
Abstract
Head and neck squamous cell carcinoma (HNSCC) is the sixth most common malignancy worldwide. There is an urgent need to develop effective therapeutic approaches to prevent and treat HNSCC. Recent deep sequencing of the HNSCC genomic landscape revealed a multiplicity and diversity of genetic alterations in this malignancy. Although a large variety of specific molecules were found altered in each individual tumor, they all participate in only a handful of driver signaling pathways. Among them, the PI3K/mTOR pathway is the most frequently activated, which plays a central role in cancer initiation and progression. In turn, targeting of mTOR may represent a precision therapeutic approach for HNSCC. Indeed, mTOR inhibition exerts potent anti-tumor activity in HNSCC experimental systems, and mTOR targeting clinical trials show encouraging results. However, advanced HNSCC patients may exhibit unpredictable drug resistance, and the analysis of its molecular basis suggests that co-targeting strategies may provide a more effective option. In addition, although counterintuitive, emerging evidence suggests that mTOR inhibition may enhance the anti-tumor immune response. These new findings raise the possibility that the combination of mTOR inhibitors and immune oncology agents may provide novel precision therapeutic options for HNSCC.
Similar content being viewed by others

1 Background
Head and neck squamous cell carcinoma (HNSCC) is the sixth most common malignancy worldwide. As a major public health concern, HNSCC arises in the oral cavity, larynx, and pharynx, affecting approximate 600,000 patients each year [1], only 40–50% of which will survive more than 5 years [2]. The leading risk factors include the use of tobacco, alcohol, and betel quid and areca nut chewing, while high-risk human papillomavirus (HPV) infection has emerged as a major risk factor, nowadays accounting for more than 20% of all HNSCC cases [3]. Currently, the main therapeutic modalities include surgery, radiation, and chemotherapy. However, these nonselective treatments may cause associated morbidity and mortality and usually have high systemic toxicities. Cetuximab, a monoclonal antibody-inhibiting EGFR, is the only cancer-targeting agent approved for HNSCC, although only ~10% of HNSCC patients respond to this agent and often for a short period of time [4, 5]. Two immune check point inhibitors targeting PD-1 have been recently approved by the FDA for HNSCC patients, albeit the rate of response is approximately 20%, lower than that of other malignancies, such as melanoma [6,7,8,9,10,11,12,13,14]. There is an urgent need to develop new effective strategies to prevent and treat HNSCC. Understanding the contribution of genomic alterations driving HNSCC initiation and progression may help explore novel precision therapeutic options.
2 Genomic landscape in head and neck cancer
Deep sequencing approaches for the study of cancer genomes have recently revolutionized medical oncology [15]. By providing an unprecedented knowledge of the multiplicity and diversity of genomic and epigenetic alterations which underlie every individual cancer lesion, these approaches deepen the understanding of dysregulated signaling circuitries and molecular mechanisms driving cancer development. Based on these information, novel druggable targets for therapeutic interventions in various human malignancies have been revealed. Several recent reports [16,17,18,19] and a landmark study from the Cancer Genome Atlas (TCGA) Network [1] has provided a comprehensive genomic characterization of HNSCC, revealing hundreds of mutations in each HNSCC lesion. This complexity of genomic alterations makes it daunting to search for molecular events driving the development of cancer, especially to differentiate driver from passenger mutations, the latter having a minimal influence on tumor progression and/or therapeutic response. However, in-depth analysis of the HNSCC oncogenome suggests that despite the complexity of the distinct molecular alterations in individual lesions, they all fall within a limited number of dysregulated molecular pathways that may contribute to most HNSCC patients [1, 3].
Specifically, the most frequently identified alterations in HNSCC participate in biologic processes regulated by the TP53 (71% mutated), FAT1 (23% mutated and 5% deleted), NOTCH1 (9% mutated and 66% signaling pathway alterations), CASP8 (10% mutated), CDKN2A (22% mutated and 60% gene copy loss) genes, and PIK3CA (~20% mutated and 30% signaling pathway alterations) [3]. This reductionist approach based on comprehensive genomic profiling may be exploited to distinguish oncogenic signaling-related subgroups from unselected cancer cohorts and facilitate the identification of actionable therapeutic targets for HNSCC patients.
3 Activation of PI3K-mTOR signaling pathway in head and neck cancer
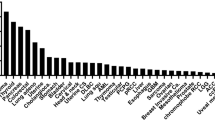
A more pathway-specific analysis of the HNSCC oncogenome suggests that most genomic alterations are involved in aberrant mitogenic signaling routes, including the PI3K, MAPK, and JAK/STAT pathways [17]. Remarkably, the PI3K-mTOR pathway is mutated in the highest percentage of the cases. In contrasts, MAPK and JAK/STAT pathways harbor mutations in less than 10% of the lesions. Specifically for PI3K, the in-depth analysis of TCGA data from 428 HPV− and 76 HPV+ HNSCC samples [20] revealed that PIK3CA is the highest mutated gene when considering all HNSCC cases (16.8%), and PI3K mutations (PIK3CA, PIK3CB, PIK3CG, and PIK3CD) are frequently mutated genes in 30% of the HPV+ lesions. Moreover, PI3K mutations are not the only genomic alterations causing the persistent activation of PI3K/AKT/mTOR pathway in HNSCC. Various genetic and epigenetic changes coordinate with PI3K mutations to sustain activation of this pathway in HNSCC (Fig. 1).

Frequent genetic alterations of PI3K/mTOR signaling pathway in HNSCC. Data was extracted from the HNSCC Cancer Genome Anatomy (TCGA) effort, including 428 HPV(−) and 76 HPV(+) HNSCC samples. Alterations identified in each key gene are shown, percentages outside and inside parentheses represent HPV(−) and HPV(+) samples, respectively. Red represents oncogene mutations and amplifications, and green represents tumor suppressor gene mutations and copy losses (copy loss refers to homozygous and heterozygous deletion of genes)
For instance, DNA copy number gain and messenger RNA (mRNA) overexpression of PIK3CA frequently occur in HNSCC (20 and 52%, respectively). Other PI3K isoforms and multiple PI3K regulatory subunits also have mutations and copy number gains (0.5–11%). Over 90% of HNSCC lesions overexpressed the epidermal growth factor receptor (EGFR), which is upstream of PI3K/AKT signaling, a major driver of epithelial cell proliferation. And a low frequency of HNSCC cases has mutations in AKT2 and mTOR or its regulatory subunits, RICTOR and RAPTOR. A network-based analysis of the HNSCC oncogene revealed that a high percentage of lesions also exhibit loss of at least one copy of a candidate PI3K/mTOR pathway tumor suppressor gene (TSG), PTEN (31%), TSC1 (11%), TSC2 (13%), STK11 (34%), and EIF4EBP1 (36%) [20]. Interestingly, co-occurrence of their gene loss is a highly statistically significant event (Table 1). Similarly, PIK3CA amplification co-occurs in a highly statistically significant fashion with gene copy gains in PI3KCB, encoding the PI3Kβ subunit. These occurrences of multiple alterations may cooperate to persistently activate PI3K/AKT/mTOR pathway in most HNSCC lesions.
HPV infection has been recently recognized as a viral etiologic agent responsible for HNSCC, more specifically in the oropharynx [21, 22]. While the overall incidence for HNSCC continues to decrease, it is observed that the incidence of HPV-associated HNSCC has a highly significant increase, predominantly among young patients [23,24,25]. Among the viral proteins encoded by high-risk HPVs (HPV16 primarily in HNSCC), E6 and E7 function as major driver oncogenic proteins. By disrupting p53 and RB tumor suppressor proteins, respectively [26,27,28], E6 and E7 induce malignant transformation. Recent findings suggest that HPV+ HNSCC has a significant enrichment of PIK3CA mutations (25% more than HPV−) and exhibit elevated mTOR activity [1, 29,30,31]. Of note, E6 and E7 oncoproteins could not be therapeutically targeted so far, making it essential to explore druggable targets for HPV+ HNSCC, in which mTOR inhibition provides suitable therapeutic options [31].
