
Abstract
Needle nematodes are polyphagous root-ectoparasites parasitizing a wide range of economically important plants not only by directly feeding on root cells, but also by transmitting nepoviruses. This study deciphers the diversity of the complex Longidorus goodeyi through integrative diagnosis method, based on a combination of morphological, morphometrical, multivariate analysis and molecular data. A new Longidorus species, Longidorus panderaltum n. sp. is described and illustrated from a population associated with the rhizosphere of asphodel (Asphodelus ramosus L.) in southern Spain. Morphologically, L. panderaltum n. sp. is characterized by having a moderately long female body (5.2-7.0 mm), lip region bluntly rounded and slightly offset by a depression with body contour, amphidial pouch with slightly asymmetrical lobes, odontostyle 80.5–101.0 µm long, tail short and conoid rounded. Longidorus panderaltum n. sp. is quite similar to L. goodeyi and L. onubensis in major morphometrics and morphology. However, differential morphology in the tail shape of first-stage juvenile, phylogeny and haplonet analyses indicate they are three distinct valid species. This study defines those three species as members of L. goodeyi complex group and reveals the taxonomical complexity of the genus Longidorus. This L. goodeyi complex group demonstrated that the biodiversity of Longidorus in this region is still not fully clarified.
Similar content being viewed by others
Introduction
Needle nematodes of the genus Longidorus Micoletzky, 1922 are a globally important group of ectoparasitic nematodes and considered a major group of plant-pathogens. These nematodes use their needle stylet to feed on the apical root cells inducing galls in the tips and reducing the yield and quality of a range of horticultural or agricultural crops and some of them are recognised as vectors of important nepovirus (Taylor and Brown 1997; Decraemer and Robbins 2007; Palomares-Rius et al. 2017a). Parasitism by Longidorus spp. have a detrimental effect on root growth by inducing hypertrophied uninucleate cells, highly active metabolically, followed by hyperplasia with synchronized cell division (Palomares et al. 2017a). This genus constitutes a great complex group of around 180 valid species (Archidona-Yuste et al. 2019a; Cai et al. 2020a, b; Amrei et al. 2020) and species delimitation is critical from a phytopathological, ecological and biogeographical point of view.
Several Longidorus spp. are distributed worldwide whilst other species have a limited distribution (Brown and Taylor 1987; Robbins and Brown 1991; Sturhan et al. 1997; Doucet et al. 1998; Coomans et al. 2001). Among them, Longidorus goodeyi Hooper, 1961 was described from turf grasses at Rothamsted, UK, and has been reported sporadically in several European countries including Belgium, Bulgaria, France, Germany, Malta, Poland, Portugal, Slovak Republic, Spain, The Netherlands (Lamberti et al. 1982; Arias et al. 1985; Tophan and Alphey 1985; De Waele and Coomans 1990; Lisková and Brown 1998; Gutiérrez-Gutiérrez et al. 2016), and Tadzhikistan (Kankina and Melitskaya 1983). In Spain, Longidorus goodeyi has only been reported in central regions (Arias et al. 1985) associated with cereals and common horehound (Marrubium vulgare L.). Surprisingly, this species was not detected in southern Spain during systematic and exhaustive nematode surveys on several crops and natural environments carried out throughout the last four decades (Arias 1977; Arias et al. 1985; Gutiérrez-Gutiérrez et al. 2011, 2013; Archidona-Yuste et al. 2016b; 2019a, b; Cai et al. 2020a, b).
The morphological convergence among Longidorus spp. and the existence of cryptic species complexes make the accurate identification of species considerably more difficult (De Luca et al. 2004; Gutiérrez-Gutiérrez et al. 2013; Archidona-Yuste et al. 2016b, 2019a; Cai et al. 2020a, b). In the family Longidoridae, morphological and morphometric studies in addition to molecular sequencing have been used simultaneously to group specimens into species, including multivariate analysis using morphometric characters in which a high number of measured individuals were analysed in order to find morphometric differences amongst them (Archidona-Yuste et al. 2016a; Cai et al. 2020b). Also, the utility of DNA barcoding and molecular species delimitation approaches in species discovery and the detection of cryptic lineages into the genus Longidorus have been demonstrated by numerous studies (Ye et al. 2004; Gutiérrez-Gutiérrez et al. 2013; Palomares-Rius et al. 2017c; Archidona-Yuste et al. 2016b, 2019a; Lazarova et al. 2019; Amrei et al. 2020). Specifically, molecular methods using different fragments of nuclear ribosomal DNA (including 28S rRNA, 18S rRNA and ITS), mitochondrial DNA (cytochrome c oxidase subunit I, coxI and nicotinamide dehydrogenase subunit 4. nad4) gene sequences have been used to provide precise identification of species and elucidate the phylogenetic relationships within the genus Longidorus (Ye et al. 2004; Neilson et al. 2004; Palomares-Rius et al. 2008; Gutiérrez-Gutiérrez et al. 2012; Kumari and Subbotin 2012; Subbotin et al. 2014; Archidona-Yuste et al. 2016a, b).
In recent years, several studies have demonstrated that the species diversity within the family Longidoridae remains as a major gap in the biodiversity of soil nematodes, particularly in the Iberian Peninsula which is considered a plausible speciation centre for this family (Coomans 1996; Archidona-Yuste et al. 2016a, b; Cai et al. 2020a, b). This suggests that the continued systematic sampling in unexplored environments in this area could lead to an increase in the overall species richness of this group of nematodes. Following this sampling strategy during the spring of 2019 in southern Spain, we observed a high density of a needle nematode morphologically resembling Longidorus goodeyi suggesting a wider geographical distribution for this nematode species or the occurrence of a new species complex within the genus Longidorus. This fact prompted us to undertake a detailed comparative morphological and molecular study with previous reported data including topotype specimens of this species. In addition, a detailed integrative approach was conducted in order to clarify the taxonomical status of the new nematode population detected where the preliminary results indicated that this population belongs to an unknown Longidorus species and therefore, the existence of a new species complex within this genus. Therefore, the objectives of this study were: (1) to discover the diversity of L. goodeyi complex through integrative taxonomy combining morphological analysis and a species delineation approach based on multivariate morphometric methods and nuclear haplonets tools; (2) to describe a new species of the genus Longidorus which belongs to the L. goodeyi complex group; (3) to characterise molecularly the sampled Longidorus sp. population using the D2-D3 expansion segments of the 28S rRNA gene, ITS1 and partial 18S rRNA gene; and (4) to study the phylogenetic relationships of the identified Longidorus species with available sequenced species.
Material and methods
Nematode population sampling, extraction and morphological identification
Specimens from the population of the unidentified Longidorus species were collected during the spring season of 2019 in a natural pasture of asphodel (Asphodelus ramosus L.) with a stony soil at 1,800 m elevation in La Pandera Mountain, Valdepeñas de Jaén, Jaén province, in Andalusia, southern Spain (Table 1). Soil samples were collected using a shovel, randomly selecting four to five cores, and considering the upper 5–50 cm depth of soil. Nematodes were extracted from a 500-cm3 sub-sample of soil by centrifugal flotation and a modification of Cobb´s decanting and sieving methods (Coolen 1979; Flegg 1967).
Specimens for study using light microscopy (LM) and morphometric studies were killed and fixed in an aqueous solution of 4% formaldehyde + 1% glycerol, dehydrated using alcohol-saturated chamber and processed to pure glycerine using Seinhorst’s method (Seinhorst 1966) as modified by De Grisse (1969). Specimens were examined using a Zeiss III compound microscope with differential interference contrast at magnifications up to 1,000x. Photomicrographs of nematodes were taken by a Nikon DM100 (Nikon, Barcelona, Spain). All measurements were expressed in micrometres (µm). For line drawings of the new species, light micrographs were imported to CorelDraw version X7 and redrawn. All other abbreviations used are as defined in Jairajpuri and Ahmad (1992).
Topotype specimens of L. goodeyi from Rothamsted, UK and a population from Yorkshire, UK were also used for morphological, morphometric and molecular analyses after verifying that their morphology was congruent with that of the original description.
