
Abstract
Background
Lubiprostone helps relieve constipation in short-term 4-week studies. There are limited data on long-term pharmacological treatment with lubiprostone for chronic idiopathic constipation.
Aims
To examine the long-term safety and effectiveness of lubiprostone in patients with chronic idiopathic constipation.
Methods
In this prospective, multicenter, open-labeled trial, 248 patients aged ≥18 years with chronic idiopathic constipation were directed to take lubiprostone 24 mcg BID as needed for 48 weeks. Patients were allowed to decrease the dose in response to the perceived severity of constipation and need for relief. Hematology and chemistry profiles and assessment of constipation symptoms and its severity were performed at all visits. Adverse events (AEs) were recorded.
Results
Of the 248 patients who entered the trial, 127 (51%) completed the trial. A dose reduction was observed in 17% of the patients, resulting in an average study medication exposure across the study of approximately 1.7 capsules (or approximately 40.8 mcg) per day. The most common treatment-related AEs were nausea (19.8%), diarrhea (9.7%), abdominal distension (6.9%), headache (6.9%), and abdominal pain (5.2%). No deaths were reported and of the 16 reported serious AEs, one was considered possibly treatment related. Average changes in serum electrolytes were not clinically relevant at any time point during the study. On average, lubiprostone significantly (p < 0.0001) reduced patient-reported constipation severity, abdominal bloating, and abdominal discomfort across 48 weeks when compared to baseline.
Conclusions
During this 48-week open-label study, lubiprostone was well tolerated. Bowel symptoms consistently improved over 48 weeks in adult patients with chronic idiopathic constipation.
Similar content being viewed by others

Introduction
Constipation is a common complaint, with prevalence estimates ranging from 2–28% in the United States; constipation is also found more commonly in women and the elderly [1–3]. In the United States, physician visits from 1958 to 1986 for constipation averaged 2.5 million per year [1]. A more recent study by Martin and colleagues, using data from 2001, found 5.7 million physician/emergency room visits were constipation related [4]. However, these data likely underestimate the problem, since only a minority of people with constipation seek medical care [5, 6].
The prevalence of chronic constipation and the growing demand for treatment dictate the need for safe and effective treatment options. Currently available interventions include lifestyle changes and a broad range of over-the-counter (OTC) and prescription medications. Studies of short-term treatment with dietary fiber supplements and exercise have demonstrated marginal effectiveness [7, 8]. Additionally, while patients may note symptomatic short-term improvement with the use of OTC medications (e.g., bulk laxatives, osmotic laxatives, and stimulant laxatives), the current approved indication for such agents does not support their long-term use in patients with chronic constipation [9–11].
Lubiprostone, an activator of chloride channels (ClC-2), is a member of a class of compounds called prostones, and has been approved by the Federal Drug Administration (FDA) as an agent for the treatment of chronic idiopathic constipation. Activation of ClC-2 results in increased chloride secretion with associated passive transport of sodium and water across gastrointestinal mucosal epithelia, thereby enhancing fluid secretion into the intestinal lumen and promoting intestinal transit [12–15].
Two recent clinical studies have demonstrated lubiprostone’s efficacy in the short-term treatment of chronic idiopathic constipation [16, 17]. In a 4-week, randomized, placebo-controlled trial involving 242 patients with chronic idiopathic constipation, lubiprostone 24 mcg twice daily (BID) significantly increased the number of spontaneous bowel movements (SBMs) compared with placebo (5.1–5.7 per week for lubiprostone vs. 2.8–3.5 for placebo [p < 0.002]) [16]. Likewise, in a second 4-week trial involving 237 patients with chronic idiopathic constipation, lubiprostone 24 mcg BID increased the number of SBMs from a pretreatment mean of 1.30 to 5.89 SBMs per week at Week 1, which was significantly greater than placebo (p < 0.0001) [17].
The aim of the current study was to assess the safety and effectiveness of lubiprostone 24 mcg BID taken open-label as needed for 48 weeks in patients with chronic idiopathic constipation.
Methods
Study Design and Patients
The study was conducted in accordance with the U.S. Code of Federal Regulations on Good Clinical Practice, consistent with the Declaration of Helsinki, and the International Conference on Harmonisation (ICH). Prior to enrollment, written informed consent was obtained from each patient. The study protocol, informed consent form, and volunteer information requirements (inclusion/exclusion criteria) were approved by the Institutional Review Boards of the participating centers.