Taken together, the above findings suggest that, although genomic alterations found in HNSCC varies and are remarkably complex, most fall within certain oncogenic pathways, most of which result in persistent aberrant activation of the mTOR signaling pathway.
4 The roles of mTOR signaling pathway in cancer
The mTOR (mechanistic target of rapamycin) pathway regulates major cellular processes involved in organismal growth and homeostasis [32,33,34]. Dysregulation of this pathway occurs in multiple human diseases, such as cancer, obesity, type II diabetes, and neurodegeneration, to name but a few [33].
In the past decades, mTOR-dependent processes have been continuously uncovered. Briefly, mTOR is an atypical serine/threonine protein kinase. By interacting with several proteins, mTOR encompasses two distinct protein complexes: mTOR complex 1 (mTORC1) (which includes raptor, pras40, deptor, and mLST8) and mTOR complex 2 (mTORC2) (which includes rictor, mSin1, protor1/2, deptor, and mLST8) [33]. Through phosphorylation of two key eukaryotic translation regulators, p70S6K (p70-S6 kinase) and EIF4EBP1 (4EBP1, short for eukaryotic translation initiation factor 4E binding protein 1), mTORC1 regulates ribosomal biogenesis and protein synthesis. In addition, mTORC1 also controls lipid synthesis, autophagy, and metabolism by targeting key effectors SREBP1/2, HIF1α, and ULK1/ATG13/FIP200, respectively [32, 33]. mTORC2 directly phosphorylates AKT at S473, and mTORC2 is required for activation of SGK1, known as serum and glucocorticoid-regulated kinase 1, and plays an essential role in multiple processes including cell survival, neuronal excitability, and renal sodium excretion [35,36,37,38]. Collectively, the mTOR pathway regulates cell growth and components of the pathway are key molecules involved in numerous pathological conditions.
Specifically for cancer pathogenesis, many studies have documented the important role of mTOR pathway. Evidence shows that deregulation of protein synthesis controlled by 4E-BP/eIF4E, downstream of mTORC1, plays a central role [39,40,41,42,43]. It is thought that mTOR phosphorylates and represses the inhibitory activity of 4E-BP1 on eIF4E, affecting the translation of mRNA coding for a subset of pro-oncogenic proteins, including cMYC and cyclin D1 [41, 43,44,45,46,47,48,49]. Lipid synthesis is characterized as a hallmark for proliferation of cancer cells [50]. SREBP1, a central pro-lipogenic factor, can be activated by mTORC1 [51]. Autophagy has both tumor suppressive and cancer cell survival protective effects. In a nutrient and oxygen deprivation environment, autophagy makes cancer cells insensitive to these stressors and provides survival advantage. Meanwhile, autophagy may cause apoptosis due to lack of energy storage. Activation of mTORC1 signaling inhibits autophagy in cancer cells and may protect against autophagy-induced apoptosis [33, 52]. In addition to mTORC1, recent studies suggest mTORC2 plays a distinct role in multiple cancer types. The mTORC2-AKT-FOXO circuit regulates proliferation, angiogenesis, and apoptosis [53,54,55].
These evidences point to the importance of mTOR pathway in cancer initiation and progression. Due to the recent characterization of molecular alterations found in HNSCC, we now know that the PI3K/mTOR signaling circuitry is the most frequent dysregulated signaling pathway in HNSCC, as described above. Thus, the use of precise molecular therapeutic approaches to reduce the activity of the mTOR pathway could have anti-cancer effects in HNSCC, and the dissection of the underlying mechanisms may help select the patient population that will benefit the most from this therapy.
5 Targeting mTOR signaling pathway in head and neck cancer
Rapamycin, also known as sirolimus, represents the first generation of mTOR inhibitors. It was firstly used as an immunosuppressant since the 1970s. Despite its anti-cancer activity being discovered in the early 1980s, the application of rapamycin for cancer therapy was not exploited until the late 1990s [32]. A class of drugs that target mTOR, termed rapamycin analogues (also known as rapalogs), were subsequently developed [56,57,58]. Rapamycin and rapalogs block primarily mTOR in its complex 1 (mTORC1) indirectly by binding to FKBP12, while a second generation of mTOR inhibitors block mTOR kinase directly, hence inhibiting both mTORC1 and mTORC2 [34, 57,58,59].
To investigate the effectiveness of mTOR inhibitors in HNSCC, a series of experimental models has been established. Our group pioneered the use of rapamycin as a single agent to treat HNSCC xenografts; rapamycin rapidly decreased mTOR activity, as indicated by its marker, pS6, and caused rapid tumor regression [60]. Since then, several groups demonstrated anti-cancer effect of rapalogs in HNSCC xenografts either as a single agent or when combined with chemotherapy/radiotherapy [61,62,63,64,65,66]. Genetically relevant cell lines are necessary for xenograft models, and in recent studies, we reported the detailed characterization of a large panel of HNSCC-derived cell lines by performing exome and transcriptome sequencing [30]. Not surprisingly, genetic alteration in PIK3CA, HRAS, PTEN, and other agents which results in PI3K-mTOR pathway activation were identified in those panels, consistent with the HNSCC genomic landscape described above. These efforts facilitated the identification of biomarkers for diagnosis and treatment, providing selective precision models for xenograft studies [30, 67]. Patient-derived tumorgraft (PDX) models maintain the tumor heterogeneity of the primary tumor; thus, they may be better for clinical outcome prediction. Many recent studies reported the effectiveness of mTOR inhibitors in HNSCC PDX models [68,69,70]. Meanwhile, chemically induced mouse SCC experimental models were also established. The DMBA-TPA two-stage chemical-induced carcinogenesis is widely used as a mouse SCC experimental model, and in genetically defined animals, such as mice conditionally expressing HPV E6/E7, or lacking Pten or Tgfbr1, DMBA-TPA induces more SCC lesions and more rapidly [71,72,73]. Another carcinogen, 4-nitroquinoline-1-oxide (4NQO) that mimics tobacco use, was optimized in our laboratory as an oral-specific chemical carcinogenesis model [74]. Using drinking water with soluble 4NQO, oral tumors could be developed in the tongue within several weeks. In both chemical carcinogenesis-induced models, persistent mTOR activation was observed, which could be blocked by administration of rapamycin, hence causing regression of SCCs [71,72,73,74]. Overall, these experimental efforts led to a rationale for using mTOR inhibitors as a novel precision therapeutic approach for HNSCC.
Several mTOR inhibitors are being used either alone or in combination with chemotherapy or radiotherapy in HNSCC clinical trials, including rapamycin (sirolimus), everolimus (RAD001), temsirolimus (CCI-779), and others, as summarized in Table 2. Most clinical trials using mTOR inhibitors in HNSCC are under evaluation or have been recently completed. Our study treating newly diagnosed HNSCC patients with rapamycin (NCT01195922) showed encouraging outcomes, as the majority of the patients (15 of 16) gained clinical improvement, including one patient with complete response [75]. Meanwhile, metformin, a drug with well-established safety profile routinely used in type II diabetes patients, has been discovered as a potential anti-cancer agent by reducing mTOR activity in experimental animal models [76, 77]. A multi-institutional phase IIa trial (NCT02581137) in patients with oral premalignant lesion (OPL) has been initiated to evaluate the effect metformin for oral cancer prevention. It is expected that the patients with HNSCC or OPL could benefit from these experimental efforts and relevant clinical trials.
6 mTOR co-targeting strategies in HNSCC to bypass drug resistance
Molecular mechanism-based precision medicine provides promising rationale for cancer therapy. However, the clinical efficacy of several agents is frequently compromised due to the emergence of drug resistance. Specifically for therapeutic drugs targeting the mTOR pathway, despite the fact that promising outcomes were achieved using mTOR inhibitors in experimental models and rapamycin showed encouraging result in the adjuvant setting in a HNSCC clinical trial (NCT01195922, Table 2), advanced HNSCC patients may still display unpredictable drug resistance. One possible reason is that HNSCC patients, similar to other cancer types, usually receive multiple rounds of radiation and chemotherapy. This may cause DNA damage; thus, the consequent genetic alterations and epigenetic regulation can induce the emergence of drug resistance.