Nematode molecular identification
To avoid mistakes in the case of mixed needle populations in the same sample, three to four live nematodes from each population were temporarily mounted in a drop of 1M NaCl containing glass beads to ensure specimens were not damaged. All necessary morphological and morphometric data by taking pictures and measurements using the above camera-equipped microscope were recorded. This was followed by DNA extraction from single female individuals and polymerase chain reaction (PCR) assays were performed as described by Castillo et al. (2003). The D2-D3 expansion segments of 28S rRNA was amplified using the D2A (5’-ACAAGTACCGTGAGGGAAAGTTG-3’) and D3B (5’-TCGGAAGGAACCAGCTACTA-3’) primers (De Ley et al. 1999). The ITS1 region was amplified using forward primer 18S (5´TTGATTACGTCCCTGCCCTTT-3´) (Vrain et al. 1992) and reverse primer rDNA1 (5´-ACGAGCCGAGTGATCCACCG-3´) (Cherry et al. 1997). The portion of 18S rRNA was amplified using primers 988F (5´-CTC AAA GAT TAA GCC ATG C-3´), 1912R (5´-TTT ACGGTC AGA ACT AGG G-30), 1813F (5´-CTG CGT GAG AGGTGA AAT-3´) and 2646R (50-GCT ACC TTG TTA CGA CTT TT-3´) (Holterman et al. 2006). Finally, the portion of the coxI gene was amplified as described by Lazarova et al. (2006) using the primers COIF (5′-GATTTTTTGGKCATCCWGARG-3′) and COIR XIPHR2 (5′-GTACATAATGAAAATGTGCCAC CWACATAATAAGTATCATG-3′).
PCR cycle conditions for ribosomal genes were: one cycle of 94 °C for 15 min, followed by 35 cycles of 94 °C for 30 s, annealing temperature of 55 °C for 45 s, 72 °C for 3 min, and finally 72 °C for 10 min. The cycle for mtDNA was as described by He et al. (2005): 95 °C for 10 min, five cycles at 94 °C for 30 s, 45 °C for 40 s, and 72 °C for 1 min, and a further 35 cycles at 94 °C for 30 s, 37 °C for 30 s, and 72 °C for 1 min, followed by an extension at 72 °C for 10 min. PCR products were purified after amplification using ExoSAP-IT (Affimetrix, USB products), and used for direct sequencing in both directions using the primers referred above. The resulting products were purified and run on a DNA multicapillary sequencer (Model 3130XL genetic analyser; Applied Biosystems, Foster City, CA, USA), using the BigDye Terminator Sequencing Kit v.3.1 (Applied Biosystems, Foster City, CA, USA), at the Stab Vida sequencing facilities (Caparica, Portugal). The newly obtained sequences were submitted to the GenBank database under accession numbers indicated on Table 1 and the phylogenetic trees.
Recognition of putative species within Longidorus goodeyi complex and species delimitation approach
This species group was identified from previous large-scale taxonomic and phylogenetic studies in the genus Longidorus (Archidona-Yuste et al. 2016b; 2019a; Cai et al. 2020a, b). From the analyses of phylogenetic relationships analyses, a well-supported clade that included the Iberian Peninsula species was identified (clade I; Archidona-Yuste et al. 2019a). Morphological comparison showed that several of the diagnosis characters defining the genus Longidorus (Chen et al. 1997; Loof and Chen 1999; Peneva et al. 2013) were characteristic of the group as a whole, highlighting a hemispherical convex-conoid tail shape. We named the group the L. goodeyi complex, after the oldest described species within the group, and used the main diagnostic features characterizing this species to ascertain morphologically closely related species (viz. L. goodeyi and L. onubensis Archidona-Yuste et al. 2016b). Additional morphological traits were then recognized as diagnostic characters of this nematode complex such as overall nematode size and shape, odontostyle length, location of dorsal and ventrosublateral gland nuclei on the terminal pharyngeal bulb or the lip region shape amongst others (Chen et al. 1997; Loof and Chen 1999; Peneva et al. 2013). The new population of Longidorus sp. detected in this study was also included in this group given the close relationship morphologically with L. goodeyi as outlined above. An iterative analysis of morphometric and molecular data using two independent strategies of species delimitation was utilised to asses described and undescribed specimens and to determine species boundaries within this newly-defined species complex.
Species delineation using morphometry was conducted with principal component analysis (PCA) in order to estimate the degree of association among species within the L. goodeyi-complex (Legendre and Legendre 2012). PCA was based upon the following morphological characters: L (body length), the ratios a, c, c', d, d', V, odontostyle and odontophore length, lip region width and hyaline region length (Table 2, Archidona-Yuste et al. 2016a, b; Jairajpuri and Ahmad 1992). Prior to the statistical analysis, diagnostic characters were tested for collinearity (Zuur et al. 2010). We used the collinearity test based on the values of the variance inflation factor (VIF) method that iteratively excludes numeric covariates showing VIF values > 10 as suggested by Montgomery and Peck (1992). PCA was performed by a decomposition of the data matrix amongst populations using the principal function implemented in the package ‘psych’ (Revelle 2019). Orthogonal varimax raw rotation was used to estimate the factor loadings. Only factors with sum of squares (SS) loadings > 1 were extracted. Finally, a minimum spanning tree (MST) based on the Euclidean distance was superimposed on the scatter plot of the L. goodeyi-specimens complex against the PCA axes. MST was performed using the ComputeMST function implemented in the package ‘emstreeR’ (Quadros 2019). All statistical analyses were performed using the R v. 3.5.1 freeware (R Core Team 2019).
Species delineation based on molecular data was performed using nuclear haplonet tools in order to determine species boundaries and to clarify putative molecular species within L. goodeyi complex. Haplotype network was constructed to each of the two separated datasets, i.e. the nuclear 28S region and the ITS1 region. Alignments were converted to the NEXUS format using DnaSP V.6 (Rozas et al. 2017); and TCS networks (Clement et al. 2002) were applied in the program PopART V.1.7 (http://popart.otago.ac.nz). In this case, no heterozygous individuals were found in nuclear 28S and ITS1 sequences, so haploweb was not suitable for analysis and individuals were simply classified as haplogroups.
Phylogenetic analyses
D2-D3 expansion segments of 28S rRNA, ITS1, partial 18S rRNA and ITS1 rRNA sequences of the unidentified Longidorus species, L. onubensis and L. goodeyi populations were obtained in this study. These sequences and other sequences from species of Longidorus spp. from GenBank were used for phylogenetic analyses. Outgroup taxa for each dataset were chosen following previously published studies (He et al. 2005; Holterman et al. 2006; Gutiérrez-Gutiérrez et al. 2013; Archidona-Yuste et al. 2019a; Cai et al. 2020a, b; Radivojevic et al. 2020). Multiple sequence alignments of the different genes were made using the FFT-NS-2 algorithm of MAFFT V.7.450 (Katoh et al. 2017). Sequence alignments were visualised using BioEdit (Hall 1999) and edited by Gblocks ver. 0.91b (Castresana 2000) in Castresana Laboratory server (http://molevol.cmima.csic.es/castresana/Gblocks_server.html) using options for a less stringent selection (minimum number of sequences for a conserved or a flanking position: 50% of the number of sequences + 1; maximum number of contiguous non-conserved positions: 8; minimum length of a block: 5; allowed gap positions: with half). Phylogenetic analyses of the sequence datasets were based on Bayesian inference (BI) using MrBayes 3.2.7a (Ronquist et al. 2012). The best-fit model of DNA evolution was obtained using JModelTest V.2.1.7 (Darriba et al. 2012) with the Akaike Information Criterion (AIC). The best-fit model, the base frequency, the proportion of invariable sites, and the gamma distribution shape parameters and substitution rates in the AIC were then given to MrBayes for the phylogenetic analyses. Unlinked general time-reversible model with invariable sites and a gamma-shaped distribution (GTR + I + G) for the D2-D3 expansion segments of 28S rRNA and the partial 18S rRNA and a transitional model with a gamma-shaped distribution (TIM2 + G) for the ITS1 region. These BI analyses were run separately per dataset using four chains for 2 × 106 generations for each molecular marker. The Markov Chains were sampled at intervals of 100 generations. Two runs were conducted for each analysis. After discarding burn-in samples and evaluating convergence, the remaining samples were retained for further analyses. The topologies were used to generate a 50% majority-rule consensus tree. Posterior probabilities (PP) are given on appropriate clades. Trees from all analyses were visualised using FigTree software V.1.4.4 (http://tree.bio.ed.ac.uk/software/figtree/).