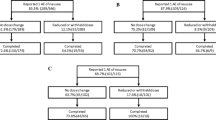
In this prospective, 48-week, multicenter, open-label study, patients were directed to take lubiprostone 24 mcg BID or a lower dose (24 mcg QD) as needed, based on their perceived severity of constipation and need for relief, or at the investigator’s discretion in response to adverse events (AEs). Following the treatment period, patients were monitored for an additional 2 weeks. Patients were enrolled either directly into the study (after a 2-week baseline washout period) or immediately after completing an independent randomized-withdrawal trial (Fig. 1). In the randomized-withdrawal study, following 4 weeks of treatment with lubiprostone 24 mcg BID, half of the patients were randomized to receive placebo and the remaining patients were continued on active treatment with lubiprostone for an additional 3 weeks [18]. Upon completion of the randomized-withdrawal study, eligible patients were enrolled into the open-label treatment phase reported here, during which all patients were instructed to take lubiprostone 24 mcg BID for 48 weeks.

Flow of participants through each stage of the study. Single asterisk One patient hospitalized before receiving study medication; one patient lost to follow-up. Double asterisk Two patients discontinued the trial before receiving study medication in the open-labeled phase
The study enrolled male and female patients aged ≥18 years with ≥3 months of constipation, defined as an average of <3 SBMs/week and ≥1 of the following symptoms with at least 25% of bowel movements: very hard (little balls) and/or hard stools, a sensation of incomplete evacuation, and/or straining at defecation. Patients were directed not to change their lifestyle or diet during the study, including exercise and fiber intake. Study exclusion criteria included: documented mechanical obstruction, a megacolon/megarectum, or a diagnosis of pseudo-obstruction; known or suspected organic small or large bowel disorders; evidence of secondary causes of constipation; hospitalization for any gastrointestinal or abdominal surgery during the 3 months prior to study commencement; or any bowel resection. Patients aged <50 years were required to have had a flexible sigmoidoscopy or colonoscopy performed within the previous 5 years. For patients aged ≥50 years and any participant for whom there was evidence of weight loss, anemia, or rectal bleeding since the previous evaluation, a barium enema with flexible sigmoidoscopy or colonoscopy was required. Female patients could not breast feed or be pregnant, as confirmed by a negative serum pregnancy test at the baseline visit and at subsequent office visits during the treatment period.
Patients were directed to take lubiprostone 24 mcg BID with food (usually breakfast and dinner) and at least 8 oz. of water. However, the daily dose could be reduced at the discretion of the investigator in response to exaggerated pharmacodynamic events (e.g., diarrhea, or other treatment-related AEs such as nausea). Patients who had been routinely using a daily fiber supplement for ≥3 months prior to baseline were allowed to continue use throughout the study but were not allowed to change the dose or administration schedule. Patients were prohibited from taking other prescription or OTC medications for constipation during the study period, including the baseline/washout period. All other medications were permitted but usage was documented; initiation and/or discontinuation of medications after study initiation were limited to the extent possible and at the discretion of the investigator. Study investigators could authorize participants to administer the following rescue medications if they had not experienced a bowel movement for ≥3 consecutive days and felt they needed relief: Dulcolax® (bisacodyl, Boehringer Ingelheim Consumer Healthcare, UK) suppository or, if unsuccessful, a Fleet® Enema (dibasic sodium phosphate and monobasic sodium phosphate, C.B. Fleet Company, Incorp., Lynchburg, VA). If neither medication was effective, another rescue medication could be prescribed for limited use at the investigator’s discretion.
Safety and Patient-Reported Outcome Assessments
At the baseline visit prior to study enrollment, patients’ medical and constipation histories were taken. Baseline constipation severity and symptoms (e.g., timing and consistency of, and straining during, bowel movements) were assessed via patient diary recordings during the preceding 2-week washout period. Patients underwent a physical examination, including vital signs and body weight, and laboratory testing (hematology, chemistries, and urinalysis) at baseline and at weeks 12, 24, 36, and 48 during the treatment period.
Safety assessments consisted of AEs (recorded from the time of the first lubiprostone dose until the follow-up visit), serious AEs (SAEs; recorded up to 7 days after the final lubiprostone dose), clinical laboratory evaluations, vital signs, and physical examinations. Intensity (mild, moderate, or severe) and relationship to study drug (unrelated, possibly, probably, or definitely related) were recorded for all AEs. Use of concomitant therapy was evaluated at every study visit, and any concomitant therapy given as treatment for a new condition or worsening of an existing condition was considered an AE.