Understanding the molecular basis of potential drug resistance has emerged to be a formidable challenge. To date, diverse mechanisms of drug resistance have been discovered, including adaptive changes impacting drug pharmacokinetics (such as absorption, distribution, metabolism, and excretion), structural changes in the drug-binding domain of targeted molecules, and (re)activation of pro-survival signaling pathway. Mechanisms of resistance vary depending on the individual drug. For example, metformin, a novel drug candidate for cancer prevention, is proved to inhibit mTOR signaling [78], and low expression of organic cation transporter 3 (OCT3/SLC22A3), a metformin uptake transporter [77, 79] causes resistance to metformin. The immunodetection of OCT3 expression levels in HNSCC cases provide a surrogate marker which may predict a favorable response to metformin, and on the other hand, it may also suggest that patients with low OCT3 expression may be excluded from metformin trials [77, 79].
Changes in the structure of drug target proteins are frequently observed in experimental models when cell lines were continuously exposed to certain precision medicines. Selective pressure leads tumor cells to develop resistant subpopulations. It is reasonable to predict that a patient with long-term use of a drug may experience similar situations. Structure-based functional design of drugs could be used to optimize existing compounds to target altered drug-insensitive proteins. For example, deep sequencing of MCF7 breast cancer cells acquiring mTOR resistance revealed the juxtaposition of the binding sites of rapamycin and AZD8055, an mTOR kinase inhibitor. Based on this knowledge, it was possible to develop a bivalent mTOR inhibitor. This third-generation mTOR inhibitor, named RapaLink-1, maintained activity in both rapamycin-resistant and AZD8055-resistant xenografts in breast cancer [80]. This drug discovery effort provides opportunities for precision medicine approaches to target mTOR for cancer therapy.
Perhaps more often, targeted agents promote the activation of adaptive survival signaling in tumor cells, either by the same or parallel pathways to mTOR. For example, rapamycin-induced feedback phosphorylation and activation of Akt signaling is frequently reported [81,82,83,84]. Also, mTOR inhibition by rapamycin and other TOR kinase inhibitors induces tyrosine receptor kinase and ERK/MAPK feedback activation [84,85,86,87]. Low expression of 4E binding protein 1 (4EBP1), a primary downstream substrate of mTOR suppressing eukaryotic translation initiation factor 4E (eIF4E), may confer resistance to mTOR inhibitors [39,40,41]. Other dysregulations include p27, PP2A, PIM, and many others, but their roles in resistance to mTOR inhibition are less characterized [33].
To date, precision therapeutic strategies, mTOR inhibition included, are designed based on the concept of oncogene addiction, in which the growth and survival of cancer cells can be often impaired once one single oncogene is inhibited [88]. However, the discoveries of drug resistance bring to attention the importance of co-targeting strategies. In fact, numerous clinical trials using mTOR inhibitors are being carried out in combination with other targeted therapy (Table 2). The knowledge of the mechanism of resistance to mTOR inhibition may provide optimized second target(s) to be combined with as a therapeutic option. For instance, compensatory increased Akt and/or ERK signaling after mTOR inhibition are responsible for tumor relapse and their targeted agents have been widely investigated [81,82,83,84,85,86,87]. Consistent with the above findings, we recently performed a synthetic lethality screen using shRNA libraries in HNSCC cell lines and found that co-targeting numerous molecules involved in ERK/MAPK pathway sensitizes the growth suppressive activities of mTOR inhibition. Indeed, trametinib, a MEK1/2 inhibitor, exhibited a synergistic effect by sensitizing HNSCC to rapamycin [89]. In another study, co-targeting mTOR with cetuximab, a monoclonal antibody targeting EGFR that acts upstream of both Akt and ERK pathway, prevented the growth of HNSCC tumor xenografts by decreasing cell proliferation and lymphangiogenesis [90]. Of importance, safety of cetuximab has been proven and its combination with mTOR inhibitors may minimize their side effects. These pre-clinical efforts have provided a rationale for using mTOR co-targeting strategies in HNSCC patients.
7 mTOR and cancer immunology
Cancers, including HNSCC, are immunosuppressive diseases. Cancer cells normally avoid immune surveillance and anti-tumor immune response by recruiting myeloid-derived suppressor cells (MDSC) and suppressive regulatory T cells (Tregs) [91], while macrophages undergo polarization toward an immune suppressive (M2) tumor-associated macrophage (TAM) phenotype [92,93,94]. In HNSCC, patients often have low absolute lymphocyte counts (ALC), impaired natural killer (NK) cell activity, and poor antigen presentation function compared to healthy volunteers [6]. To recognize cancer cells as foreign instead of self and effectively attack them by the immune system, three general categories, known as immuno-oncology (IO) therapies, could be applied, including checkpoint inhibitors, immune stimulatory cytokines, and cancer vaccines [6, 95]. Recently, revolutionary therapeutic strategies have been used to restore T cell-mediated anti-tumor immunity in HNSCC by targeting immune checkpoint molecules, such a PD-L1 and PD-1. These studies demonstrated immune modulation and durable remissions and led to the recent approval by the FDA of anti-PD-1 antibodies, nivolumab and pembrolizumab, for use in HNSCC treatment [6,7,8,9,10,11]. Numerous trials in HNSCC are being evaluated. However, the overall response rate to these IO therapies in HNSCC is only ~20% [6,7,8,9,10,11]. There is a clear need to identify therapeutic options to enhance the response to these IO agents in HNSCC.
Both precision therapies and IO therapies are novel therapeutic modalities being under clinical evaluation for cancer treatment. This provides numerous potential opportunities for synergistic treatment strategies. Specifically for mTOR inhibition, the mTOR pathway was early considered to be a target of immunosuppressive therapy, and rapamycin has been used in renal transplant patients who were also taking cyclosporine and corticosteroids [96]. Nonetheless, rapamycin-treated patients have similar numbers of myeloid dendritic cells (DCs) and plasmacytoid dendritic cells (pDCs), suggesting that at the dose used, rapamycin does not compromise the DC compartments in patients [97]. Moreover, in cancer patients, multiple trials using single-agent rapamycin (or rapalogs) have shown no evidence of increased incidence of immunosuppression [98,99,100]. Paradoxically, recent basic and clinical studies have associated rapamycin with increased immune responses and potentiation of the activity of IO agents in cancer models [101,102,103,104,105,106,107,108,109]. Thus, it is possible that co-targeting mTOR may potentially enhance rather than reduce the anti-tumor activity of IO agents.
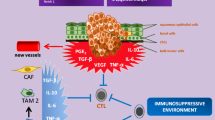
The generation of anti-tumor immune response usually requires multiple steps, including (1) tumor antigen (peptide epitope) capture, (2) effector T cell differentiation, and (3) evasion of negative regulation [91]. Firstly, mTOR inhibitors, similar to many other cancer precision therapeutic options, may cause programmed cancer cell death [110]. The apoptotic tumor cells killed in situ can expose multiple antigens. Similar to a variety of cancer vaccines, those antigens can be processed into major histocompatibility complex (MHC) class I and class II pathways, to activate CD8+ T cells and CD4+ T cells, respectively [91]. Secondly, mTOR has double-edged sword effects during the progress of T cell differentiation. While mTOR activation programs their differentiation into functionally distinct lineages [111], mTOR inhibition drives T cell toward long-lived tumor specific memory T cells [91]. Thirdly, the checkpoint proteins CTLA4 and programmed cell death protein 1 (PD1) are among the major inhibitory molecules suppressing activated T cells [112,113,114,115]. We reported in a HNSCC mouse model that an increase in programmed death-ligand 1 (PD-L1) expression can be elicited by Pten gene deletion [116], suggesting PI3K/mTOR pathway activation contributes to PD-L1/PD1 stimulation in HNSCC. A recent study showed that co-targeting mTOR and PD-L1 enhances tumor control by increasing the IFNγ production capacity in peripheral and tumor-infiltrating CD8 T cells in a syngeneic oral cavity cancer model [109]. Meanwhile, tolerogenic cytokines secreted by regulatory T (Treg) cells and MDSCs also inhibit anti-tumor immune responses [117, 118]. The expression of inhibitory molecules, such as interleukin-10 (IL-10) and transforming growth factor-β (TGFβ) could be decreased by mTOR inhibition [119,120,121,122] (Fig. 2).

