic that has adversely affected the livelihood of millions of people and the economy of many nations. As of October 24, 2020, SARS-CoV-2 has infected 42,584,296 million people with 1,150,769 deaths worldwide [2]. According to the US Center for Disease Control, more than 8,387,047 million American residents carry the disease, which has resulted in 222,447 deaths at the time of revising this review (October 24, 2020) [3].
The virus that causes COVID-19 is transmitted through respiratory aerosols or physical contact with contaminated droplets [4]. SARS-CoV-2 virus then finds its way into the respiratory system, where they induce a cascade of inflammatory reactions [4]. SARS-CoV-2-induced COVID-19 has caused a large variety of complications resulting in high morbidity and mortality. COVID-19-induced pathologies are not limited to the respiratory system alone, but they are also seen in other body systems and organs such as the brain [5], eye [4], gastrointestinal tract [6], renal and urinary structures [7], hemopoietic [8], and cardiovascular [9] system. Recent reports show that COVID-19 causes several cardiovascular complications including myocardial infarction (MI), myocarditis, heart failure (HF), cardiogenic shock, and sudden cardiac death (SCD) [10].
In spite of the fact that COVID-19 is commonly associated with HF in which the heart is unable to pump blood efficiently around the body to meet its demand, the biological mechanisms leading to the development of COVID-19-induced HF are still points of discussion. Knowledge of the possible mechanisms by which COVID-19 patients develop HF would help in the understanding and the management of HF in this cohort of patients.
Methods
The authors review the status of COVID-19-induced HF with emphasis on pathways leading to the development of HF. Data and information were collected from PubMed, Scopus, Centre for Disease Control (USA), Worldometer and Science Direct using the terms “SARS-CoV-2/COVID-19 and heart,” “SARS-CoV-2/COVID-19 and heart failure,” and “SARS-CoV-2/COVID-19 and cardiovascular complications”.
Response to SARS-CoV-2 invasion
Immediately after the invasion of either the conjunctiva, oral, or nasal mucosas by optimal viral load, the following pathophysiological changes occur in the susceptible individual.
-
a
Extensive and generalized SARS-CoV-2 replication. SARS-CoV-2 starts with a fast and expeditious division after entry into host cells via angiotensin-converting enzyme receptor 2 (ACE2) located on cell surfaces in the lung, heart, and epithelium of several other organ systems [9].
-
b
Virus-induced infiltration of the peri-, myo-, and endocardium by inflammatory cells. Immune cells such as neutrophils, pro-inflammatory monocyte/macrophages, and lymphocytes assemble around the areas of myocardium infiltrated by the virus [11, 12]. SARS-CoV-2 particles have been identified in the myocardium of patients with COVID-19 [13]
-
c
Activation of cytokines: neutrophils, pro-inflammatory monocyte/macrophages, and lymphocytes infiltrating the myocardium around the viral (SARS-CoV-2) inclusions release a myriad of cytokines. These cytokines, including but not limited to monocyte chemoattractant protein-1, interleukin-1β, interleukin-6, and tumor necrosis factor-α, create a “cytokine storm” in COVID-19 patients [14].
-
d
Endothelial injury and micro-thrombosis are induced in the body of the victim. Several reports showed that the endothelium of several organs contains ACE2 receptor (SARS-CoV-2 receptor) and invasion of these epithelial membranes by SARS-CoV-2 results in immediate inflammatory response with the activation of the complement and thrombin systems. In addition, the platelet and white blood cells congregate at the site of SARS-CoV-2-induced lesions in the endothelium. All of these pathophysiological reactions culminate in the development of coagulopathy with elevated levels of D-dimer and fibrin degradation products (FDPs) [15] and eventual development of micro-thrombi [16].
-
e
Acute respiratory distress syndrome (ARDS)/respiratory failure is the most lethal insult to the body of the patient. In addition to SARS-CoV-2-induced myocardial lesions, endothelial injury and micro-thrombosis, the impact of poor oxygen supply to the heart cannot be ignored. ARDS/Respiratory failure leads to hypoxia, which in turn could lead to the development of HF. Although it is known that the most common outcome of COVID-19 is respiratory failure [17], the hypoxia induced by respiratory failure may put additional strain on the heart.