Patient-reported outcome assessments were comprised of patient global assessments (severity of constipation; treatment effectiveness) and abdominal assessments (perceptions of bloating and discomfort upon waking in the morning). Patients were assessed at baseline, enrollment, and each subsequent study visit. Study visits occurred every 6 weeks and alternated between office and phone visits for a total of 11 study visits over a 48-week treatment period, with the last follow-up visit occurring at week 50. Severity of constipation and abdominal symptoms were rated on a 5-point scale: 0 = absent; 1 = mild; 2 = moderate; 3 = severe; 4 = very severe. Treatment effectiveness was also evaluated using a 5-point scale: 0 = not at all effective; 1 = a little bit effective; 2 = moderately effective; 3 = quite a bit effective; 4 = extremely effective. These same scales were used for the pivotal, 4-week randomized, placebo-controlled lubiprostone studies [16, 17].
Statistical Analyses
All patients who received ≥1 dose of lubiprostone during the 48-week treatment period were evaluated for safety; a subset of this population was analyzed for effectiveness. Data from the most recent study visit were analyzed per the study protocol. Approximately 300 patients with constipation were planned for enrollment into the study with the goal of completing 100 patients with 1 year of exposure to lubiprostone. This sample size goal was based on ICH recommendations [19] and was estimated to yield a 95% chance of detecting events occurring at a rate of ≥1% in a population.
Patient demographics and baseline characteristics were summarized including constipation data using descriptive statistics. Demographic differences between patients who completed the study versus those who did not were analyzed using Fisher’s exact test, and changes in constipation and abdominal symptom severity were assessed using Wilcoxon matched-pair signed-rank tests.
Cumulative exposure to lubiprostone was expressed as the sum of all dose administrations provided from the start of treatment until the next visit, when capsule counts were conducted. Average daily exposure was calculated as the cumulative exposure divided by the number of days of treatment in this interval.
AEs were summarized in terms of incidence, with the incidence rates of AEs expressed as a percentage of the number of safety-evaluable patients experiencing at least one episode of the AE during the safety window (time from first to last dose of study medication plus 7 days). Incidence rates of AEs, AEs assessed as at least possibly related to the study medication, SAEs, and AEs reported by ≥5% of patients were calculated. Patient-days were calculated using date of last visit minus date of first dose plus 1 day. Nausea event rate was calculated using number of nausea events divided by total patient days. Laboratory test results were tabulated using descriptive statistics. Descriptive statistics were used to evaluate mean changes of continuous vital sign data at each visit. Paired t tests were used to analyze the significance of observed changes in vital signs from baseline. For patient-reported parameters, global assessments (severity of constipation and treatment effectiveness) and abdominal assessments (bloating and discomfort upon waking in the morning) were summarized. Missing values for patient-reported variables, including patient withdrawals, were not imputed. Analyses of each time point were performed on the observed cases for that time point. Analyses are broken down by enrollment group and all subjects combined. Changes in severity of constipation and abdominal symptoms from baseline through 48 weeks were analyzed via Wilcoxon matched-pairs signed-ranks tests.
Results
Patients
Of the 250 patients enrolled in the study, 80 were recruited from the randomized withdrawal study (4 weeks of treatment followed by 3 weeks of randomized withdrawal) [18], while 170 (lubiprostone-naive) were enrolled directly into the current open-label treatment study. Of the 248 patients who began the study, 127 (51%) completed the trial. The most common reasons for discontinuation were a lack of effectiveness (17.7%), AEs (13.3%), and voluntary withdrawal (9.3%; Fig. 1).
Two patients withdrew from the study prior to taking the first dose of lubiprostone. A comparison of the demographics revealed the two patient groups to be similar, and the combined data are summarized in Table 1.
The mean daily dose of lubiprostone was consistent throughout the trial (approximately 1.7 capsules/day), with a mean daily intake of 1.67 capsules at Week 48. Of the patients, 17% of patients required a dose reduction of lubiprostone, as determined by the investigator. There were no statistically significant demographic differences (i.e., gender, age, weight, and ethnicity) between the patients who required a dose adjustment and those who did not.