mTOR inhibition may enhance the anti-tumor immune response. A DCs capture tumor antigens and present them to T cells through MHC (class I and class II) pathways. mTOR inhibition induces apoptotic cells, which may contribute as vaccination in situ. B mTOR inhibition drives T cells toward long-lived tumor specific memory T cells. C The inhibitory molecule PDL1 from tumor cells can bind PD1 in T cells and weaken effector T cell’s function. Co-targeting mTOR may reduce PD-L1 expression, restraining PD-L1/PD1 mediated inhibition. Effector T cells refer to as cytotoxic T cells (CD8+) and helper T cells (CD4+). D Immunosuppressive cytokines secreted by Tregs and MDSCs inhibit anti-tumor response. mTOR inhibition may prevent cytokine secretion by regulation of their translational control. DCs dendritic cells, MHC major histocompatibility complex, Tregs regulatory T cell, MDSCs myeloid-derived suppressor cells
Therefore, although counterintuitive, emerging evidence supports that PI3K/mTOR inhibition can be optimized to enhance, rather than suppress, the anti-tumor immune response by overcoming immune evasion in a context-dependent fashion. Thus co-targeting the mTOR signaling circuitry based on the genetic stratification of PI3K/mTOR network subtypes with IO agents, may represent a novel precision immune therapeutic approach for HNSCC.
8 Conclusion
In summary, the importance of mTOR signaling circuitry in HNSCC has been well documented, and targeting mTOR as a precision therapy approach in HNSCC has been widely investigated in experimental models, and recently tested in clinical trials. Newly developed genetic approaches could be applied to evaluate the status of mTOR activation which may predict individuals’ clinical response to this precision therapy. This mechanism-based therapeutic approach may help select patient populations that may benefit the most from the concomitant administration of mTOR inhibitors, and also provide improved therapeutic options, namely co-targeting strategies to circumvent innate and acquired resistance to mTOR inhibitors. Furthermore, there is now a strong rationale for co-targeting mTOR with IO agents to enhance their anti-tumor activity. Overall, we can expect that the development of novel mTOR co-targeting strategies may achieve durable responses and cancer remission, hence increasing the life expectancy and quality of life of HNSCC patients.
Change history
05 September 2018
The author’s name was tagged incorrectly in XML. “Silvio Gutkind J” should be tagged as “J. Silvio Gutkind”. Given name is “J. Silvio” and the Family name is “Gutkind”.
05 September 2018
The author���s name was tagged incorrectly in XML. ���Silvio Gutkind J��� should be tagged as ���J. Silvio Gutkind���. Given name is ���J. Silvio��� and the Family name is ���Gutkind���.
References
The Cancer Genome Atlas, N. (2015). Comprehensive genomic characterization of head and neck squamous cell carcinomas. [Article]. Nature, 517(7536), 576–582. doi:10.1038/nature14129.
Leemans, C. R., Braakhuis, B. J., & Brakenhoff, R. H. (2011). The molecular biology of head and neck cancer. Nature Reviews. Cancer, 11(1), 9–22. doi:10.1038/nrc2982.
Iglesias-Bartolome, R., Martin, D., & Gutkind, J. S. (2013). Exploiting the head and neck cancer oncogenome: widespread PI3K-mTOR pathway alterations and novel molecular targets. [Research Support, N.I.H., Intramural]. Cancer Discovery, 3(7), 722–725. doi:10.1158/2159-8290.CD-13-0239.
Vermorken, J. B., Mesia, R., Rivera, F., Remenar, E., Kawecki, A., Rottey, S., et al. (2008). Platinum-based chemotherapy plus cetuximab in head and neck cancer. The New England Journal of Medicine, 359(11), 1116–1127. doi:10.1056/NEJMoa0802656.
Bonner, J. A., Harari, P. M., Giralt, J., Azarnia, N., Shin, D. M., Cohen, R. B., et al. (2006). Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. The New England Journal of Medicine, 354(6), 567–578. doi:10.1056/NEJMoa053422.
Ferris, R. L. (2015). Immunology and immunotherapy of head and neck cancer. Journal of Clinical Oncology, 33(29), 3293–3304. doi:10.1200/JCO.2015.61.1509.
Ferris, R. L., Blumenschein Jr., G., Fayette, J., Guigay, J., Colevas, A. D., Licitra, L., et al. (2016). Nivolumab for recurrent squamous-cell carcinoma of the head and neck. The New England Journal of Medicine, 375(19), 1856–1867. doi:10.1056/NEJMoa1602252.
Shayan, G., Srivastava, R., Li, J., Schmitt, N., Kane, L. P., & Ferris, R. L. (2017). Adaptive resistance to anti-PD1 therapy by Tim-3 upregulation is mediated by the PI3K-Akt pathway in head and neck cancer. Oncoimmunology, 6(1), e1261779. doi:10.1080/2162402X.2016.1261779.
Economopoulou, P., Perisanidis, C., Giotakis, E. I., & Psyrri, A. (2016). The emerging role of immunotherapy in head and neck squamous cell carcinoma (HNSCC): anti-tumor immunity and clinical applications. Ann Transl Med, 4(9), 173. doi:10.21037/atm.2016.03.34.
Chow, L. Q., Haddad, R., Gupta, S., Mahipal, A., Mehra, R., Tahara, M., et al. (2016). Antitumor activity of pembrolizumab in biomarker-unselected patients with recurrent and/or metastatic head and neck squamous cell carcinoma: results from the phase Ib KEYNOTE-012 expansion cohort. Journal of Clinical Oncology. doi:10.1200/JCO.2016.68.1478.
Ott, P. A., Bang, Y. J., Berton-Rigaud, D., Elez, E., Pishvaian, M. J., Rugo, H. S., et al. (2017). Safety and antitumor activity of pembrolizumab in advanced programmed death ligand 1-positive endometrial cancer: results from the KEYNOTE-028 study. Journal of Clinical Oncology, JCO2017725952. doi:10.1200/JCO.2017.72.5952.
Larkin, J., Hodi, F. S., & Wolchok, J. D. (2015). Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. The New England Journal of Medicine, 373(13), 1270–1271. doi:10.1056/NEJMc1509660.
Ribas, A., Puzanov, I., Dummer, R., Schadendorf, D., Hamid, O., Robert, C., et al. (2015). Pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory melanoma (KEYNOTE-002): a randomised, controlled, phase 2 trial. The Lancet Oncology, 16(8), 908–918. doi:10.1016/S1470-2045(15)00083-2.
Robert, C., Schachter, J., Long, G. V., Arance, A., Grob, J. J., Mortier, L., et al. (2015). Pembrolizumab versus ipilimumab in advanced melanoma. The New England Journal of Medicine, 372(26), 2521–2532. doi:10.1056/NEJMoa1503093.
Garraway, L. A., & Lander, E. S. (2013). Lessons from the cancer genome. Cell, 153(1), 17–37. doi:10.1016/j.cell.2013.03.002.
Pickering, C. R., Zhang, J., Yoo, S. Y., Bengtsson, L., Moorthy, S., Neskey, D. M., et al. (2013). Integrative genomic characterization of oral squamous cell carcinoma identifies frequent somatic drivers. [Research Support, N.I.H., Extramural]. Cancer Discovery, 3(7), 770–781. doi:10.1158/2159-8290.CD-12-0537.
Lui, V. W., Hedberg, M. L., Li, H., Vangara, B. S., Pendleton, K., Zeng, Y., et al. (2013). Frequent mutation of the PI3K pathway in head and neck cancer defines predictive biomarkers. Cancer Discovery, 3(7), 761–769.