Results and descriptions
Species delimitation was carried out using two independent methods based on morphometric (multivariate analysis) and molecular data using ribosomal sequences (haplonet). Multivariate morphometric and haplonet methods were performed on the studied populations to verify species identifications. The integration of this procedure with the analysis of nematode morphology allowed us to verify Longidorus panderaltum n. sp. as valid new species within the L. goodeyi complex. Additionally, we maintained a consensus approach for the different species delimitation methods, including concordant results in phylogenetic trees inferred from nuclear markers and/or different morphological or morphometric characteristics.
Multivariate morphometric analyses of Longidorus goodeyi complex
In the principal component analysis (PCA), the first two components (sum of squares (SS) loadings > 1) accounted for 73.20% of the total variance in the morphometric characters of the L. goodeyi-complex (Table 2). Table 2 includes the SS loadings for the three extracted factors, which were a linear combination of all characters in the analysis. The eigenvectors for each character were used to interpret the biological meaning of the factors. First, principal component 1 (PC1) was mainly dominated by nematode body length and the a ratio with a high positive weight (eigenvector = 0.418 and 0.449), relating this component with the overall nematode size and shape. PC2 was mainly dominated by high positive weight for the lip region width and the c’ ratio (eigenvectors = 0.510 and 0.523, respectively) as well as similar but negative weight for the c ratio (eigenvector = -0.535) (Table 2). This component was therefore related with the lip and tail shape. Overall, these results suggest that all of the extracted PCs were related to the overall size and shape of nematode populations. The results of the PCA were represented graphically in Cartesian plots in which populations of the L. goodeyi-species complex were projected on the plane of the x- and y-axes, respectively, as pairwise combination of PC1 and 2 (Fig. 1). In the graphic representation of the L. goodeyi complex, the specimens of L. goodeyi and L. panderaltum n. sp. were projected showing a clustered distribution pattern owing to their low morphometric variation within population (Tables 3 and 4). However, the specimens of L. onubensis were projected showing an expanded distribution owing to the wide morphometric variation detected in this species (Archidona-Yuste et al. 2016b) (Fig. 1). We observed that all species were clearly separated amongst them, being this spatial distribution dominated by the two extracted principal components (PC1 and 2, 73.20% of the total of variance) (Table 2 and Fig. 1). The spatial separation dominated by PC1 grouped species according to the nematode body length and the maximum body width as derived by the ratio a (Table 2). Thus, L. panderaltum n. sp. specimens having shorter and wider nematode body were located at the left side, and on the opposite side was L. onubensis, which is characterized by longer and narrower nematode body (Fig. 1). However, specimens of L. goodeyi were located in the middle part of the plane and clearly grouped among the specimens of L. panderaltum n. sp. and L. onubensis, having an intermediate nematode body length and maximum body width between these two species (Fig. 1). Likewise, the spatial separation dominated by PC2 grouped species according to the lip region width and female tail length as derived by the ratios c and c’ (Table 2). In this case, L. goodeyi specimens having a longer tail and wider lip region were located on the top side (above y = 0), clearly separating from the specimens of L. panderaltum n. sp. and L. onubensis which showed similar values for these diagnostic characters (Fig. 1; Table 3; Archidona-Yuste et al. 2016b). A minimum spanning tree (MST) superimposed on the plot of the first two principal components showed that the morphometric variation of the L. goodeyi-specimens complex located L. goodeyi as link connecting for L. panderaltum n. sp. and L. onubensis, indicating a wider morphological separation between these last two species (Fig. 1). These results support the denomination of this species complex using L. goodeyi not only because it is the oldest described species but also for its central position in the morphometric variation of this species complex.

Principal component on morphometric characters to characterize Longidorus goodeyi-complex with a superimposed minimum spanning tree (based on Euclidean distance).
Haplotype network analyses
Species delimitation using haplonet methods in L. goodeyi-complex species revealed that the 28S rRNA and ITS1 alignments contained 24 and 17 sequences with three and 6 different haplotypes, respectively (Fig. 2). The TCS haplotype analysis inferred from the nuclear 28S region showed three well-differentiated haplogroups, corresponding to three different main lineages (clade I-III) (Fig. 2). Twelve 28S sequences of L. panderaltum n. sp. were refined to one haplotype (28S-pan1), and only one haplotype (28S-goo1) was found from three populations of L. goodeyi (Yorkshire: MT271709-MT271710, Peebles: AY601581 and Topotype: MT271706-MT271708). Six 28S sequences from L. onubensis also showed one haplotype (28S-onu1). Moreover, for L. panderaltum n. sp. six ITS1 sequences were classified into one haplotype (I-pan1). L. goodeyi from Rothamsted, UK (topotypes) and Yorkshire, UK were divided into two different haplotypes (Table 1, I-goo1 and I-goo2, respectively). Longidorus onubensis was collected in one sampling spot, but three haplotypes were found (I-onu1, I-onu2, and I-onu3). Therefore, the 28S rRNA and ITS1 haplonets clearly resolved L. panderaltum n. sp., L. goodeyi and L. onubensis as separate and genetically isolated lineages. Besides, no heterozygous individuals were found in nuclear 28S and ITS sequences, so haploweb was not suitable for analysis and individuals were simply classified as haplogroups.

Haplonets for L. goodeyi species-complex. A 28S rRNA haploweb of Longidorus panderaltum n. sp. with close related species, B ITS1 haploweb of Longidorus panderaltum n. sp. with close related species. Coloured circles represent haplotypes and their diameters are proportional to the number of individuals sharing the same haplotype. Black short lines on the branches indicate the number of mutated positions in the alignment that separate each haplotype. Co-occurring haplotypes are enclosed in dashes.
Difficulties to obtain coxI sequences from L. goodeyi prevent to carry out this analysis for mitochondrial genes.
Longidorus panderaltumFootnote 1n. sp. (Figs. 3, 4, and 5, Table 3)

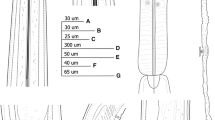
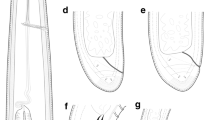
Line drawings of Longidorus panderaltum n. sp. from the rhizosphere of asphodel (Asphodelus ramosus L.) from Valdepeñas de Jaén, Jaén, Spain (A-G). A: Pharyngeal region of holotype; B, C: Lip region of paratypes; D: Tail region of holotype; E, F: Tail region of paratypes; G: Tail of first-stage juvenile paratype (J1).

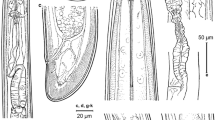
Light micrographs of Longidorus panderaltum n. sp. female specimens from the rhizosphere of asphodel (Asphodelus ramosus L.) from Valdepeñas de Jaén, Jaén, Spain (A-S). A-B: Female anterior regions; C-F: Lip region of paratypes showing amphidial fovea (arrowed); G, H: Detail of basal bulb of paratypes; I: Vulval region of holotype; J: First-stage juvenile anterior region; K: Tail region of holotype; L-N, S: Tail regions of paratypes; O-R: Tail of J1, J2, J3 and J4, respectively. Abbreviations: a = anus; af = amphidial fovea; dn = dorsal nucleus; gr = guiding ring; Rost = replacement odontostyle; svn = subventral nucleus; V = vulva. (Scale bars: A, B = 30 µm; C-H, J-S = 15 µm; H = 50 µm)

Relationship of body length to length of functional and replacement odontostyle (=Odontostyle and =Replacement odontostyle) length in all developmental stages from first-stage juveniles (J1) to mature females of Longidorus panderaltum n. sp.