Safety
Overall, 75.4% (n = 187) of the patients reported at least one AE and of the total AEs, most were mild (49%) or moderate (47%) in intensity, and 42.3% of the total were considered by the investigator to be related to lubiprostone (Table 2 summarizes the AEs reported by ≥5% of patients). Of the total AEs broken down by body system, the most common were gastrointestinal disorders (49.6%), followed by infections/infestations (26.6%), disorders of the nervous system (13.3%), and musculoskeletal and connective-tissue disorders (12.9%). The AEs occurring as severe in intensity and reported by ≥2 patients overall were abdominal distension, abdominal pain, arthralgia, back pain, neck pain, and nausea.
The majority of treatment-related AEs reported by patients were of mild (50%) or moderate (44%) intensity. Of the two most commonly reported AEs, nausea (19.8%) and diarrhea (9.7%), >95% of these events were reported as either mild or moderate in severity and none were serious adverse events. In addition, the total number of patient days during the study was 62,325 days, and nausea and diarrhea event rates were 1.08 and 0.61 per 1,000 patient days, respectively.
Overall, a total of 33 patients (13.3%) withdrew from the study due to AEs. Nausea accounted for 13 (5.2%) withdrawals, of which approximately nine patients (~70%) withdrew within the first 12 weeks, ten (>75%) by week 14, and all 13 (100%) by week 27. Other common AEs that led to discontinuation were abdominal distension (n = 5; 2.0%), headache (n = 4; 1.6%), abdominal pain (n = 4; 1.6%), diarrhea (n = 3; 1.2%), and vomiting (n = 3; 1.2%).
Eleven patients reported a total of 16 SAEs, of which only one, a normal pregnancy resulting in a baby with bilateral clubfoot, was considered possibly related to the study drug by the investigator. This SAE is further described in the discussion section. There were no deaths during this study.
The average changes in vital signs, physical examination, and urinalysis parameters were not clinically significant. The mean hematology and biochemistry values and the shifts in measurements did not show any clinically significant trends (data on file). Most notably, there were no clinically meaningful trends in creatine kinase, alanine transaminase (ALT), aspartate transaminase (AST), and electrolyte (such as sodium, calcium, potassium, chloride, and magnesium) concentrations [20]. Electrocardiographic (ECG) changes were not measured in this study. Prior work demonstrated no significant ECG abnormalities or prolongation of the QTc interval [21].
Patient-Reported Outcomes
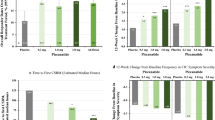
Patients who received ≥1 dose of lubiprostone and had at least 1 outcome evaluation (n = 248) were eligible for outcome assessment. The mean patient-reported constipation severity score was 2.94 at baseline (0 = absent; 4 = very severe). Following initiation of lubiprostone treatment, mean constipation severity scores were <2, and remained at this level for the duration of treatment (Fig. 2). Overall, the mean reduction in constipation severity at all post-baseline time points was statistically significant (p < 0.0015). Similar post-baseline reductions were reported for mean abdominal bloating and discomfort scores. Mean baseline scores for abdominal bloating and discomfort were 2.10 and 1.88, respectively. After treatment initiation, mean abdominal bloating scores were ≤1.13 at all visits and <1 for weeks 18 and 30–48 (Fig. 2), increasing to 1.15 at study end. Mean abdominal discomfort scores were <1 at every post-baseline assessment, with a score of 0.98 at study end. Decreases (improvements) in both abdominal bloating and discomfort scores, at all time points post-baseline, were statistically significant (p ≤ 0.011).

Mean symptom ratings for constipation severity, abdominal bloating, and abdominal discomfort with lubiprostone 24 mcg BID. End of treatment is defined as the last non-missing, post-baseline treatment period value. Follow-up is 2 weeks after end of treatment
Mean patient global assessments of treatment effectiveness (1 = a little bit effective; 4 = extremely effective) remained above 2 (moderately effective) for the entire study, with median values ranging from 2–3 for all treatment weeks.
Discussion
The short-term (4-week) effectiveness and safety of lubiprostone had been previously established in two placebo-controlled trials in patients with chronic constipation [16, 17]. However, chronic constipation typically requires long-term treatment. Therefore, it is important to establish the long-term efficacy and safety profile of lubiprostone.