Stransky, N., Egloff, A. M., Tward, A. D., Kostic, A. D., Cibulskis, K., Sivachenko, A., et al. (2011). The mutational landscape of head and neck squamous cell carcinoma. Science, 333(6046), 1157–1160. doi:10.1126/science.1208130.
Agrawal, N., Frederick, M. J., Pickering, C. R., Bettegowda, C., Chang, K., Li, R. J., et al. (2011). Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science, 333(6046), 1154–1157. doi:10.1126/science.1206923.
Amornphimoltham, P., Roth, S. J., Ideker, T., & Silvio Gutkind, J. (2017). Targeting the mTOR signaling circuitry in head and neck cancer. In S. Warnakulasuriya & Z. Khan (Eds.), Squamous cell carcinoma: molecular therapeutic targets (pp. 163–181). Dordrecht: Springer Netherlands.
D'Souza, G., Kreimer, A. R., Viscidi, R., Pawlita, M., Fakhry, C., Koch, W. M., et al. (2007). Case-control study of human papillomavirus and oropharyngeal cancer. The New England Journal of Medicine, 356(19), 1944–1956. doi:10.1056/NEJMoa065497.
Gillison, M. L., & Shah, K. V. (2001). Human papillomavirus-associated head and neck squamous cell carcinoma: mounting evidence for an etiologic role for human papillomavirus in a subset of head and neck cancers. [Review]. Current Opinion in Oncology, 13(3), 183–188.
Chaturvedi, A. K., Engels, E. A., Anderson, W. F., & Gillison, M. L. (2008). Incidence trends for human papillomavirus-related and -unrelated oral squamous cell carcinomas in the United States. [Research Support, N.I.H., Intramural]. Journal of Clinical Oncology, 26(4), 612–619. doi:10.1200/JCO.2007.14.1713.
Ryerson, A. B., Peters, E. S., Coughlin, S. S., Chen, V. W., Gillison, M. L., Reichman, M. E., et al. (2008). Burden of potentially human papillomavirus-associated cancers of the oropharynx and oral cavity in the US, 1998-2003. [Research Support, U.S. Gov’t, P.H.S.] Cancer, 113(10 Suppl), 2901–2909. doi:10.1002/cncr.23745.
Shiboski, C. H., Schmidt, B. L., & Jordan, R. C. (2005). Tongue and tonsil carcinoma: Increasing trends in the U.S. population ages 20–44 years. Cancer, 103(9), 1843–1849. doi:10.1002/cncr.20998.
Scheffner, M., Werness, B. A., Huibregtse, J. M., Levine, A. J., & Howley, P. M. (1990). The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell, 63(6), 1129–1136.
Werness, B. A., Levine, A. J., & Howley, P. M. (1990). Association of human papillomavirus types 16 and 18 E6 proteins with p53. Science, 248(4951), 76–79.
Huang, P. S., Patrick, D. R., Edwards, G., Goodhart, P. J., Huber, H. E., Miles, L., et al. (1993). Protein domains governing interactions between E2F, the retinoblastoma gene product, and human papillomavirus type 16 E7 protein. Molecular and Cellular Biology, 13(2), 953–960.
Nichols, A. C., Palma, D. A., Chow, W., Tan, S., Rajakumar, C., Rizzo, G., et al. (2013). High frequency of activating PIK3CA mutations in human papillomavirus-positive oropharyngeal cancer. JAMA Otolaryngology. Head & Neck Surgery, 139(6), 617–622. doi:10.1001/jamaoto.2013.3210.
Martin, D., Abba, M. C., Molinolo, A. A., Vitale-Cross, L., Wang, Z., Zaida, M., et al. (2014). The head and neck cancer cell oncogenome: a platform for the development of precision molecular therapies. Oncotarget, 5(19), 8906–8923. doi:10.18632/oncotarget.2417.
Molinolo, A. A., Marsh, C., El Dinali, M., Gangane, N., Jennison, K., Hewitt, S., et al. (2012). mTOR as a molecular target in HPV-associated oral and cervical squamous carcinomas. Clinical Cancer Research, 18(9), 2558–2568. doi:10.1158/1078-0432.CCR-11-2824.
Guertin, D. A., & Sabatini, D. M. (2007). Defining the role of mTOR in cancer. Cancer Cell, 12(1), 9–22. doi:10.1016/j.ccr.2007.05.008.
Laplante, M., & Sabatini, D. M. (2012). mTOR signaling in growth control and disease. Cell, 149(2), 274–293. doi:10.1016/j.cell.2012.03.017.
Thoreen, C. C., & Sabatini, D. M. (2009). Rapamycin inhibits mTORC1, but not completely. Autophagy, 5(5), 725–726.
Garcia-Martinez, J. M., & Alessi, D. R. (2008). mTOR complex 2 (mTORC2) controls hydrophobic motif phosphorylation and activation of serum- and glucocorticoid-induced protein kinase 1 (SGK1). The Biochemical Journal, 416(3), 375–385. doi:10.1042/BJ20081668.
Heikamp, E. B., Patel, C. H., Collins, S., Waickman, A., Oh, M. H., Sun, I. H., et al. (2014). The AGC kinase SGK1 regulates TH1 and TH2 differentiation downstream of the mTORC2 complex. Nature Immunology, 15(5), 457–464. doi:10.1038/ni.2867.
Yan, L., Mieulet, V., & Lamb, R. F. (2008). mTORC2 is the hydrophobic motif kinase for SGK1. The Biochemical Journal, 416(3), e19–e21. doi:10.1042/BJ20082202.
Pearce, L. R., Sommer, E. M., Sakamoto, K., Wullschleger, S., & Alessi, D. R. (2011). Protor-1 is required for efficient mTORC2-mediated activation of SGK1 in the kidney. The Biochemical Journal, 436(1), 169–179. doi:10.1042/BJ20102103.
Hara, K., Yonezawa, K., Kozlowski, M. T., Sugimoto, T., Andrabi, K., Weng, Q. P., et al. (1997). Regulation of eIF-4E BP1 phosphorylation by mTOR. The Journal of Biological Chemistry, 272(42), 26457–26463.
Gingras, A. C., Kennedy, S. G., O'Leary, M. A., Sonenberg, N., & Hay, N. (1998). 4E-BP1, a repressor of mRNA translation, is phosphorylated and inactivated by the Akt(PKB) signaling pathway. Genes & Development, 12(4), 502–513.
Gingras, A. C., Gygi, S. P., Raught, B., Polakiewicz, R. D., Abraham, R. T., Hoekstra, M. F., et al. (1999). Regulation of 4E-BP1 phosphorylation: a novel two-step mechanism. Genes & Development, 13(11), 1422–1437.
Hinnebusch, A. G. (2012). Translational homeostasis via eIF4E and 4E-BP1. Molecular Cell, 46(6), 717–719. doi:10.1016/j.molcel.2012.06.001.
Faller, W. J., Jackson, T. J., Knight, J. R., Ridgway, R. A., Jamieson, T., Karim, S. A., et al. (2015). mTORC1-mediated translational elongation limits intestinal tumour initiation and growth. Nature, 517(7535), 497–500. doi:10.1038/nature13896.
Hay, N., & Sonenberg, N. (2004). Upstream and downstream of mTOR. Genes & Development, 18(16), 1926–1945. doi:10.1101/gad.1212704.
Choo, A. Y., Yoon, S. O., Kim, S. G., Roux, P. P., & Blenis, J. (2008). Rapamycin differentially inhibits S6Ks and 4E-BP1 to mediate cell-type-specific repression of mRNA translation. Proceedings of the National Academy of Sciences of the United States of America, 105(45), 17414–17419. doi:10.1073/pnas.0809136105.
Dilling, M. B., Germain, G. S., Dudkin, L., Jayaraman, A. L., Zhang, X. W., Harwood, F. C., et al. (2002). 4E-binding proteins, the suppressors of eukaryotic initiation factor 4E, are down-regulated in cells with acquired or intrinsic resistance to rapamycin. Journal of Biological Chemistry, 277(16), 13907–13917. doi:10.1074/jbc.M110782200.