Female
Body long, slightly tapering towards anterior end, open C-shape when heat relaxed. Cuticle 3.4 ± 0.7 (2.5–4.5) µm thick at mid body, and 12.8 ± 1.8 (11.0-16.5) µm thick at tail hyaline part. Lip region bluntly rounded, one sixth of mid-body-width wide, and slightly offset by a depression with body contour. Amphidial aperture pore-like and amphidial fovea more or less pouch-shaped and almost slightly asymmetrically bilobed, lobes slightly unequal in length, and extending about 1/2 part of oral aperture-guiding ring distance. Guiding ring single, located approximately ca. 2 times the lip region width from anterior end. Odontostyle moderately long, 1.5 times as long as odontophore, straight or slightly arcuate. Odontophore weakly developed, with rather weak swollen base. Pharynx short, 495.5 ± 52.7 (430.0-651.0) µm, extending to the terminal pharyngeal bulb with one dorsal nucleus (DN) and two ventrosublateral gland nuclei (SVN) separately located at 42.2 ± 5.6 (36.0-46.7) %, 56.1 ± 7.6 (49.2–64.3) % and 52.9 ± 4.1 (50.0-55.8) % of distance from anterior end of pharyngeal bulb (Loof and Coomans 1972). Glandularium 119.5 ± 7.8 (114.0-125.0) µm long. Reproductive system didelphic-amphidelphic, anterior branch 709.3 ± 34.2 (672.0-739.0) µm long and posterior branch 691.7 ± 30.0 (667.0-725.0) µm long, each branch composed of a reflexed ovary 225–311 µm long. Pars dilatata oviductus and uterus of about equal length (225.0-311.0 µm long), separated by a strong and muscularized sphincter. Vulva slit-like, situated at 51.8–57.7% of body length. Vagina perpendicular to body axis, 34.9 ± 1.2 (33.5–36.5) µm long, occupying 32.2–35.8% of corresponding body diameter and surrounded by well-developed constrictor muscles; pars distalis vaginae 13.5–16.0 µm long, pars proximalis vaginae measuring 16.5–20.0 × 19.5–22.0 µm. Prerectum 394.0 ± 118.3 (272.0-533.0) µm long, rectum 31.1 ± 5.6 (23.5–38.0) µm long. Tail short and conoid rounded, with two or three pairs of caudal pores present on each side.
Male
Not found.
Juveniles
Four developmental juvenile stages were detected based on body, odontostyle and replacement odontostyle length, and tail shape (Figs. 4 and 5). Habitus more or less an open C-shape. Morphologically similar to female, except for their size and sexual characteristics (Table 3). The 1st-stage juveniles were characterized by the replacement odontostyle inserted into odontophore base with a tail convex-conoid shape with a short subdigitate terminus (Figs. 3 and 4). J1-J4 tail length 28.2, 30.0, 33.8, and 30.8 µm long, respectively.
Type habitat and locality
Rhizosphere of Asphodel (Asphodelus ramosus L.) at 1,858 m elevation in La Pandera Mountain, Valdepeñas de Jaén, Jaén province, in Andalusia, southern Spain (GPS coordinates: 37º 37' 56.31" N; 3°46′24.57″W) collected by R. Cai on June 9, 2019.
Type material and nomenclatural registration
Holotype female (slide PAND-02) and two female paratypes (PAND-03-PAND-06) were deposited in the Nematode Collection of Institute for Sustainable Agriculture, IAS-CSIC, Córdoba, Spain. One female paratype deposited at each of the following collections: Istituto per la Protezione delle Piante (IPP) of Consiglio Nazionale delle Ricerche (C.N.R.), Sezione di Bari, Bari, Italy (PAND-07); USDA Nematode Collection (T-7367p); and Nematode Collection of Zhejiang University, Hangzhou, China (ZJU-28-1). Specific D2-D3, ITS1, partial 18S rRNA, and partial coxI sequences deposited in GenBank with accession numbers MT271694-MT271705, MT271721-MT271726, MT271715-MT271716, and MT270783-MT270786, respectively.
The new species binomial has been registered in the ZooBank database (zoobank.org) under the identifier: urn:lsid:zoobank.org:act:A78BFC45-8FA4-4704-B918-73782B034B7A. The LSID for the publication is: urn:lsid:zoobank.org:pub:667C796E-13BF-4147-BFDB-C8A80DAFE391.
Diagnosis and relationships
Longidorus panderaltum n. sp. is an apomictic species characterized by a moderately long body (5.2-7.0 mm); lip region 15.5–18.0 µm wide and slightly marked by depression from body contour; amphidial fovea pouch-shaped, slightly asymmetrically bilobed, and extending about 1/2 part of oral aperture-guiding ring distance; relatively long odontostyle (80.5–101.0 µm), and total stylet (144.0-164.0 µm); guiding ring located at 30.0-34.5 µm from anterior end; vulva located at 51.8–57.7% of body length; female tail short and bluntly conoid (28.0-35.5 µm long, c’ = 0.6–0.7), with two or three pairs of caudal pores. No males found. Four developmental juvenile stages were identified, the tail of the first-stage juvenile convex-conoid shape with a short subdigitate terminus (c’ = 1.3–1.5); J2 characterised by a conoid-rounded tail, curved dorsally and slightly concave ventrally with a dorsal-ventral depression at hyaline region level; J2-J4 tail similar to that of female but becoming progressively shorter and stouter in each moult. According to the polytomous key by Chen et al. 1997, supplement by Loof and Chen 1999 and the addition of some characters by Peneva et al. 2013, codes for the new species are (codes in parentheses are exceptions): A3(24)-B3-C3(2)-D3-E2-F3-G1-H1-I1-J1-K4; and specific D2-D3 expansion segments of 28S rRNA, ITS1, partial 18S-rRNA and partial coxI sequences.
According to the body and odontostyle length, distance of guiding ring from anterior body end, lip region and tail shape (in this order of main features), the new species is most similar to L. goodeyi and L. onubensis Archidona-Yuste et al. 2016b. The new species differs from paratypes and topotypes of L. goodeyi by having a lower a ratio (48.7–63.6 vs 67.0-117.0, 60.4–79.5, respectively), higher c ratio (164.5-235.8 vs 99.0-154.0, 123.7-172.9, respectively), lower c’ ratio (0.6–0.7 vs about 1.0, 0.8–0.9, respectively), amphids shape and size (slightly asymmetrically bilobed, lobes slightly unequal in length, and extending about 1/2 part of oral aperture-guiding ring distance vs large, asymmetrically bilobed, ventral lobe twice the length of dorsal lobe and reaching to guide ring), a slightly shorter odontostyle (80.5–101.0 µm vs 96.0-109.0 µm), and the tail of the 1st -stage juvenile convex-conoid shape with a short subdigitate terminus vs elongate digitate terminus (J1 c’ ratio, 1.3–1.5 vs 3.0, 2.1–2.9, respectively). From L. onubensis, it can be separated by shorter body length (5.2-7.0 mm vs 7.4–9.6 mm), lower a ratio (48.7–63.6 vs 75.9-107.5), shorter odontostyle length (80.5–101.0 µm vs 103.0-121.0 µm), and the absence vs presence of males.
Longidorus goodeyi Hooper 1961 (Fig. 6, Table 4)

Light micrographs of Longidorus goodeyi Hooper 1961 female topotypes from the rhizosphere of perennial ryegrass (Lolium perenne L.) from Rothamsted Research, Harpenden, UK (A-P). A-D: Female anterior regions; E, F: Detail of basal bulb; G: Vulval region; H-J: Female tail regions; K-M: Tail region of J1; N-P: Tail region of J2, J3 and J4, respectively. Abbreviations: a = anus; af = amphidial fovea; dn = dorsal nucleus; gr = guiding ring; svn = subventral nucleus. (Scale bars: A-D = 15 µm; E-P = 25 µm)
The studied topotype population from turfgrass at Rothamsted agree closely with original description (Hooper 1961) in morphology and morphometry, except for only minor differences in a and c ratios (72.7 (60.4–79.5), 143.4 (123.7-172.9) vs 58.1 (48.7–63.6), 193.9 (164.5-235.8), respectively), which may be due to intraspecific variability (Fig. 6, Table 4). This population was characterized by a lip region bluntly rounded, one fifth of mid-body-width wide, a large amphid fovea asymmetrically bilobed, ventral lobe twice the length of dorsal lobe and reaching to guide ring (Fig. 6). Reproductive system amphidelphic, with both branches equally developed (G1 = 9.2–12.7, G2 = 8.4–12.9), each composed of a reflexed ovary, oviductus with a well-developed pars dilatata oviductus, tubular uterus, vagina 44.0 to 47.0 µm or 42 to 53% of the corresponding body width, pars distalis ca. 22.0 µm long, pars proximalis vaginae about as high as its width (19.0–28.0 µm long, 27.0–36.0 µm wide), and a transverse slit vulva. Tail dorsally convex, bluntly conoid, about as long as anal-body-width.