The mean daily capsule intake of 1.7/day indicated that patients were generally compliant with BID dosing throughout the 48-week study period. The study further demonstrated that lubiprostone 24 mcg BID was generally well tolerated by adult patients with chronic constipation. The results were consistent with those observed during the 4-week chronic constipation trials [16, 17]. Nearly all treatment-related AEs were of mild (51%) to moderate (42%) intensity. The most common treatment-related AE was nausea (19.8%), followed by diarrhea (9.7%), with event rates of 1.08 and 0.61 per 1,000 patient-days, respectively. Thirty-three patients (13.3%) withdrew from the study due to AEs, of which 13 were due to nausea (5.2%). It is notable that, although the current study comprised 48 treatment weeks, the percentage of patients withdrawing from this study due to nausea (5.2%) was similar to the percentages of patients discontinuing treatment in the 4-week studies (approximately 5%) [16, 17]. Furthermore, since 70% of discontinuations due to nausea in this long-term study were within the first 12 weeks, these results might suggest that patients who can tolerate the drug initially may demonstrate successful long-term outcomes. The mechanism responsible for nausea is unknown, but has been hypothesized to involve an exaggerated pharmacodynamic effect from secreted fluid in the small intestine or a direct gastric effect resulting from a lubiprostone-related modest delay in gastric emptying (mean t 1/2, 132.4 min lubiprostone vs. 106.1 min placebo) [15]. In the current study, there were no clinically relevant consequences from the patients who experienced diarrhea.
One SAE (out of 16 total in the trial) was assessed to be possibly related to the study drug. The patient, a 27-year-old female with no relevant medical history (concomitant medications included diphenhydramine, paroxetine hydrochloride, bisacodyl, cortisone, and hydroxyzine) became pregnant, approximately 8-1/2 months after enrollment into the study. Because of her pregnancy, the patient discontinued participation in the study. Seven months later, she gave birth to a healthy infant with bilateral clubfoot.
Including this case, there have been a total of six reported pregnancies in clinical trials with lubiprostone. Five of the six pregnancies were carried to term (the other one was electively terminated) and no other fetal abnormalities were reported.
On average, the changes in serum electrolyte levels were not clinically significant. This finding suggests a more physiologic laxation effect with lubiprostone when compared to reports of clinically significant electrolyte shifts with some commercially available laxatives [7].
Long-term, open-label studies have inherent limitations relating to the assessment of treatment efficacy, particularly in the areas of generalizability of the results to unselected populations and patient attrition during the study period. Nevertheless, there is some value in examining the patient-reported outcomes in this long-term study population. Patients reported significant improvements from baseline in constipation severity and abdominal symptoms. Significant relief of these symptoms was observed at the first evaluation and was sustained throughout all 48 weeks of the study, suggesting that lubiprostone provided sustained relief of these symptoms without leading to tachyphylaxis. Furthermore, evaluation of patient satisfaction indicated that most patients rated lubiprostone therapy as moderately effective or quite effective; this result is likely reflected by the fact that a significant proportion of patients appeared satisfied with their treatment as >50% of patients continued treatment for the entire study period. These results are consistent with other lubiprostone trials of shorter duration [16, 17]. Results from this long-term trial suggest that lubiprostone remains safe and effective and provides consistent symptom relief for 1 year in adult patients with chronic constipation.
A limitation of the current study design was the lack of blinding or a placebo control group. Placebo-controlled studies of long duration are difficult to perform due to the high dropout rate of patients in the placebo arm [22]. However, despite the absence of a placebo arm, these results indicate that the 48-week safety and effectiveness results were consistent with those reported in the shorter-term (i.e., 4 weeks) double-blinded, placebo-controlled studies [16, 17].
In conclusion, the present study demonstrates that lubiprostone may be considered safe and generally well tolerated, with effectiveness consistently maintained across 48 weeks in adult patients with chronic constipation.
References
Sonnenberg A, Koch TR. Epidemiology of constipation in the United States. Dis Colon Rectum. 1989;32:1–8.
Stewart W, Liberman J, Sandler RS, et al. Epidemiology of constipation (EPOC) study in the United States: relation of clinical subtypes to sociodemographic features. Am J Gastroenterol. 1999;94:3530–3540.
Higgins PD, Johanson JF. Epidemiology of constipation in North America: a systematic review. Am J Gastroenterol. 2004;99:750–759.
Martin BC. National estimates of office and emergency room constipation-related visits in the United States. Am J Gastroenterol. 2004;99:S244. Abstract.
Talley NJ, Zinsmeister AR, Van Dyke C, Melton LJ III. Epidemiology of colonic symptoms and the irritable bowel syndrome. Gastroenterology. 1991;101:927–934.
Talley NJ. Differentiating functional constipation from constipation-predominant irritable bowel syndrome: management implications. Rev Gastroenterol Disord. 2005;5:1–9.