Lynch, M., Fitzgerald, C., Johnston, K. A., Wang, S., & Schmidt, E. V. (2004). Activated eIF4E-binding protein slows G1 progression and blocks transformation by c-myc without inhibiting cell growth. The Journal of Biological Chemistry, 279(5), 3327–3339. doi:10.1074/jbc.M310872200.
Rosenwald, I. B., Lazaris-Karatzas, A., Sonenberg, N., & Schmidt, E. V. (1993). Elevated levels of cyclin D1 protein in response to increased expression of eukaryotic initiation factor 4E. Molecular and Cellular Biology, 13(12), 7358–7363.
Rousseau, D., Kaspar, R., Rosenwald, I., Gehrke, L., & Sonenberg, N. (1996). Translation initiation of ornithine decarboxylase and nucleocytoplasmic transport of cyclin D1 mRNA are increased in cells overexpressing eukaryotic initiation factor 4E. Proceedings of the National Academy of Sciences of the United States of America, 93(3), 1065–1070.
Menendez, J. A., & Lupu, R. (2007). Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nature Reviews. Cancer, 7(10), 763–777. doi:10.1038/nrc2222.
Duvel, K., Yecies, J. L., Menon, S., Raman, P., Lipovsky, A. I., Souza, A. L., et al. (2010). Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Molecular Cell, 39(2), 171–183. doi:10.1016/j.molcel.2010.06.022.
Inoki, K., Li, Y., Xu, T., & Guan, K. L. (2003). Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes & Development, 17(15), 1829–1834. doi:10.1101/gad.1110003.
Hay, N. (2011). Interplay between FOXO, TOR, and Akt. Biochimica et Biophysica Acta, 1813(11), 1965–1970. doi:10.1016/j.bbamcr.2011.03.013.
Chen, C. C., Jeon, S. M., Bhaskar, P. T., Nogueira, V., Sundararajan, D., Tonic, I., et al. (2010). FoxOs inhibit mTORC1 and activate Akt by inducing the expression of Sestrin3 and Rictor. Developmental Cell, 18(4), 592–604. doi:10.1016/j.devcel.2010.03.008.
Zhang, X. B., Tang, N. M., Hadden, T. J., & Rishi, A. K. (2011). Akt, FoxO and regulation of apoptosis. Biochimica et Biophysica Acta-Molecular Cell Research, 1813(11), 1978–1986. doi:10.1016/j.bbamcr.2011.03.010.
Vilar, E., Perez-Garcia, J., & Tabernero, J. (2011). Pushing the envelope in the mTOR pathway: the second generation of inhibitors. Molecular Cancer Therapeutics, 10(3), 395–403. doi:10.1158/1535-7163.MCT-10-0905.
Ballou, L. M., & Lin, R. Z. (2008). Rapamycin and mTOR kinase inhibitors. Journal of Chemical Biology, 1(1–4), 27–36. doi:10.1007/s12154-008-0003-5.
Wander, S. A., Hennessy, B. T., & Slingerland, J. M. (2011). Next-generation mTOR inhibitors in clinical oncology: how pathway complexity informs therapeutic strategy. The Journal of Clinical Investigation, 121(4), 1231–1241. doi:10.1172/JCI44145.
Janes, M. R., Vu, C., Mallya, S., Shieh, M. P., Limon, J. J., Li, L. S., et al. (2013). Efficacy of the investigational mTOR kinase inhibitor MLN0128/INK128 in models of B-cell acute lymphoblastic leukemia. Leukemia, 27(3), 586–594. doi:10.1038/leu.2012.276.
Amornphimoltham, P., Patel, V., Sodhi, A., Nikitakis, N. G., Sauk, J. J., Sausville, E. A., et al. (2005). Mammalian target of rapamycin, a molecular target in squamous cell carcinomas of the head and neck. Cancer Research, 65(21), 9953–9961. doi:10.1158/0008-5472.CAN-05-0921.
Ekshyyan, O., Rong, Y., Rong, X., Pattani, K. M., Abreo, F., Caldito, G., et al. (2009). Comparison of radiosensitizing effects of the mammalian target of rapamycin inhibitor CCI-779 to cisplatin in experimental models of head and neck squamous cell carcinoma. Molecular Cancer Therapeutics, 8(8), 2255–2265. doi:10.1158/1535-7163.mct-08-1184.
Shin, D. H., Min, H. Y., El-Naggar, A. K., Lippman, S. M., Glisson, B., & Lee, H. Y. (2011). Akt/mTOR counteract the antitumor activities of cixutumumab, an anti-insulin-like growth factor I receptor monoclonal antibody. Molecular Cancer Therapeutics, 10(12), 2437–2448. doi:10.1158/1535-7163.mct-11-0235.
Cassell, A., Freilino, M. L., Lee, J., Barr, S., Wang, L., Panahandeh, M. C., et al. (2012). Targeting TORC1/2 enhances sensitivity to EGFR inhibitors in head and neck cancer preclinical models. Neoplasia, 14(11), 1005–1014.
Zhong, R., Pytynia, M., Pelizzari, C., & Spiotto, M. (2014). Bioluminescent imaging of HPV-positive oral tumor growth and its response to image-guided radiotherapy. Cancer Research, 74(7), 2073–2081. doi:10.1158/0008-5472.can-13-2993.
D'Amato, V., Rosa, R., D'Amato, C., Formisano, L., Marciano, R., Nappi, L., et al. (2014). The dual PI3K/mTOR inhibitor PKI-587 enhances sensitivity to cetuximab in EGFR-resistant human head and neck cancer models. British Journal of Cancer, 110(12), 2887–2895. doi:10.1038/bjc.2014.241.
Coppock, J. D., Vermeer, P. D., Vermeer, D. W., Lee, K. M., Miskimins, W. K., Spanos, W. C., et al. (2016). mTOR inhibition as an adjuvant therapy in a metastatic model of HPV+ HNSCC. Oncotarget, 7(17), 24228–24241. doi:10.18632/oncotarget.8286.
Fadlullah, M. Z., Chiang, I. K., Dionne, K. R., Yee, P. S., Gan, C. P., Sam, K. K., et al. (2016). Genetically-defined novel oral squamous cell carcinoma cell lines for the development of molecular therapies. Oncotarget, 7(19), 27802–27818. doi:10.18632/oncotarget.8533.
Klinghammer, K., Raguse, J. D., Plath, T., Albers, A. E., Joehrens, K., Zakarneh, A., et al. (2015). A comprehensively characterized large panel of head and neck cancer patient-derived xenografts identifies the mTOR inhibitor everolimus as potential new treatment option. International Journal of Cancer, 136(12), 2940–2948. doi:10.1002/ijc.29344.
Mazumdar, T., Byers, L. A., Ng, P. K., Mills, G. B., Peng, S., Diao, L., et al. (2014). A comprehensive evaluation of biomarkers predictive of response to PI3K inhibitors and of resistance mechanisms in head and neck squamous cell carcinoma. Molecular Cancer Therapeutics, 13(11), 2738–2750. doi:10.1158/1535-7163.MCT-13-1090.
Tentler, J. J., Tan, A. C., Weekes, C. D., Jimeno, A., Leong, S., Pitts, T. M., et al. (2012). Patient-derived tumour xenografts as models for oncology drug development. Nature Reviews. Clinical Oncology, 9(6), 338–350. doi:10.1038/nrclinonc.2012.61.
Amornphimoltham, P., Leelahavanichkul, K., Molinolo, A., Patel, V., & Gutkind, J. S. (2008). Inhibition of mammalian target of rapamycin by rapamycin causes the regression of carcinogen-induced skin tumor lesions. Clinical Cancer Research, 14(24), 8094–8101. doi:10.1158/1078-0432.ccr-08-0703.
Callejas-Valera, J. L., Iglesias-Bartolome, R., Amornphimoltham, P., Palacios-Garcia, J., Martin, D., Califano, J. A., et al. (2016). mTOR inhibition prevents rapid-onset of carcinogen-induced malignancies in a novel inducible HPV-16 E6/E7 mouse model. Carcinogenesis, 37(10), 1014–1025. doi:10.1093/carcin/bgw086.
