
Abstract
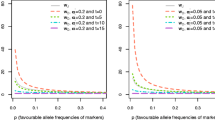
Genomic selection refers to the use of dense markers covering the whole genome to estimate the breeding value of selection candidates for a quantitative trait. This paper considers prediction of breeding value based on a linear combination of the markers. In this case the best estimate of each marker’s effect is the expectation of the effect conditional on the data. To calculate this requires a prior distribution of marker effects. If the marker effects are normally distributed with constant variance, BLUP can be used to calculate the estimated effects of the markers and hence the estimated breeding value (EBV). In this case the model is equivalent to a conventional animal model in which the relationship matrix among the animals is estimated from the markers instead of the pedigree. The accuracy of the EBV can approach 1.0 but a very large amount of data is required. An alternative model was investigated in which only some markers have non-zero effects and these effects follow a reflected exponential distribution. In this case the expected effect of a marker is a non-linear function of the data such that apparently small effects are regressed back almost to zero and consequently these markers can be deleted from the model. The accuracy in this case is considerably higher than when marker effects are normally distributed. If genomic selection is practiced for several generations the response declines in a manner that can be predicted from the marker allele frequencies. Genomic selection is likely to lead to a more rapid decline in the selection response than phenotypic selection unless new markers are continually added to the prediction of breeding value. A method to find the optimum index to maximise long term selection response is derived. This index varies the weight given to a marker according to its frequency such that markers where the favourable allele has low frequency receive more weight in the index.




Similar content being viewed by others


References
Beavis WD (1994) QTL analysis: power, precision and accuracy. In: Paterson AH (ed) Molecular dissection of complex traits. CRC, Boca Raton, pp 145–162
Boichard D, Fritz S, Rossignol MN, Guillaume F, Colleau JJ, Druet T (2006) Implementation of marker assisted selection: practical lessons from dairy cattle. 8th World congress on genetics applied to livestock production, August 13–18, 2006, Belo Horizonte, MG, Brasil
Bouvenhuis H, Weller JI (1994) Mapping and analysis of dairy cattle quantitative trait loci by maximum likelihood methodology using milk protein genes as genetic markers. Genetics 137:267–280
Bulmer MG (1971) The effect of selection on genetic variability. Am Nat 105:201–211. doi:10.1086/282718
Chamberlain AJ, Bowman PJ, Meuwissen THE, McPartlan HM, Goddard ME (2007) Estimating the distribution of QTL effects for milk production traits in dairy cattle. Genetics (submitted)
Dekkers JCM (2004) Commercial application of marker- and gene-assisted selection in livestock: strategies and lessons. J Anim Sci. 82 E-Suppl:E313–E328
Dekkers JCM, van Arendonk JAM (1998) Optimizing selection for quantitative traits with information on an identified locus in outbred populations. Genet Res 71:257–275. doi:10.1017/S0016672398003267
Eyre-Walker A, Keightley PD (2007) The distribution of fitness effects of new mutations. Nat Rev Genet 8:610–618. doi:10.1038/nrg2146
Fernando RL, Gianola D (1986) Optimal properties of the conditional mean as a selection criterion. Theor Appl Genet 72:822–825
Franklin IR (1977) The distribution of the proportion of the genome which is homozygous by descent in inbred animals. Theor Popul Biol 11:60–80. doi:10.1016/0040-5809(77)90007-7
Goddard ME (1983) Selection indices for non-linear profit functions. Theor Appl Genet 64:339–344. doi:10.1007/BF00274177
Goddard ME, Hayes BJ (2007) Genomic selection. J Anim Breed Genet 124:323–330
Goddard ME, Wiggans G (1999) Genetic improvement of dairy cattle. In: Fries R, Ruvinsky A (eds) Genetics of cattle. CAB International, Oxon, UK
Grisart B, Coppieters W, Farnir F, Karim L, Ford C, Berzi P et al (2001) Positional candidate cloning of a QTL in dairy cattle: Identification of a missense mutation in the bovine DGAT1 gene with major effect on milk yield and composition. Genome Res 12:222–231. doi:10.1101/gr.224202
Habier D, Fernando RL, Dekkers JCM (2007) The impact of genetic relationship information on genome-assisted breeding values. Genetics 177:2389–2397
Hayes BJ, Goddard ME (2001) The distribution of the effects of genes affecting quantitative traits in livestock. Genet Sel Evol 33:209–229. doi:10.1051/gse:2001117
Hayes BJ, Chamberlain A, Goddard ME (2006) Use of linkage markers in linkage disequilibrium with QTL in breeding programs. Proc 8th World Congr Genet Appl Livest Prod Belo Horizonte, Brazil