Correlation between COVID-19 and heart failure
Myocardial injury is particularly common in patients with COVID-19. In a retrospective study of 191 COVID patients in Wuhan, China, HF is the 4th most common outcome of the disease [17]. In another study of 131 patients who have died of COVID-19, 49% of all deaths were attributed to HF in patients without a previous history of cardiovascular diseases [18]. It is thus evident that after sepsis, respiratory failure including acute respiratory distress syndrome (ARDS), cardiac injury, and HF are the most common sequelae of COVID-19. Since COVID-19 is still evolving, the extent of the degree of cardiac involvement is still elusive. It will take time before we know how common or severe cardiac complications of COVID-19 are.
Putative mechanisms by which COVID-19 induces heart failure
Infiltration of heart by inflammatory cells
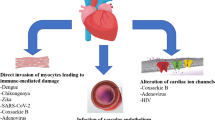
Reports on 61 retrospective cases of children who died of COVID-19 show that SARS-CoV-2 infection is not limited to the lung alone but also observed in the heart. The SARS-CoV-2-induced infection leads to the formation of viral inclusions with subsequent attraction of monocytes/macrophages, neutrophils, and lymphocytes [19, 20]. The virus-induced infiltration by inflammatory cells is associated with edema. All of these factors may have an adverse impact on the function of the heart, resulting in the development of HF. In fact, a virus-induced cell-mediated autoimmune response leads to myocarditis. This type of virus-induced myocarditis causes edema of the myocardial interstitium coupled with necrosis of myocardium and interstitial connective tissue. Myocarditis may progress to dilated cardiomyopathy (DCM) [21, 22]. Moreover, DCM patients who harbor cardiotropic viruses coupled with myocarditis have a much poorer prognosis [23]. Epidemiological studies showed that myocarditis is present in about 20–30% of patient suffering from COVID-19 [24]. Whether HF develops or not depends on the severity of the cardiac lesion, comorbidities, age, and many other factors.
Mitigation of the number and severity of infiltration by pro-inflammatory cells with a reduction in the associated edema may help in the prevention of COVID-19-induced cardiac lesions and HF. This may be effective with the administration of anti-inflammatory drugs and diuretics. Indeed, diuretics are included in the regimen for the treatment of COVID-19 patients [3]. The putative route by which SARS-CoV-2 is transmitted from the air to the lungs and through pulmonary capillaries to the heart is shown in Fig. 1.

This schematic diagram illustrates the passage of SARS-CoV-2 from the air via the nostrils and then into the pulmonary alveoli. From here, the viral particles make their way into the alveolar capillaries and into both the pulmonary and systemic circulations. SARS-CoV-2 particles from coronary arteries get into the myocardium via smaller arteries. SARS-CoV-2 particles are surrounded by inflammatory cells (monocyte/microphage, neutrophils, and lymphocytes). These pro-inflammatory cells release cytokines such as interleukin-1β, interleukin-6 and monocyte chemoattractant protein-1, and tumor necrosis factor-α. LV = left ventricle, RV = right ventricle
Pro-inflammatory cytokines
The body system of SARS-CoV-2-infected individual reacts to viral infiltration by releasing a large variety of cytokines [15, 25]. Monocyte chemoattractant protein-1 is one of the cytokines that increases significantly after the onset of COVID-19. It is also a major regulator for the migration and infiltration of the monocytes/macrophages system [26] to the site of SARS-CoV-2 infection. Therefore, the accumulation of macrophages around viral inclusions may impose a mechanical disruption of the function of the heart.
Another important member of the cytokine family associated with COVID-19 is interleukin-1β, a key regulator of inflammatory response, with the ability to stimulate the release of other cytokines such as IL-17, IL-21, and IL-22. Interleukin-1β and tumor necrosis factor alpha both contribute to pain associated with inflammation via the induction of cyclooxygenase-2 in the nervous system [27]. This may play a role in the severe headache associated with COVID-19 infection. This COVID-19 associated pain, which can occur in joints and as headache, may persist for several weeks after infection [28]. Interleukin-1β also increases cell proliferation and differentiation [29]. The ability of interleukin-1β to promote cell proliferation may contribute to the thickening of the layers of the heart leading to cardiomyopathy. Tumor necrosis factor alpha (TNF-α) is secreted by many cells including macrophage, neutrophil, mast cell and even by cardiomyocytes [30]. TNF-α has been implicated in a variety of inflammatory processes including cell proliferation and fever [31]. These pro-inflammatory cytokines have adverse effect on the outcome of virus-induced infection such as dengue fever [32].