Sun, Z. J., Zhang, L., Hall, B., Bian, Y., Gutkind, J. S., & Kulkarni, A. B. (2012). Chemopreventive and chemotherapeutic actions of mTOR inhibitor in genetically defined head and neck squamous cell carcinoma mouse model. Clinical Cancer Research, 18(19), 5304–5313. doi:10.1158/1078-0432.ccr-12-1371.
Czerninski, R., Amornphimoltham, P., Patel, V., Molinolo, A. A., & Gutkind, J. S. (2009). Targeting mammalian target of rapamycin by rapamycin prevents tumor progression in an oral-specific chemical carcinogenesis model. Cancer Prevention Research (Philadelphia, Pa.), 2(1), 27–36. doi:10.1158/1940-6207.capr-08-0147.
Shirai, K., Day, T. A., Szabo, E., Waes, C. V., O'Brien, P. E., Matheus, M. G., et al. (2015). A pilot, single arm, prospective trial using neoadjuvant rapamycin prior to definitive therapy in head and neck squamous cell carcinoma. Journal of Clinical Oncology, 33(15_suppl), 6071–6071. doi:10.1200/jco.2015.33.15_suppl.6071.
Vitale-Cross, L., Molinolo, A. A., Martin, D., Younis, R. H., Maruyama, T., Patel, V., et al. (2012). Metformin prevents the development of oral squamous cell carcinomas from carcinogen-induced premalignant lesions. Cancer Prevention Research, 5(4), 562–573. doi:10.1158/1940-6207.CAPR-11-0502.
Madera, D., Vitale-Cross, L., Martin, D., Schneider, A., Molinolo, A. A., Gangane, N., et al. (2015). Prevention of tumor growth driven by PIK3CA and HPV oncogenes by targeting mTOR signaling with metformin in oral squamous carcinomas expressing OCT3. Cancer Prevention Research, 8(3), 197–207. doi:10.1158/1940-6207.CAPR-14-0348.
Viollet, B., Guigas, B., Sanz Garcia, N., Leclerc, J., Foretz, M., & Andreelli, F. (2012). Cellular and molecular mechanisms of metformin: an overview. Clinical Science (London, England), 122(6), 253–270. doi:10.1042/CS20110386.
Chen, L., Pawlikowski, B., Schlessinger, A., More, S. S., Stryke, D., Johns, S. J., et al. (2010). Role of organic cation transporter 3 (SLC22A3) and its missense variants in the pharmacologic action of metformin. Pharmacogenetics and Genomics, 20(11), 687–699. doi:10.1097/FPC.0b013e32833fe789.
Rodrik-Outmezguine, V. S., Okaniwa, M., Yao, Z., Novotny, C. J., McWhirter, C., Banaji, A., et al. (2016). Overcoming mTOR resistance mutations with a new-generation mTOR inhibitor. Nature, 534(7606), 272–276. doi:10.1038/nature17963.
Xue, Q., Nagy, J. A., Manseau, E. J., Phung, T. L., Dvorak, H. F., & Benjamin, L. E. (2009). Rapamycin inhibition of the Akt/mTOR pathway blocks select stages of VEGF-A164-driven angiogenesis, in part by blocking S6Kinase. Arteriosclerosis, Thrombosis, and Vascular Biology, 29(8), 1172–1178. doi:10.1161/ATVBAHA.109.185918.
Rodrik-Outmezguine, V. S., Chandarlapaty, S., Pagano, N. C., Poulikakos, P. I., Scaltriti, M., Moskatel, E., et al. (2011). mTOR kinase inhibition causes feedback-dependent biphasic regulation of AKT signaling. Cancer Discovery, 1(3), 248–259. doi:10.1158/2159-8290.CD-11-0085.
Wan, X., Harkavy, B., Shen, N., Grohar, P., & Helman, L. J. (2007). Rapamycin induces feedback activation of Akt signaling through an IGF-1R-dependent mechanism. Oncogene, 26(13), 1932–1940. doi:10.1038/sj.onc.1209990.
Chen, X. G., Liu, F., Song, X. F., Wang, Z. H., Dong, Z. Q., Hu, Z. Q., et al. (2010). Rapamycin regulates Akt and ERK phosphorylation through mTORC1 and mTORC2 signaling pathways. Molecular Carcinogenesis, 49(6), 603–610. doi:10.1002/mc.20628.
Carracedo, A., Ma, L., Teruya-Feldstein, J., Rojo, F., Salmena, L., Alimonti, A., et al. (2008). Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. The Journal of Clinical Investigation, 118(9), 3065–3074. doi:10.1172/JCI34739.
Sunayama, J., Matsuda, K., Sato, A., Tachibana, K., Suzuki, K., Narita, Y., et al. (2010). Crosstalk between the PI3K/mTOR and MEK/ERK pathways involved in the maintenance of self-renewal and tumorigenicity of glioblastoma stem-like cells. Stem Cells, 28(11), 1930–1939. doi:10.1002/stem.521.
Chappell, W. H., Steelman, L. S., Long, J. M., Kempf, R. C., Abrams, S. L., Franklin, R. A., et al. (2011). Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR inhibitors: rationale and importance to inhibiting these pathways in human health. Oncotarget, 2(3), 135–164. doi:10.18632/oncotarget.240.
Weinstein, I. B., & Joe, A. (2008). Oncogene addiction. Cancer Research, 68(9), 3077–3080; discussion 3080, doi:10.1158/0008-5472.CAN-07-3293.
Yamaguchi, K., Iglesias-Bartolome, R., Wang, Z., Callejas-Valera, J. L., Amornphimoltham, P., Molinolo, A. A., et al. (2016). A synthetic-lethality RNAi screen reveals an ERK-mTOR co-targeting pro-apoptotic switch in PIK3CA+ oral cancers. Oncotarget, 7(10), 10696–10709. doi:10.18632/oncotarget.7372.
Wang, Z., Martin, D., Molinolo, A. A., Patel, V., Iglesias-Bartolome, R., Degese, M. S., et al. (2014). mTOR co-targeting in cetuximab resistance in head and neck cancers harboring PIK3CA and RAS mutations. Journal of the National Cancer Institute, 106(9). doi:10.1093/jnci/dju215.
Vanneman, M., & Dranoff, G. (2012). Combining immunotherapy and targeted therapies in cancer treatment. Nature Reviews. Cancer, 12(4), 237–251. doi:10.1038/nrc3237.
Noy, R., & Pollard, J. W. (2014). Tumor-associated macrophages: from mechanisms to therapy. Immunity, 41(1), 49–61. doi:10.1016/j.immuni.2014.06.010.
Sica, A., Schioppa, T., Mantovani, A., & Allavena, P. (2006). Tumour-associated macrophages are a distinct M2 polarised population promoting tumour progression: potential targets of anti-cancer therapy. European Journal of Cancer, 42(6), 717–727. doi:10.1016/j.ejca.2006.01.003.
Chen, J. J., Lin, Y. C., Yao, P. L., Yuan, A., Chen, H. Y., Shun, C. T., et al. (2005). Tumor-associated macrophages: the double-edged sword in cancer progression. Journal of Clinical Oncology, 23(5), 953–964. doi:10.1200/JCO.2005.12.172.
Farkona, S., Diamandis, E. P., & Blasutig, I. M. (2016). Cancer immunotherapy: the beginning of the end of cancer? BMC Medicine, 14, 73. doi:10.1186/s12916-016-0623-5.
Haydar, A. A., Denton, M., West, A., Rees, J., & Goldsmith, D. J. (2004). Sirolimus-induced pneumonitis: three cases and a review of the literature. American Journal of Transplantation, 4(1), 137–139.
Weichhart, T., Hengstschlager, M., & Linke, M. (2015). Regulation of innate immune cell function by mTOR. Nature Reviews. Immunology, 15(10), 599–614. doi:10.1038/nri3901.