Hayes BJ, Chamberlain AC, McPartlan H, McLeod I, Sethuraman L, Goddard ME (2007) Accuracy of marker assisted selection with single markers and marker haplotypes in cattle. Genet Res 89:215–220. doi:10.1017/S0016672307008865
Hill WG (l982) Predictions of response to artificial selection from new mutations. Genet Res 40:255–278
Hill WG (1993) Variation in genetic composition in backcrossing programs. J Hered 84:212–213
Hill WG, Robertson A (1966) The effects of linkage on limits to artificial selection. Genet Res 8:269–294
Jeon JT, Carlborg O, Torsten A, Giuffra E, Amarger V, Chardon P et al (1999) A paternally expressed QTL affecting skeletal and cardiac muscle in pigs maps to the IGF2 locus. Nat Genet 21:157–158. doi:10.1038/5938
Lande R, Thompson R (1990) Efficiency of marker-assisted selection in improvement of quantitative traits. Genetics 124:734–756
Meuwissen THE, Goddard ME (1996) The use of marker haplotypes in animal breeding. Genet Sel Evol 28:161–176. doi:10.1051/gse:19960203
Meuwissen THE, Goddard ME (1999) Marker assisted estimation of breeding values when marker information is missing on many animals. Genet Sel Evol 31:375–394. doi:10.1051/gse:19990405
Meuwissen THE, Sonesson A (2004) Genotype-assisted optimum contribution selection to maximise response over a specified time period. Genet Res 84:109–116. doi:10.1017/S0016672304007050
Meuwissen THE, Hayes BJ, Goddard ME (2001) Prediction of total genetic value using genome wide dense marker maps. Genetics 157:1819–1829
Moffatt MF, Kabesch M, Liang L, Dixon AL, Strachan D, Heath S et al (2007) Genetic variants regulating ORMDL3 expression contribute to the risk of childhood asthma. Nature 448:470–473. doi:10.1038/nature06014
Muir WM (2007) Comparison of genomic and traditional BLUP-estimated breeding value accuracy and selection response under alternative trait and genomic parameters. J Anim Breed Genet 124:342–355
Sanchez L, Caballero A, Santiago E (2006) Palliating the impact of fixation of a major gene on the genetic variation of artificially selected polygenes. Genet Res 88:105–118. doi:10.1017/S0016672306008421
Schaeffer LR (2006) Strategy for applying genome-wide selection in dairy cattle. J Anim Breed Genet 123:218–223. doi:10.1111/j.1439-0388.2006.00595.x
Stam P (1980) The distribution of the fraction of the genome identical by descent in finite random mating populations. Genet Res 35:131–155
Sved J (1971) Linkage disequilibrium and homozygosity of chromosome segments in finite populations. Theor Popul Biol 2:125–141. doi:10.1016/0040-5809(71)90011-6
Tibshirani R (1996) Regression shrinkage and selection via the Lasso. J Roy Statist Soc Ser B Methodol 58:267–288
Visscher PM, Medland SE, Ferreira MAR, Morley KI, Zhu G, Cornes B et al (2006) Assumption-free estimation of heritability from genome-wide indentity-by-descent sharing between full siblings. PLoS Genet 2:e41. doi:10.1371/journal.pgen.0020041
Wilson T, Wu XY, Juengel JL, Ross IK, Lumsden JM, Lord EA et al (2001) Highly prolific Booroola sheep have a mutation in the intracellular kinase domain of bone morphogenetic protein IB receptor (ALK-6) that is expressed in both oocytes and granulosa cells. Biol Reprod 64:1225–1235. doi:10.1095/biolreprod64.4.1225
Wray NR, Goddard ME, Visscher PM (2007) Prediction of individual risk to disease from genome-wide association studies. Genome Res 17:1520–1528. doi:10.1101/gr.6665407
Zhang X-S, Hill WG (2005) Predictions of patterns of response to artificial selection in lines derived from natural populations. Genetics 169:411–425. doi:10.1534/genetics.104.032573
Acknowledgements
I acknowledge support from the Australian Research Council (grant DP0770096), and thank Bill Hill and a referee for helpful comments.
Author information
Authors and Affiliations
Corresponding author
Appendix 1
Appendix 1
Effective number of Loci (M e )
The equivalence of the animal model and the genomic selection model shows that the ability to estimate the breeding value of animals without a phenotype depends on the variation between pairs of animals in their realised relationship (i.e. the proportion of genes that they share identical by descent). This is so because, if all pairs of animals had the same relationship, all animals without a phenotype would receive the same EBV. For instance, in a population of full sibs, all the relationships based only on the pedigree are 0.5 and all animals receive the same EBV (i.e. the family mean). Therefore we will consider the effect of the number of loci on the variance of the relationship between pairs of animals or pairs of gametes. In the main text, I use a simple model in which the genome consists of M independent loci. In reality, the loci are not independent due to linkage and pedigree relationship, so I will define the effective number of loci (M e ) as the number of independent loci that gives the same variance of realised relationship as obtained in the more realistic situation. This is the same definition as used by Visscher et al. (2006).