These cytokines can adversely affect the function of the heart by facilitating the infiltration of the myocardium with monocyte/macrophages, neutrophils, and lymphocytes. The deposition of these cells within the myocardium could impair the function of the heart leading to HF. Moreover, the action of the cytokines could also have a direct effect on cardiac function.
Prevention of the inflammation caused by COVID-19 following treatment with either NSAID or specific monoclonal antibodies against these cytokines could help in the prevention of the pathological events leading to cardiac injury and HF. In fact, anti-inflammatory medications [33] and monoclonal antibodies [34] are part of the therapeutic regimens for COVID-19 in addition to other adjunct drugs and anti-viral medications. The putative pathway by which pro-inflammatory cytokines contribute to the development of hypoxia and HF after infection with SARS-CoV-2 is shown in Fig. 2.

This schematic diagram depicts the inclusion of SARS-CoV-2 in the myocardium. Pro-inflammatory cells initially surround SARS-CoV-2 particles to form an inclusion. Thereafter, the inflammatory cells around viral particles release cytokines such as monocyte chemoattractant protein-1 (MCP-1), interleukin-1β, and tumor necrosis factor-α (TNF-α) which act as noxious factors to the heart resulting in hypoxia and if severe, sudden cardiac death
Endothelial injury and micro-thrombosis
In patients who died of COVID-19, capillaritis coupled with micro-thrombosis was observed very early during the course of the disease. Large thrombi, microangiopathy, and signs of disseminated intravascular coagulation were observed in chronic and more severe cases of COVID-19 [16, 35]. The dysfunction of the vascular endothelium in patients with COVID-19 due to coagulopathy and thrombosis may cause myocardial infarction and HF, in addition to loss of function in other organ systems. Other reports show that in addition to coagulopathy, endothelial injury may lead to increased vascular permeability and low nitric oxide level in the internal layer of capillary coat [36]. All of these factors could lead to severe cardiac injury culminating in HF.
Some reports have shown that a combination of thrombolytic (recombinant tissue plasminogen) and anti-inflammatory (tocilizumab) medications can ameliorate the signs and symptoms of COVID-19 [37].
Acute respiratory distress syndrome (ARDS)/Respiratory failure
Patients with ARDS and respiratory failure will have less oxygen to supply the heart. It is imperative therefore that a failing lung will have adverse effect on the function of the heart. In order to save the heart, the lung should also be saved, as these two vital organs depend on each other to function perfectly. Previous reports have shown that the more severe the respiratory failure the more likely the heart will be deprived of oxygen leading to early heart failure [17, 18]. Hypoxia is due to a sequelae of respiratory failure, which in turn is a risk factor for survival in COVID-19 patients [38].
Markers of cardiac function
Biomarkers of cardiac function are crucial to monitoring the progression of injury in the myocardium of COVID-19 patients. One of the most common biomarkers examined is troponin T. The higher the level of troponin T, the higher the risk of mortality in patients suffering from COVID-19 [39]. Troponin T is released into circulation during injury to cardiac myocyte. Cardiomyocytes show deranged myofibrils in a failing heart [40, 41]. Other biomarkers used in the assessment of cardiac function in COVID-19 patients include N-terminal pro-B-type natriuretic peptide, D-dimer, ferritin, interleukin-6, creatinine kinase-myocardial band, and lactate dehydrogenase. These biomarkers correlate well with the level of troponin T. In a study of more than 400 hospitalized patients in Wuhan, China, all of the cardiac biomarkers measured were high in patients with COVID-19. The report also concluded that the higher these values, the higher the risk of HF and sudden cardiac deaths (SCD) [42].
It is therefore important to monitor cardiac functions as early as possible to prevent severe and irreversible myocardial lesions and heart failure.