According to the polytomous key by Chen et al. (1997), the supplement by Loof and Chen (1999), and additional codes (Peneva et al. 2013; Archidona et al. 2016b), this species has the following codes (codes in parentheses are exceptions): A3(4)-B3-C3-D2-E3-F3(4)-G1(2)-H1-I1-J1–K7.
Molecular characterization and phylogenetic relationships of Longidorus panderaltum n. sp.within the genus Longidorus
The amplification of D2-D3 segments of 28S rRNA, ITS1, 18S rRNA, and partial coxI regions yielded single fragments of ca 900 bp, 1100 bp and 1800 bp, and 500 bp, respectively, based on gel electrophoresis.
The D2-D3 sequence divergence was significant between L. panderaltum n. sp. (MT271694-MT271705) with other congeneric species, except for L. goodeyi showing 98.5% similarity (AY601581, 10 different nucleotides and 0 indels), but 96.4% similarity with L. crataegi Roca and Bravo, 1996 (JX445114, 28 different nucleotides, 7 indels), and 96.2% with L. onubensis (KT308857, 29 different nucleotides and 8 indels). No intraspecific variation for D2-D3 expansion segments was detected in L. panderaltum n. sp. (MT271694-MT271705), L. goodeyi (MT271706-MT271710) and L. onubensis (MT271711-MT271714, KT308857-KT308858). The ITS1 of L. panderaltum n. sp. (MT271721-MT271726) showed 94.2–94.0% similarity with L. goodeyi topotype and Yorkshire populations (MT271727-MT271732, 54–57 different nucleotides and 24 indels), 85.5% similarity with L. vinearum Bravo and Roca, 1995 (KT308892, 145 different nucleotides and 56 indels) and L. onubensis (MT271733-MT271735, KT308882-KT308883, 143 different nucleotides and 51 indels). No intraspecific variation for ITS1 was observed in this population of L. panderaltum n. sp., whereas, L. goodeyi topotype and Yorkshire populations differed in 5 nucleotides and no indels. The 18S rRNA sequences of L. panderaltum n. sp. (MT271715-MT271716) closely matched with several species, such as L. goodeyi (MT271717-MT271720), L. wicuolea Archidona-Yuste et al. 2016b (KT308900) and L. cf. olegi (MH430010). Intraspecific variation for 18S rRNA was only for 2 nucleotides and 0 indels in L. goodeyi topotype and Yorkshire populations. Finally, the partial coxI sequences of L. panderaltum n. sp. (MT270783-MT270786) showed 80.3% similarity with L. onubensis (KY816695, 67 different nucleotides and 0 indels) and 80.1% similarity with L. vinearum (KY816713, 68 different nucleotides and 2 indels). No intraspecific variation for coxI sequences of L. panderaltum n. sp. was observed. Unfortunately, repeated difficulties were experienced with the partial coxI sequences from L. goodeyi and it not could be sequenced.
Phylogenetic relationships among Longidorus species inferred from analyses of D2-D3 expansion segments of 28S rRNA, partial 18S rRNA, and ITS1 gene sequences using BI are given in Figs. 7, 8 and 9, respectively. Since scarce similarity was detected for ITS1 sequences from L. panderaltum n. sp. (MT271721-MT271726) and L. goodeyi (MT271727-MT271732) with those deposited in GenBank, only closer species were included in the analyses of this region.

Phylogenetic relationships of Longidorus panderaltum n. sp. and Longidorus goodeyi within the genus Longidorus. Bayesian 50% majority rule consensus trees as inferred from D2-D3 expansion segments of 28S rRNA sequences alignments under the GTR + I + G model. Posterior probabilities more than 70% are given for appropriate clades. Newly obtained sequences in this study are in bold.

Phylogenetic relationships of Longidorus panderaltum n. sp. and Longidorus goodeyi with closer species within the genus Longidorus. Bayesian 50% majority rule consensus trees as inferred from ITS1 rRNA sequences alignments under the TIM2 + G model. Posterior probabilities more than 70% are given for appropriate clades. Newly obtained sequences in this study are in bold.

Phylogenetic relationships of Longidorus panderaltum n. sp. and Longidorus goodeyi within the genus Longidorus. Bayesian 50% majority rule consensus trees as inferred from 18S rRNA sequences alignments under the GTR + I + G model. Posterior probabilities more than 70% are given for appropriate clades. Newly obtained sequences in this study are in bold.
The D2-D3 expansion segments of 28S rRNA tree of Longidorus spp. based on a multiple edited alignment including 121 sequences and 727 total characters revealed 5 major clades (marked with roman numerals from I to V) (Fig. 7). Longidorus panderaltum n. sp. (MT271694-MT271705) and L. goodeyi (MT271706-MT271710) clustered in a moderately supported clade (PP = 0.94) with L. crataegi (JX445114), together with a moderately supported clade (PP = 0.85) by L. onubensis (MT271711-MT271714, KT308857-KT308858) (Fig. 7). Clade I was well-supported (PP = 1.00) including 33 species, comprising the three species of L. goodeyi complex and other species mainly described or reported from the Iberian Peninsula (Fig. 7). Clade I species shared some morphological traits such as a hemispherical to bluntly conoid tail (Fig. 7) (c’< 1.0, except some species as L. wicuolea and L. andalusicus Gutiérrez-Gutiérrez, Cantalapiedra-Navarrete, Montes-Borrego, Palomares-Rius and Castillo, 2013 with c’ 1.0-1.5), and lip region anteriorly rounded, continuous or slightly depressed with body contour (Fig. 7) (except L. oleae with an anteriorly concave lip region and L. lusitanicus Macara, 1985 with an anteriorly flattened lip region).
For the ITS1 region sequences, the 50% majority rule consensus BI tree of a multiple sequence alignment containing 23 sequences and 964 characters is showed in Fig. 8. Longidorus panderaltum n. sp. (MT271721-MT271726) clustered with L. goodeyi (MT271727-MT271732) in a high supported clade (PP = 1.00), and forming a well-supported major clade (PP = 0.99) with L. vinearum (KT308892-KT308893), L. magnus Lamberti, Bleve-Zacheo and Arias, 1982 (HM921340) and L. lusitanicus (KT308891), whereas L. onubensis (MT271733-MT271735, KT308882-KT308883) clustered separately in a high supported clade (PP = 1.00). Finally, for partial 18S rRNA gene sequences, the 50% majority rule consensus BI tree was based on a multiple sequence alignment containing 89 sequences and 1681 characters. Longidorus panderaltum n. sp. (MT271715-MT271716) clustered with L. goodeyi (MT271717-MT271720) in a well-supported clade (PP = 1.00). However, L. onubensis (KT308897) did not cluster with other species of L. goodeyi complex (Fig. 9), but clustered inside a major highly supported clade (PP = 1.00) including other species from the Iberian Peninsula.
Discussion
The primary objective of this study was to unravel the diversity of the L. goodeyi complex by using integrative approaches including morphology, morphometry and molecular analyses. Our results confirmed that the needle nematode L. goodeyi it is composed by a complex group of three distinct species (L. goodeyi, L. onubensis and L. panderaltum n. sp.) that can be separated using integrative approaches. Consequently, we have described a new species of the genus Longidorus that can be separated from L. goodeyi complex by a combination of morphological, allometrical, and molecular analyses. Our results demonstrated that the use of integrative taxonomy may help to distinguish very similar species and to unravel the biodiversity in this complex group of plant-parasitic nematodes. These analyses supported the separation of L. panderaltum n. sp. from other species in the L. goodeyi complex and reinforced the importance of these analyses to decipher species boundaries that are essential for agronomic management, ecological analyses and implications in food security. Our results suggested that other populations of L. goodeyi reported from central Spain (Arias 1977; Arias et al. 1985), from The Netherlands that differed from the type description in some morphological characters (Seinhorst and Van Hoof 1981), or those from France, that were smaller than that of paratypes (Dalmasso 1969), need to be confirmed by integrative taxonomy in order to clarify their identity. The application of multivariate analysis may help to differentiate these closely related species. In particular, detailed morphology of J1 tail shape of these L. goodeyi populations may also help to clarify the identification of these records. In Longidoridae, J1 individuals can be identified by the position of the replacement odontostyle, which lies mostly within the odontophore, with the anterior tip near the base of the functional odontostyle, and have practical significance when distinguishing closely related species (Hunt 1993; Robbins et al 1996).