Consider a model of the genetic value (g) of gametes (i.e. a haploid model) controlled by M QTL, g = w′a = (x − p)′a where a ~ N(0, I σ 2a ), p i = the allele frequency of the ith locus and x i = 0 or 1 indicating whether the gamete contains allele Q1 or Q2 at the ith locus whose gene effect is a i . The covariance between two gametes j and k is
The variance of this covariance among random pairs of gametes is σ 4a V(w j ′w k ). If gametes j and k are drawn at random from a population with allele frequency of locus i = p i , then for a single locus i, V(w ji * w ki ) = p i 2 (1 − p i )2 with average value across loci = H 2.
If all gametes were equally related by pedigree and if all loci were unlinked, then w ji * w ki would be independent from one locus to another and so V(w j ′w k ) = H 2 M. Therefore
In a more realistic model, with linkage disequilibria, the terms w ji * w ki are not independent from one locus to another. Therefore,
As the number of loci in these sums increases they are dominated by the terms in which i is not equal to l, so we can ignore terms where i = l. The terms such as w ji * w jl would be zero were it not for linkage disequilibrium between loci i and l. In fact, E(w ji * w jl ) = Cov(w i , w l ) = D il , where D il is the conventional measure of LD between loci i and l. Therefore
Franklin (1977) also showed that this summation of all pairs of loci gives the variance of the proportion of the genome that is IBD.
Thus the variance of the relationship, based on a large number of loci, would be zero except for LD. LD and variation in relationship occur due to two reasons. Firstly, even in a random mating population, animals vary in pedigree and some pairs share more recent common ancestors than other pairs. We can evaluate the effect of this on the variance of relationship by dropping the assumption that all gametes have the same pedigree relationship while maintaining the assumption that all loci are unlinked. In a random mating population, the E(D il 2) = p i (1 − p i )p l (1 − p l )/(1 + 4N e c) where N is effective population size and c is the recombination rate between loci i and l (Sved 1971). However, this approximation breaks down for unlinked loci with c = 0.5, because it neglects one generation of recombination which is important when c is so high. A better approximation is E(D il 2) = p i (1 − p i )p l (1 − p l )/(6N) so
where H = the average value of p(1 − p).
Consequently,
Comparing this formula with that for the simplified model,
the effective number of loci due to variation is pedigree is M e = 6N e .
This conclusion can be reached by an alternative argument. The probability that two gametes share a common ancestor in the last generation is 4/(2N e ) because each gamete has two ‘parents’. If two gametes do share a common parent, their relationship is 0.25. This variation between pairs of gametes that share a parent (relationship = 0.25) and pairs that do not (relationship = 0) causes a variance of relationship of 1/(8N e ). The probability that they share a common ancestor in the previous generation is 16/(2N e ) and if they do their relationship is 1/16, so this probability adds 1/(32N e ) to the variance. Adding all previous generation to the variance leads to 1/(6N e ).
The second reason for LD and why pairs of gametes vary in their relationship is linkage. Consider the situation with loci spread continuously along the chromosome, then the sum ΣΣ E(D il 2) can be evaluated as an integral. Using
where c is the distance between loci i and l in Morgan, and c = |x 1 − x 2 |,
where L is the length of the chromosome in Morgans and both integrals are evaluated from 0 to L (Hill 1993). Thus
Comparing this formula with that for the simplified model,
the effective number of loci in a gamete of length L is M e = (2N e L)/log(4N e L).
An alternative way to reach this conclusion is informative. Consider comparing two gametes; trace the first locus of each gamete back to their first common ancestor; the coalescence of a second locus, very close to the first, will be the same, leading to the same common ancestor. However, as you move along the chromosome, a recombination will be reached so that the locus being traced has a different common ancestor to previous loci in these two gametes. By continuing this process along the chromosome one can see that the two gametes can be divided into segments that coalesce. Stam (1980) gives the pdf of the length (x) of these segments as
and the mean length as 1/(4N e ). Thus in a chromosome of length L the average number of segments is 4N e L. However, these segments are not of equal length. If we calculate the probability (two loci chosen at random fall on the same segment), it is approximately log(4N e L)/(2N e L) implying the number of effective segments or loci as M e = 2N e L/log(4N e L) as before. However, the simple number of segments (4N e L) may be important in determining the accuracy of genomic selection and further simulation will be needed to assess this.
Thus, for any two gametes, we can think of the genome as being broken up into Me segments of equal size. This is a consequence of linkage. However, some segments although they coalesce separately, coalesce in the same common ancestor. This happens more often than expected by chance if these two gametes are more related than average i.e. there is a similarity in their pedigrees due to a common ancestor.
The picture of two gametes divided into segments which coalesce without recombining thus explains the two sources of variation in relationship that have been quantified above—variation in pedigree relationship and additional variation in realised relationship due to linkage. If one is predicting the increase in accuracy of EBVs due to markers, above that achieved using the known pedigree, it is the effective number of loci due to linkage that is relevant.
Rights and permissions
About this article
Cite this article
Goddard, M. Genomic selection: prediction of accuracy and maximisation of long term response. Genetica 136, 245–257 (2009). https://doi.org/10.1007/s10709-008-9308-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10709-008-9308-0