Conclusion
Cardiac lesions are common complications of COVID-19. They are caused by factors arising from the body’s reaction to SARS-CoV-2 (viral inclusions, infiltrating immune cells, the effect of cytokines released by macrophages, lymphocytes and neutrophils, ARDS/respiratory failure-induced hypoxia, coagulopathy resulting from endothelial injury). The approach to preventing the development of cardiac lesions, HF, and SCD is multifaceted in nature. The first and foremost approach is to prevent the virus from getting into the body either by social distancing or virus elimination by drug therapy or vaccination. A schematic illustration of the putative mechanisms by which COVID-19 induces HF is depicted in Fig. 3.

This schematic diagram illustrates the putative mechanisms by which COVID-19 induces heart failure (HF). SARS-CoV-2 inclusions within the myocardium may cause infiltrative restrictive cardiomyopathy leading to HF. Moreover, pro-inflammatory cells surrounding SARS-CoV-2 inclusions release cytokines such as tumor necrosis alpha (TNF-α), monocyte chemoattractant protein-1 (MCP-1), interleukin-1β (IL-1β), interleukin-6 (IL-6) and many others. These cytokines increase the tissue levels of matrix metalloproteinases (MMPs), protease-activated receptor (PAR), nitric oxide synthase (NOS), severe oxidative stress, apoptosis, and edema in both the heart and lungs. All of these could lead to cardiac and pulmonary fibrosis and necrosis, eventually resulting in HF. In addition, SARS-CoV-2-induced lesion of the cardiovascular endothelium leads to the activation of thrombin and the complement systems. This activation causes disseminated intravascular coagulopathy (DIC) and many thrombi, which could block coronary arteries resulting in myocardial infarction and subsequently HF. Lung fibrosis and hypoxia stimulate further recruitment of pro-inflammatory cells, thereby initiating a vicious cycle
References
Bostanciklioglu M (2020) Severe acute respiratory syndrome coronavirus 2 is penetrating to dementia research. Curr Neurovasc Res. https://doi.org/10.2174/1567202617666200522220509 (Online)
Worldometers.info (2020). COVID-19 Coronavirus pandemic. Dover, Delaware, USA. https://www.worldometers.info/coronavirus/?utm_campaign=homeAdvegas1?(Accessed 24 October, 2020)
Centers for Disease Control (2020). Coronavirus Disease 2019 (COVID-19). https://covid.cdc.gov/covid-data-tracker/#cases_casesper100klast7days (Accessed 24 October 2020)
Güemes-Villahoz N, Burgos-Blasco B, Vidal-Villegas B et al (2020) Novel insights into the transmission of SARS-CoV-2 through the ocular surface and its detection in tears and conjunctival secretions: a review. Adv Ther, 1–10. Advance online publication. https://doi.org/10.1007/s12325-020-01442-7
Le Guennec L, Devianne J, Jalin L et al (2020) Orbitofrontal involvement in a neuroCOVID-19 patient. Epilepsia. Jun 26.https://doi.org/10.1111/epi.16612 (Online)
Kant R, Chandra L, Antony MA, Verma V (2020) Case of COVID-19 presenting with gastrointestinal symptoms. World J Virol 25(9):1–4. https://doi.org/10.5501/wjv.v9.i1.1
Gabarre P, Dumas G, Dupont T, Darmon M, Azoulay E, Zafrani L (2020) Acute kidney injury in critically ill patients with COVID-19. Intensive Care Med 12:1–10. https://doi.org/10.1007/s00134-020-06153-9
Liu X, Zhang R, He G (2020) Hematological findings in coronavirus disease 2019: indications of progression of disease. Ann Hematol 99(7):1421–1428. https://doi.org/10.1007/s00277-020-04103-5
Zheng YY, Ma YT, Zhang JY, Xie X (2020) COVID-19 and the cardiovascular system. Nat Rev Cardiology 17(5):259–260. https://doi.org/10.1038/s41569-020-0360-5