Phylogenetic inferences based on the D2-D3 expansion domains of 28S, ITS1 and 18S rRNA genes suggest that L. panderaltum n. sp. and L. goodeyi are closely related species (Figs. 7, 8 and 9). Results of all analyses on the three species of L. goodeyi complex were consistent and clearly separated them by phylogenetic and species delimitation methods. In all cases these species clustered in a major clade comprising the majority of Longidorus species reported in the Iberian Peninsula with a characteristic tail (hemispherical convex-conoid tail shape) as reported in previous studies (Gutiérrez-Gutiérrez et al. 2013; Archidona-Yuste et al. 2016b; 2019a; Cai et al. 2020a, b). Sequences of nuclear ribosomal RNA genes, particularly D2-D3 expansion segments of the 28S rRNA gene and ITS1 region, have proven to be a powerful tool for providing accurate species identification of Longidoridae (Palomares-Rius et al. 2017b; Archidona-Yuste et al. 2016b; 2019a; Cai et al. 2020a, b). By contrast, the low nucleotide variability found in partial 18S rRNA makes it difficult to identify individuals to the species level as previously described in other studies (Gutiérrez-Gutiérrez et al. 2013; Archidona-Yuste et al. 2016b; a).
The 28S rRNA and ITS1 haplonet analyses of L. panderaltum n. sp. showed it as a unique species, and clearly different from L. goodeyi and L. onubensis. No heterozygous individuals were found in those three species.
The description of L. panderaltum n. sp. suggests that the biodiversity of needle nematodes in Southern Europe is still not completely deciphered and requires further research. Interestingly, the phylogenetic relationships among Iberian Peninsula species could provide insight into the speciation of some of these species specifically to the Iberian Peninsula, additionally of other main centres of origin in other parts of the world, as suggested by Coomans (1985). However, this hypothesis regarding the evolutionary patterns in the genus Longidorus must be analysed using biogeographical models and a higher number of sequences from other Longidorus spp. given the increasing diversity of this genus in the samplings at the Iberian Peninsula (Archidona-Yuste et al. 2016b; a). These results enlarge the diversity of Longidorus in Spain and agree with previous data obtained for the phylogeny and biogeography of the genus Longidorus in the Mediterranean Basin (Navas et al. 1990; Navas et al. 1993; Gutiérrez-Gutiérrez et al. 2011; Gutiérrez-Gutiérrez et al. 2013; Archidona-Yuste et al. 2016b; 2019a; Cai et al. 2020a, b).
In summary, the present study extends the biodiversity of the genus Longidorus by integrating morphological, morphometrical and molecular characterizations and elucidates phylogenetic relationships with other Longidorus spp. of the new species described. The molecular markers obtained could be used for precise and unequivocal diagnosis of this species, which may help for effective and appropriate phytopathological or ecological studies.
Notes
The species epithet refers to the compound name from the word Pandera (the mountain where the species was detected) and the Latin name altum = high.
References
Amrei, S. B., Peneva, V., Rakhshandehroo, F., & Pedram, M. (2020). Molecular and morphological description of Longidorus behshahrensis n. sp. (Nematoda: Longidoridae) in natural forests of Abbas Abad, northern Iran. European Journal of Plant Pathology, 156, 387–398.
Archidona-Yuste, A., Cantalapiedra-Navarrete, C., Castillo, P., & Palomares-Rius, J. E. (2019a). Molecular phylogenetic analysis and comparative morphology reveals the diversity and distribution of needle nematodes of the genus Longidorus (Dorylaimida: Longidoridae) from Spain. Contributions to Zoology, 88, 1–41.
Archidona-Yuste, A., Navas-Cortés, J. A., Cantalapiedra-Navarrete, C., Palomares-Rius, J. E., & Castillo, P. (2016). Cryptic diversity and species delimitation in the Xiphinema americanum-group complex (Nematoda: Longidoridae) as inferred from morphometrics and molecular markers. Zoological Journal of the Linnean Society, 176, 231–265.
Archidona-Yuste, A., Navas-Cortés, J. A., Cantalapiedra-Navarrete, C., Palomares-Rius, J. E., & Castillo, P. (2016b). Unravelling the biodiversity and molecular phylogeny of needle nematodes of the genus Longidorus (Nematoda: Longidoridae) in olive and a description of six new species. PloS One, 11, e0147689. https://doi.org/10.1371/journal.pone.0147689
Archidona-Yuste, A., Wiegand, T., Castillo, P., & Navas-Cortés, J. A. (2019b). Dataset on the diversity of plant-parasitic nematodes in cultivated olive trees in southern Spain. Data in Brief, 27, 104658.
Arias, M. (1977). Distribución del género Longidorus (Micoletzky, 1922) Filipjev, 1934 (Nematoda: Dorylaimida) en España. Nematologia Mediterranea, 5, 45–50.
Arias, M., Navas, A., & Bello, A. (1985). Nematodos ectoparásitos y transmisores de virus de la familia Longidoridae. Su distribución en España continental. Boletín Servicio de Plagas, 11, 275–337.
Bravo, M. A., & Roca, F. (1995). Observations on Longidorus africanus Merny from Portugal with description of L. vinearum n. sp. (Nematoda: Longidoridae). Fundamental and Applied Nematology, 18, 81–88.
Brown, D. J. F., & Taylor, C. E. (1987). Comments on the occurrence and geographical distribution of longidorid nematodes in Europe and the Mediterranean region. Nematologia Mediterranea, 15, 333–373.
Cai, R., Archidona-Yuste, A., Cantalapiedra-Navarrete, C., Palomares-Rius, J. E., & Castillo, P. (2020a). Integrative descriptions and molecular phylogeny of two new needle nematodes of the genus Longidorus (Nematoda: Longidoridae) from Spain. European Journal of Plant Pathology, 156, 67–86.
Cai, R., Archidona-Yuste, A., Cantalapiedra-Navarrete, C., Palomares-Rius, J. E., & Castillo, P. (2020b). New evidence of cryptic speciation in needle and dagger nematodes of the genera Longidorus and Xiphinema (Nematoda: Longidoridae). Journal of Zoological Systematics and Evolutionary Research, 58, in press, https://doi.org/10.1111/jzs.12393.
Castillo, P., Vovlas, N., Subbotin, S., & Troccoli, A. (2003). A new root-knot nematode, Meloidogyne baetica n. sp. (Nematoda: Heteroderidae), parasitizing wild olive in Southern Spain. Phytopathology, 93, 1093–1102.
Castresana, J. (2000). Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Molecular Biology and Evolution, 17, 540–552.
Chen, Q. W., Hooper, D. J., Loof, P. A. A., & Xu, J. (1997). A revised polytomous key for the identification of species of the genus Longidorus Micoletzky, 1922 (Nematoda: Dorylaimoidea). Fundamental and Applied Nematology, 20, 15–28.
Cherry, T., Szalanski, A. L., Todd, T. C., & Powers, T. O. (1997). The internal transcribed spacer region of Belonolaimus (Nemata: Belonolaimidae). Journal of Nematology, 29, 23–29.
Clement, M., Snell, Q., Walker, P., Posada, D., & Crandall, K. (2002). TCS: Estimating gene genealogies. Parallel and Distributed Processing Symposium, International Proceedings, 2, 184.
Coolen, W. A. (1979). Methods for extraction of Meloidogyne spp. and other nematodes from roots and soil. In F. Lamberti & C. E. Taylor (Eds.), Root-knot nematodes (Meloidogyne species). Systematics, biology and control (pp. 317–329). New York: Academic.
Coomans, A. (1985). A phylogenetic approach to the classification of the Longidoridae (Nematoda: Dorylaimida). Agriculture, Ecosystems and Environment, 12, 335–354.