O'Donnell, A., Faivre, S., Burris 3rd, H. A., Rea, D., Papadimitrakopoulou, V., Shand, N., et al. (2008). Phase I pharmacokinetic and pharmacodynamic study of the oral mammalian target of rapamycin inhibitor everolimus in patients with advanced solid tumors. Journal of Clinical Oncology, 26(10), 1588–1595. doi:10.1200/JCO.2007.14.0988.
Bissler, J. J., McCormack, F. X., Young, L. R., Elwing, J. M., Chuck, G., Leonard, J. M., et al. (2008). Sirolimus for angiomyolipoma in tuberous sclerosis complex or lymphangioleiomyomatosis. The New England Journal of Medicine, 358(2), 140–151. doi:10.1056/NEJMoa063564.
Hahnel, P. S., Thaler, S., Antunes, E., Huber, C., Theobald, M., & Schuler, M. (2008). Targeting AKT signaling sensitizes cancer to cellular immunotherapy. Cancer Research, 68(10), 3899–3906. doi:10.1158/0008-5472.CAN-07-6286.
Dao, V., Liu, Y., Pandeswara, S., Svatek, R. S., Gelfond, J. A., Liu, A., et al. (2016). Immune-stimulatory effects of rapamycin are mediated by stimulation of antitumor gammadelta T cells. Cancer Research, 76(20), 5970–5982. doi:10.1158/0008-5472.CAN-16-0091.
Rao, R. R., Li, Q., Odunsi, K., & Shrikant, P. A. (2010). The mTOR kinase determines effector versus memory CD8+ T cell fate by regulating the expression of transcription factors T-bet and Eomesodermin. Immunity, 32(1), 67–78. doi:10.1016/j.immuni.2009.10.010.
Mannick, J. B., Del Giudice, G., Lattanzi, M., Valiante, N. M., Praestgaard, J., Huang, B., et al. (2014). mTOR inhibition improves immune function in the elderly. Science Translational Medicine, 6(268), 268ra179. doi:10.1126/scitranslmed.3009892.
Pollizzi, K. N., Patel, C. H., Sun, I. H., Oh, M. H., Waickman, A. T., Wen, J., et al. (2015). mTORC1 and mTORC2 selectively regulate CD8(+) T cell differentiation. The Journal of Clinical Investigation, 125(5), 2090–2108. doi:10.1172/JCI77746.
Jiang, Q., Weiss, J. M., Back, T., Chan, T., Ortaldo, J. R., Guichard, S., et al. (2011). mTOR kinase inhibitor AZD8055 enhances the immunotherapeutic activity of an agonist CD40 antibody in cancer treatment. Cancer Research, 71(12), 4074–4084. doi:10.1158/0008-5472.CAN-10-3968.
Wang, Y., Wang, X. Y., Subjeck, J. R., Shrikant, P. A., & Kim, H. L. (2011). Temsirolimus, an mTOR inhibitor, enhances anti-tumour effects of heat shock protein cancer vaccines. British Journal of Cancer, 104(4), 643–652. doi:10.1038/bjc.2011.15.
Li, Q., Rao, R., Vazzana, J., Goedegebuure, P., Odunsi, K., Gillanders, W., et al. (2012). Regulating mammalian target of rapamycin to tune vaccination-induced CD8(+) T cell responses for tumor immunity. Journal of Immunology, 188(7), 3080–3087. doi:10.4049/jimmunol.1103365.
Lastwika, K. J., Wilson 3rd, W., Li, Q. K., Norris, J., Xu, H., Ghazarian, S. R., et al. (2016). Control of PD-L1 expression by oncogenic activation of the AKT-mTOR pathway in non-small cell lung cancer. Cancer Research, 76(2), 227–238. doi:10.1158/0008-5472.CAN-14-3362.
Moore, E. C., Cash, H. A., Caruso, A. M., Uppaluri, R., Hodge, J. W., Van Waes, C., et al. (2016). Enhanced tumor control with combination mTOR and PD-L1 inhibition in syngeneic oral cavity cancers. Cancer Immunology Research, 4(7), 611–620. doi:10.1158/2326-6066.CIR-15-0252.
Dormond, O., Madsen, J. C., & Briscoe, D. M. (2007). The effects of mTOR-Akt interactions on anti-apoptotic signaling in vascular endothelial cells. The Journal of Biological Chemistry, 282(32), 23679–23686. doi:10.1074/jbc.M700563200.
Chi, H. (2012). Regulation and function of mTOR signalling in T cell fate decisions. Nature Reviews. Immunology, 12(5), 325–338. doi:10.1038/nri3198.
Parry, R. V., Chemnitz, J. M., Frauwirth, K. A., Lanfranco, A. R., Braunstein, I., Kobayashi, S. V., et al. (2005). CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Molecular and Cellular Biology, 25(21), 9543–9553. doi:10.1128/MCB.25.21.9543-9553.2005.
Mahoney, K. M., Rennert, P. D., & Freeman, G. J. (2015). Combination cancer immunotherapy and new immunomodulatory targets. Nature Reviews. Drug Discovery, 14(8), 561–584. doi:10.1038/nrd4591.
Buchbinder, E. I., & Desai, A. (2016). CTLA-4 and PD-1 pathways: similarities, differences, and implications of their inhibition. American Journal of Clinical Oncology, 39(1), 98–106. doi:10.1097/COC.0000000000000239.
Kaufmann, D. E., & Walker, B. D. (2009). PD-1 and CTLA-4 inhibitory cosignaling pathways in HIV infection and the potential for therapeutic intervention. Journal of Immunology, 182(10), 5891–5897. doi:10.4049/jimmunol.0803771.
Lyford-Pike, S., Peng, S., Young, G. D., Taube, J. M., Westra, W. H., Akpeng, B., et al. (2013). Evidence for a role of the PD-1:PD-L1 pathway in immune resistance of HPV-associated head and neck squamous cell carcinoma. Cancer Research, 73(6), 1733–1741. doi:10.1158/0008-5472.CAN-12-2384.
Zamarron, B. F., & Chen, W. (2011). Dual roles of immune cells and their factors in cancer development and progression. International Journal of Biological Sciences, 7(5), 651–658.
Lindau, D., Gielen, P., Kroesen, M., Wesseling, P., & Adema, G. J. (2013). The immunosuppressive tumour network: myeloid-derived suppressor cells, regulatory T cells and natural killer T cells. Immunology, 138(2), 105–115. doi:10.1111/imm.12036.
Weichhart, T., Costantino, G., Poglitsch, M., Rosner, M., Zeyda, M., Stuhlmeier, K. M., et al. (2008). The TSC-mTOR signaling pathway regulates the innate inflammatory response. Immunity, 29(4), 565–577. doi:10.1016/j.immuni.2008.08.012.
Song, K., Wang, H., Krebs, T. L., & Danielpour, D. (2006). Novel roles of Akt and mTOR in suppressing TGF-beta/ALK5-mediated Smad3 activation. The EMBO Journal, 25(1), 58–69. doi:10.1038/sj.emboj.7600917.
Zhang, Y. E. (2009). Non-Smad pathways in TGF-beta signaling. Cell Research, 19(1), 128–139. doi:10.1038/cr.2008.328.
Zhang, Y. E. (2017). Non-Smad signaling pathways of the TGF-beta family. Cold Spring Harbor Perspectives in Biology, 9(2). doi:10.1101/cshperspect.a022129.
Acknowledgements
We thank Dr. Panomwat Amornphimoltham, Dr. Joseph Califano, and Dr. Scott Lippman for insightful suggestions.
Funding
This work was supported by the following: National Institute of Dental and Craniofacial Research (NIH/NIDCR) 1R01DE026644-01, National Natural Science Foundation of China (81520108009, 81621062, 81602376), and 111 Project of MOE (B14038), China.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Wang, Z., Valera, J.C., Zhao, X. et al. mTOR co-targeting strategies for head and neck cancer therapy. Cancer Metastasis Rev 36, 491–502 (2017). https://doi.org/10.1007/s10555-017-9688-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10555-017-9688-7