Boukhris M, Hillani A, Moroni F et al (2020) Cardiovascular Implications of the COVID-19 pandemic: a global perspective. Can J Cardiol 16:1068–1080
Gu J, Gong E, Zhang B, Zheng J, Gao Z, Zhong Y et al (2005) Multiple organ infection and the pathogenesis of SARS. J Exp Med 202:415–424
Nicholls JM, Poon LLM, Lee KC, Ng WF, Lai ST, Leung CY et al (2003) Lung pathology of fatal severe acute respiratory syndrome. Lancet 361:1773–1778. https://doi.org/10.1016/S0140-6736(03)13413-7
Tavazzi G, Pellegrini C, Maurelli M (2020) Myocardial localization of coronavirus in COVID-19 cardiogenic shock. Eur J Heart Fail 22:911–915
Huang C, Wang Y, Li X (2020) Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395:497–506. https://doi.org/10.1002/ejhf.1828
Colling ME, Kanthi Y (2020) COVID-19-associated coagulopathy: an exploration of mechanisms. Vasc Med 25(5):471-478 19:1358863X20932640. https://doi.org/10.1177/1358863X20932640. PMID: 32558620; PMCID: PMC7306998 (Online)
McFadyen JD, Stevens H, Peter K (2020) The emerging threat of micro thrombosis in COVID-19 and its therapeutic implications. Circ Res 127(4):571–587. https://doi.org/10.1161/CIRCRESAHA.120.317447
Zhou F, Yu T, Du R (2020) Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet 395:1054–1062
Chen T, Wu D, Chen H et al (2020) Clinical characteristics of 113 deceased patients with coronavirus disease 2019: retrospective study. BMJ Mar 26;368:m1091. https://doi.org/10.1136/bmj.m1091. Erratum in: BMJ. 2020 Mar 31;368:m1295. PMID: 32217556; PMCID: PMC7190011 (Online)
Chen XB, Du SH, Lu JC, Tan XH, Li DR, Yue X, Wang Q, Wang HJ, Qiao DF (2020) Retrospective analysis of 61 cases of children died of viral pneumonia. Fa Yi Xue Za Zhi 36:164–168. English, Chinese. https://doi.org/10.12116/j.issn.1004-5619.2020.02.003
Bavishi C, Bonow RO, Trivedi V, Abbott JD, Messerli FH, Bhatt DL (2020) Acute myocardial injury in patients hospitalized with COVID-19 infection: a review. Prog Cardiovasc Dis Jun 5:S0033–0620(20)30123–7. https://doi.org/10.1016/j.pcad.2020.05.013 (Online)
Imanaka-Yoshida K (2020) Inflammation in myocardial disease: From myocarditis to dilated cardiomyopathy. Pathol Int 70:1–11. https://doi.org/10.1111/pin.12868
Xu HF, Meng L, Yao J, Gu ZY, Liu GQ, Shen YW, Zhao ZQ (2013) Myocardial expression of Spry1 and MAPK proteins of viral myocarditis. Fa Yi Xue Za Zhi 29:164–167
Kažukauskienė I, Baltrūnienė V, Jakubauskas A et al (2020) Prevalence and prognostic relevance of myocardial inflammation and cardiotropic viruses in non-ischemic dilated cardiomyopathy. Cardiol J Jun 22. https://doi.org/10.5603/CJ.a2020.0088 (Online)
Akhmerov A, Marbán E (2020) COVID-19 and the heart. Circ Res 126(10):1443–1455
Guzik TJ, Mohiddin SA, Dimarco A et al (2020) COVID-19 and the cardiovascular system: implications for risk assessment, diagnosis, and treatment options. Cardiovasc Res Apr 30:116(10):1666-1687 cvaa106. https://doi.org/10.1093/cvr/cvaa106 (Online)
Deshmane SL, Kremlev S, Amini S, Sawaya BE (2009) Monocyte chemoattractant protein-1 (MCP-1): an overview. J Interferon Cytokine Res 29:313–326. https://doi.org/10.1089/jir.2008.0027
Chen P, Zhao Y, Gao H, Huang J, Lu Y, Song J, Lin L, Lin Z, Ou C, Sun H, Li Y, Zeng C, Cooper DKC, Zhan Y, Deng X, Mou L (2019) Selective inhibition of cyclooxygenase-2 protects porcine aortic endothelial cells from human antibody-mediated complement-dependent cytotoxicity. Xenotransplantation 26(6):e12536. https://doi.org/10.1111/xen.12536 (Online)