Muller-Lissner SA, Kamm MA, Scarpignato C, et al. Myths and misconceptions about chronic constipation. Am J Gastroenterol. 2005;100:232–242.
Xing JH, Soffer EE. Adverse effects of laxatives. Dis Colon Rectum. 2001;44:1201–1209.
Brandt LJ, Prather CM, Quigley EM, Schiller LR, Schoenfeld P, Talley NJ. Systematic review on the management of chronic constipation in North America. Am J Gastroenterol. 2005;100:S5–S21.
Lacy BE, Chey WD. Lubiprostone: Chronic constipation and irritable bowel syndrome with constipation. Expert Opin Pharmacother. 2009;10:143–152.
MiraLAX. Package insert, Schering-Plough HealthCare Products Inc. 2011.
Jentsch TJ, Poet M, Fuhrmann JC, Zdebik AA. Physiological functions of ClC Cl- channels gleaned from human genetic disease and mouse models. Annu Rev Physiol. 2005;67:779–807.
Cuppoletti J, Malinowska DH, Tewari KP, et al. SPI-0211 activates T84 cell chloride transport and recombinant human ClC-2 chloride currents. Am J Physiol Cell Physiol. 2004;287:C1178–C1183.
Ueno R, Osama H, Habe T, et al. Oral spi-0211 increases intestinal fluid secretion and chloride concentration without altering serum electrolyte levels (abstract). Gastroenterology. 2004;126:A-100.
Camilleri M, Bharucha AE, Ueno R, et al. Effect of a selective chloride channel activator, lubiprostone, on gastrointestinal transit, gastric sensory, and motor functions in healthy volunteers. Am J Physiol Gastrointes Liver Physiol. 2006;290:G942–G947.
Johanson JF, Morton D, Geenen J, Ueno R. Multicenter, 4-week, double-blind, randomized, placebo-controlled trial of lubiprostone, a locally acting type-2 chloride channel activator, in patients with chronic constipation. Am J Gastroenterol. 2008;103:170–177.
Barish CF, Drossman D, Johanson JF, Ueno R. Efficacy and safety of lubiprostone in patients with chronic constipation. Dig Dis Sci. 2010;55:1090–1097.
Johanson JF, Gargano MA, Holland PC, et al. Phase III, randomized withdrawal study of RU-0211, a novel chloride channel activator for the treatment of constipation. Gastroenterology. 2004;126:A-100. Abstract.
International Conference on Harmonization. The extent of population exposure to assess clinical safety for drugs for long-term treatment of non-life-threatening conditions. 1994. Available at: http://wwwichorg/cache/compo/475-272-1html#E1.
Rivera E, Wahle A, Joswick TR, et al. Lubiprostone, a novel type-2 chloride channel (ClC-2) activator, does not affect serum electrolyte balance in elderly and non-elderly patients with chronic idiopathic constipation. Gastroenterology. 2007;132:A191–A192. Abstract.
Sprenger C, Copa A, Morganroth J, et al. Effect of lubiprostone, a unique agent for the treatment of chronic idiopathic constipation, on clinical electrocardiographic results. Gastroenterology. 2007;132:A325. Abstract.
Corazziari E, Badiali D, Bazzocchi G, et al. Long-term efficacy, safety, and tolerability of low daily doses of isosmotic polyethylene glycol electrolyte balanced solution (PMF-100) in the treatment of functional chronic constipation. Gut. 2000;46:522–526.
Acknowledgments
Writing and editorial support in the preparation of the manuscript draft and subsequent revisions was provided by Brian G. Shearer, PhD, Takeda Pharmaceuticals North America, Inc. Editorial assistance was also provided by Susan Ruffalo, PharmD of MedWrite, Inc. Newport Coast, California. Amitiza® is a registered trademark of Sucampo Pharma Americas, Inc. Dulcolax® is a registered trademark of Boehringer Ingelheim GmbH. Fleet Enema® is a registered trademark of C. B. Fleet Company. SAS® is a registered trademark of SAS Institute Inc. Research supported by Sucampo Pharma Americas Inc., 4520 East–West Highway, Bethesda, Maryland, USA.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Lembo, A.J., Johanson, J.F., Parkman, H.P. et al. Long-Term Safety and Effectiveness of Lubiprostone, a Chloride Channel (ClC-2) Activator, in Patients with Chronic Idiopathic Constipation. Dig Dis Sci 56, 2639–2645 (2011). https://doi.org/10.1007/s10620-011-1801-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10620-011-1801-0