Coomans, A. (1996). Phylogeny of the Longidoridae. Russian Journal of Nematology, 4, 51–60.
Coomans, A., Huys, R., Heyns, J., & Luc, M. (2001). Character analysis, phylogeny, and biogeography of the genus Xiphinema Cobb, 1973 (Nematoda, Longidoridae) (Vol. 287). Tervuren: Annales du Musée Royal de l’Afrique Centrale (Zoologie).
Dalmasso, A. (1969). Etude anatomique et taxonomique des genres Xiphinema, Longidorus et Paralongidorus (Nematoda: Dorylaimidae). Memoires du Museum National d'Histoire Naturelle Nouvelle serie, Serie A Zoologie, 61, 82 pp.
Darriba, D., Taboada, G. L., Doallo, R., & Posada, D. (2012). jModelTest 2: more models, new heuristics and parallel computing. Nature Methods, 9, 772.
De Grisse, A. T. (1969). Redescription ou modifications de quelques techniques utilisées dans l’étude de nématodes phytoparasitaires. Mededelingen Rijksfaculteit Landbouwwetenschappen Gent, 34, 315–359.
De Ley, P., Félix, M. A., Frisse, L. M., Nadler, S. A., Sternberg, P. W., & Thomas, K. W. (1999). Molecular and morphological characterisation of two reproductively isolated species with mirror-image anatomy (Nematoda: Cephalobidae). Nematology, 1, 591–612.
De Luca, F., Reyes, A., Grunder, J., Kunz, P., Agostinelli, A., De Giorgi, C., & Lamberti, F. (2004). Characterization and sequence variation in the rDNA region of six nematode species of the genus Longidorus (Nematoda). Journal of Nematology, 36, 147–152.
De Waele, D., & Coomans, A. (1990). Occurrence and ecology of longidorid nematodes in Belgium. Revue de Nematologie, 13, 197–202.
Decraemer, W., & Robbins, R. T. (2007). The who, what and where of Longidoridae and Trichodoridae. Journal of Nematology, 39, 295–297.
Doucet, M. E., Ferraz, L., Magunacelaya, J. C., & Brown, D. J. F. (1998). The occurrence and distribution of longidorid nematodes in Latin America. Russian Journal of Nematology, 6, 111–128.
Flegg, J. J. M. (1967). Extraction of Xiphinema and Longidorus species from soil by a modification of Cobb´s decanting and sieving technique. Annals of Applied Biology, 60, 429–437.
Gutiérrez-Gutiérrez, C., Bravo, M. A., Santos, M. T., Vieira, P., & Mota, M. (2016). An update on the genus Longidorus, Paralongidorus and Xiphinema (Family Longidoridae) in Portugal. Zootaxa, 4189, 99–114.
Gutiérrez-Gutiérrez, C., Cantalapiedra-Navarrete, C., Montes-Borrego, M., Palomares-Rius, J. E., & Castillo, P. (2013). Molecular phylogeny of the nematode genus Longidorus (Nematoda: Longidoridae) with description of three new species. Zoological Journal of the Linnean Society, 167, 473–500.
Gutiérrez-Gutiérrez, C., Rius, P., Cantalapiedra-Navarrete, J. E., Landa, C., Castillo, B. B., P (2011). Prevalence, polyphasic identification, and molecular phylogeny of dagger and needle nematodes infesting vineyards in southern Spain. European Journal of Plant Pathology, 129, 427–453.
Gutiérrez-Gutiérrez C, Cantalapiedra-Navarrete C, Decraemer W, Vovlas N, Prior T, Rius JEP, Castillo P. (2012). Phylogeny, diversity, and species delimitation in some species of the Xiphinema americanum-group complex (Nematoda: Longidoridae), as inferred from nuclear and mitochondrial DNA sequences and morphology. European Journal of Plant Pathology 134: 561–597.
Hall, T. A. (1999). BioEdit: a user-friendly biological sequence alignment editor and analysis program for windows 95/98/NT. Nucleic Acids Symposium Series, 41, 95–98.
He, Y., Subbotin, S., Rubtsova, T. V., Lamberti, F., Brown, D. J. F., & Moens, M. (2005). A molecular phylogenetic approach to Longidoridae (Nematoda: Dorylaimida). Nematology, 7, 111–124.
Holterman, M., Van Der Wurff, A., Van Den Elsen, S., Van Megen, H., Bongers, T., Holovachov, O., Bakker, J., & Helder, J. (2006). Phylum-wide analysis of SSU rDNA reveals deep phylogenetic relationships among nematodes and accelerated evolution toward crown clades. Molecular Phylogenetics and Evolution, 23, 1792–1800.
Hooper, D. J. (1961). A Redescription of Longidorus elongatus (De Man, 1876) Thorne Swanger, 1936, (Nematoda, Dorylaimidae) and descriptions of five new species of Longidorus from Great Britain. Nematologica, 6, 237–257.
Hunt, D. J. (1993). Aphelenchida, longidoridae and trichodoridae: Their systematics and bionomics. Wallingford: CAB International.
Jairajpuri, M. S., & Ahmad, W. (1992). Dorylaimida. Freeliving, predaceous and plant-parasitic nematodes. New Delhi: Oxford & IBH Publishing Co., 458 pp.
Kankina, V. K., & Metlitskaya, K. V. (1983). Parasitic nematodes of the family Longidoridae (Thorne, 1935) Meyl, 1961, on currant and raspberry. Izvestiya Akademii Nauk Tadzhikskoi ssr Otdelenie Biologicheskikh Nauk, 4, 50–52.
Katoh, K., Rozewicki, J., & Yamada, K. D. (2017). MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Briefings in Bioinformatics, 20, 1160–1166.
Kumari, S., & Subbotin, S. A. (2012). Characterization of Longidorus helveticus (Nematoda: Longidoridae) from the Czech Republic. European Journal of Plant Pathology, 133, 923–933.
Lamberti, F., Bleve-Zacheo, T., & Arias, M. (1982). The Longidoridae of the Maltese Islands with the description of Longidorus magnus sp. n. and Xiphinema melitense sp. n. Nematologia Mediterranea, 10, 183–200.
Lazarova, S. S., Elshishka, M., Radoslavov, G., Lozanova, L., Hristov, P., Mladenov, A., Zheng, J., Fanelli, E., De Luca, F., & Peneva, V. K. (2019). Molecular and morphological characterisation of Longidorus polyae sp. n. and L. pisi Edward, Misra & Singh, 1964 (Dorylaimida, Longidoridae) from Bulgaria. ZooKeys, 830, 75–98.
Lazarova, S. S., Malloch, G., Oliveira, C. M. G., Hübschen, J., & Neilson, R. (2006). Ribosomal and mitochondrial DNA analyses of Xiphinema americanum-group populations. Journal of Nematology, 38, 404–410.
Legendre, P., & Legendre, L. (2012). Numerical ecology. Amsterdam: Elsevier.
Lisková, M., & Brown, D. J. F. (1998). Longidoridae (Nematoda) associated with walnut trees (Juglans regia L.) in Slovak Republic. Helminthologia, 35, 93–99.
Loof, P. A. A., & Chen, Q. (1999). A revised polytomous key for the identification of the species of the genus Longidorus Micoletzky, 1922 (Nematoda: Dorylaimoidea). Supplement 1. Nematology, 1, 55–59.
Loof, P. A. A., & Coomans, A. (1972). The oesophageal gland nuclei of Longidoridae (Dorylaimida). Nematologica, 18, 213–233.
Macara, A. M. (1985). Two new species of Longidorus (Nematoda: Longidoridae) associated with forest plants in Portugal. Nematologica, 31, 410–423.
Micoletzky, H. (1922). Die freilebenden Erd-Nematoden. Archiv für Naturgeschichte Berlin Abt, 87, 1–650.
Montgomery, D. C., & Peck, E. A. (1992). Introduction to linear regression analysis. Hoboken: Wiley.
Navas, A., Andres, M. F., & Arias, M. (1990). Biogeography of Longidoridae in the Euromediterranea area. Nematologia Mediterranea, 18, 103–112.
Navas, A., Baldwin, J. G., Barrios, L., & Nombela, G. (1993). Phylogeny and biogeography of Longidorus (Nematoda: Longidoridae) in Euromediterranea. Nematologia Mediterranea, 21, 71–88.