Shiers S, Ray PR, Wangzhou A et al (2020) ACE2 and SCARF expression in human DRG nociceptors: implications for SARS-CoV-2 virus neurological effects. Pain.161(11):2494-2501 https://doi.org/10.1097/j.pain.0000000000002051
Chen H, Wang C, Li J, Huandike M, Liu J, Huang Q, Li X, Zhang H, Zhou J, Chai L (2020) Chinese herbal formula, Huayu Tongbi Fang, Attenuates inflammatory proliferation of rat synoviocytes induced by IL-1β by regulating proliferation and differentiation of T lymphocytes. Evid Based Complement Alternat Med 2020:1706837. https://doi.org/10.1155/2020/1706837 (Online)
Chen Y, Pat B, Zheng J (2010) Tumor necrosis factor-alpha produced in cardiomyocytes mediates a predominant myocardial inflammatory response to stretch in early volume overload. J Mol Cell Cardiol 49(1):70–78
Walsh LJ, Trinchieri G, Waldorf HA, Whitaker D, Murphy GF (1991) Human dermal mast cells contain and release tumor necrosis factor alpha, which induces endothelial leukocyte adhesion molecule 1. Proc Natl Acad Sci USA 88(10):4220–4224. https://doi.org/10.1073/pnas.88.10.4220
Tuyen TT, Viet NT, Hang NT et al (2020) Pro-inflammatory cytokines are modulated in Vietnamese patients with dengue fever. Viral Immunol. May 22. 33(7):514-520 https://doi.org/10.1089/vim.2020.0023
Harikrishnan S, Mohanan PP, Chopra VK et al (2020) Cardiological society of India position statement on COVID-19 and heart failure. Version 2. Indian Heart J. 2020;72(2):75–81.https://doi.org/10.1016/j.ihj.2020.04.012
Atal S, Fatima Z (2020) IL-6 inhibitors in the treatment of serious COVID-19: a promising therapy? Pharmaceut Med 13:1–9. https://doi.org/10.1007/s40290-020-00342-z
Bösmüller H, Traxler S, Bitzer M, Häberle H, Raiser W, Nann D, Frauenfeld L, Vogelsberg A, Klingel K, Fend F (2020) The evolution of pulmonary pathology in fatal COVID-19 disease: an autopsy study with clinical correlation. Virchows Arch 477(3):349–357.https://doi.org/10.1007/s00428-020-02881-x
Amraei R, Rahimi N (2020) COVID-19. Renin-angiotensin system and endothelial dysfunction. Cells 9(7):1652. https://doi.org/10.3390/cells90716521
Papamichalis P, Papadogoulas A, Katsiafylloudis P et al (2020) Combination of thrombolytic and immunosuppressive therapy for coronavirus disease 2019: a case report. Int J Infect Dis 97:90–93. hhttps://doi.org/10.1016/j.ijid.2020.05.118. Epub ahead of print. PMID: 32497796; PMCID: PMC7263262
Mikami T, Miyashita H, Yamada T et al (2020) Risk factors for mortality in patients with COVID-19 in New York City. J Gen Intern Med. Jun 30. https://doi.org/10.1007/s11606-020-05983-z. Epub ahead of print. PMID: 32607928 (Online)
Chapman AR, Bularga A, Mills NL (2020) High-sensitivity cardiac troponin can be an ally in the fight against COVID-19. Circulation 141:1733–1735. https://doi.org/10.1161/CIRCULATIONAHA.120.047008
Adeghate E (2004) Molecular and cellular basis of the aetiology and management of diabetic cardiomyopathy: a short review. Mol Cell Biochem 261:187–191. https://doi.org/10.1023/b:mcbi.0000028755.86521.11
Adeghate E, Singh J (2014) Structural changes in the myocardium during diabetes-induced cardiomyopathy. Heart Fail Rev 19:15–23. https://doi.org/10.1007/s10741-013-9388-5
Shi S, Qin M, Shen B et al (2020) Association of cardiac injury with mortality in hospitalized patients with COVID-19 in Wuhan, China. JAMA Cardiol. Mar 25:e200950. https://doi.org/10.1001/jamacardio.2020.0950 (Online)
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Adeghate, E.A., Eid, N. & Singh, J. Mechanisms of COVID-19-induced heart failure: a short review. Heart Fail Rev 26, 363–369 (2021). https://doi.org/10.1007/s10741-020-10037-x
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10741-020-10037-x