Neilson, R., Ye, W., Oliveira, C. M. G., Hübschen, J., Robbins, R. T., Brown, D. J. F., & Szalanski, A. L. (2004). Phylogenetic relationships of Longidoridae species (Nematoda: Dorylaimida) from North America inferred from 18S rDNA sequence data. Helminthologia, 41, 209–215.
Palomares-Rius, J. E., Escobar, C., Cabrera, J., Vovlas, A., & Castillo, P. (2017a). Anatomical alterations in plant tissues induced by plant-parasitic Nematodes. Frontiers in Plant Sciences, 8, 1987. https://doi.org/10.3389/fpls.2017.01987.
Palomares-Rius, J. E., Cantalapiedra-Navarrete, C., Archidona-Yuste, A., Blok, V. C., & Castillo, P. (2017b). Mitochondrial genome diversity in dagger and needle nematodes (Nematoda: Longidoridae). Scientific Reports, 7, 41813.
Palomares-Rius, J. E., Cantalapiedra-Navarrete, C., Archidona-Yuste, A., Subbotin, S. A., & Castillo, P. (2017c). The utility of mtDNA and rDNA for barcoding and phylogeny of plant-parasitic nematodes from Longidoridae (Nematoda, Enoplea). Scientific Reports, 7, 10905.
Palomares-Rius, J. E., Escobar, C., Cabrera, J., Vovlas, A., & Castillo, P. (2017). Anatomical alterations in plant tissues induced by plant-parasitic Nematodes. Frontiers in Plant Sciences, 8, 1987. https://doi.org/10.3389/fpls.2017.01987
Palomares-Rius, J. E., Subbotin, S. A., Landa, B. B., Vovlas, N., & Castillo, P. (2008). Description and molecular characterisation of Paralongidorus litoralis sp. n. and P. paramaximus Heyns, 1965 (Nematoda: Longidoridae) from Spain. Nematology, 10, 87–101.
Peneva, V. K., Lazarova, S. S., De Luca, F., & Brown, D. J. F. (2013). Description of Longidorus cholevae n. sp. (Nematoda, Dorylaimida) from a riparian habitat in the Rila Mountains, Bulgaria. ZooKeys, 330, 1–26.
Quadros, A. (2019). emstreeR: Tools for Fast Computing and Plotting Euclidean Minimum Spanning Trees. R package version 2.2.0. https://CRAN.R-project.org/package=emstreeR.
R Core Team, (2019). R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing.
Radivojević, M., Barsi, L., Fanelli, E., & De Luca, F. (2020). Description of Longidorus barsii Radivojević & De Luca sp. n. (Nematoda: Longidoridae) from Serbia and observations on some taxonomic characters. Nematology, 22, in press. 10.1163/15685411-00003323.
Revelle, W. (2019). psych: Procedures for Psychological, Psychometric, and Personality Research.
Robbins, R. T., & Brown, D. J. F. (1991). Comments on the taxonomy, occurrence and distribution of longidoridae (Nematoda) in North America. Nematologica, 37, 395–419.
Robbins, R. T., Brown, D. J. F., Halbrendt, J. M., & Vrain, T. C. (1996). Compendium of juvenile stages of Xiphinema species (Nematoda: Longidoridae). Russian Journal of Nematology, 4, 163–171.
Roca, F., & Bravo, M. A. (1996). Description of Longidorus crataegi sp. n. from Portugal and observations on L. vineacola Sturhan & Weischer (Nematoda: Longidoridae). Fundamental and Applied Nematology, 19, 529–535.
Ronquist, F., Teslenko, M., van der Mark, P., Ayres, D. L., Darling, A., Höhna, S., Larget, B., Liu, L., Suchard, M. A., & Huelsenbeck, J. P. (2012). MRBAYES 3.2: Efficient Bayesian phylogenetic inference and model selection across a large model space. Systematic Biology, 61, 539–542.
Rozas, J., Ferrer-Mata, A., Sanchez-Delbarrio, J. C., Librado, G., Ramos-Onsins, P. S. E. & Sanchez-Gracia, A. (2017). DnaSP v6: DNA Sequence Polymorphism Analysis of Large Datas. Molecular Biology and Evolution, 34, 3299–3302.
Seinhorst, J. W. (1966). Killing nematodes for taxonomic study with hot f.a. 4:1. Nematologica, 12, 178.
Seinhorst, J. W., & Van Hoof, H. A. (1981). Atlas of plant parasitic nematodes of The Netherlands (p. 33). Dundee: Scottish Crop Research Institute.
Sturhan, D., Wouts, W., Grandison, M., G. S. & Barber, C. J. (1997). Nematode vectors of plant viruses in New Zealand. New Zealand Journal of Zoology, 24, 309–322.
Subbotin, S. A., Rogozhin, E. A., & Chizhov, V. N. (2014). Molecular characterisation and diagnostics of some Longidorus species (Nematoda: Dorylaimida) from Russia and other countries using rRNA genes. European Journal of Plant Pathology, 138, 377–390.
Taylor, C. E., & Brown, D. J. F. (1997). Nematode vectors of plant viruses (296 pp). Wallingford: CABI.
Tophan, P. B., & Alphey, T. J. W. (1985). Faunistic analysis of Longidorid nematodes in Europe. Journal of Biogeography, 12, 165–174.
Van Den Berg, E., Palomares-Rius, J. E., Vovlas, N., Tiedt, L. R., Castillo, P., & Subbotin, S. A. (2016). Morphological and molecular characterisation of one new and several known species of the reniform nematode, Rotylenchulus Linford & Oliveira, 1940 (Hoplolaimidae: Rotylenchulinae), and a phylogeny of the genus. Nematology, 18, 67–107.
Vrain, T. C., Wakarchuk, D. A., Levesque, A. C., & Hamilton, R. I. (1992). Intraspecific rDNA Restriction Fragment Length Polymorphism in the Xiphinema americanum group. Fundamental and Applied Nematology, 15, 563–573.
Ye, W., Szalanski, A. L., & Robbins, R. T. (2004). Phylogenetic relationships and genetic variation in Longidorus and Xiphinema species (Nematoda: Longidoridae) using ITS1 sequences of nuclear ribosomal DNA. Journal of Nematology, 36, 14–19.
Zuur, A. F., Ieno, E. N., & Elphick, C. S. (2010). A protocol for data exploration to avoid common statistical problems. Methods in Ecology and Evolution, 1, 3–14.
Acknowledgements
This research was financially supported by grant 201740E042, “Análisis de diversidad molecular, barcoding, y relaciones filogenéticas de nematodos fitoparásitos en cultivos mediterráneos” from Spanish National Research Council (CSIC), and by the Humboldt Research Fellowship for Postdoctoral Researchers awarded for the corresponding author. Authors thank G. León Ropero and J. Martín Barbarroja (IAS-CSIC) for the excellent technical assistance. The first author acknowledges the China Scholarship Council (CSC) for financial support. The fifth author acknowledges Spanish Ministry of Economy and Competitiveness for the “Ramon y Cajal” Fellowship RYC-2017-22228. The corresponding author is a recipient of Humboldt Research Fellowship for Postdoctoral Researchers at Helmholtz Centre for Environmental Research-UFZ, Leipzig, Germany.
Funding
Open Access funding provided by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All authors certify that 1) they do not have any actual or potential conflict of interest, 2) the study described is original and has not been published previously, and is not under consideration for publication elsewhere, 3) all prevailing local, national and international regulations and conventions, and normal scientific ethical practices, have been respected. We also certify that all authors have reviewed the manuscript and approved the final version of manuscript before submission.
Research involving human participants and/or animals
No specific permits were required for the described fieldwork studies. Permission for sampling the forests was granted by the landowner. The sites are not protected in any way.
Informed consent
All the authors certify that the work carried out in this research followed the principles of ethical and professional conduct have been followed. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cai, R., Prior, T., Lawson, B. et al. An integrative taxonomic study of the needle nematode complex Longidorus goodeyi Hooper, 1961 (Nematoda: Longidoridae) with description of a new species.. Eur J Plant Pathol 158, 59–81 (2020). https://doi.org/10.1007/s10658-020-02055-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10658-020-02055-0