
Abstract
One of the fundamental questions in invasion biology is to understand the genetic mechanisms behind success or failure during the establishment of a species. However, major limitations to understanding are usually a lack of spatiotemporal population data and information on the populations’ colonisation history. In a large-scale, detailed study on the bush-cricket Metrioptera roeselii 70 groups of founders were introduced in areas outside the species’ distribution range. We examined how (1) the number of founders (2–32 individuals), (2) the time since establishment (7 or 15 years after introduction) and (3) possible gene flow affected establishment success and temporal genetic changes of the introduced populations. We found higher establishment success in introductions with larger propagule sizes but genetic diversity indices were only partly correlated to propagule size. As expected, introduced populations were more similar to their founder population the larger the propagule size was. However, even if apparent at first, most of the differentiation in the small propagule introductions disappeared over time. Surprisingly, genetic variability was regained to a level comparable to the large and outbreeding founder population only 15 generations after severe demographic bottlenecks. We suggest that the establishment of these populations could be a result of several mechanisms acting in synergy. Here, rapid increase in genetic diversity of few introductions could potentially be attributed to limited gene flow from adjacent populations, behavioural adaptations and/or even increased mutation rate. We present unique insights into genetic processes that point towards traits that are important for understanding species’ invasiveness.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
With expanding trade routes, species are given additional ways to colonise new areas. Much easier than before, organisms can hitchhike on transports and this offers opportunities to reach regions that would not have been possible by natural dispersal (Westphal et al. 2008; Banks et al. 2015; Douma et al. 2016). After arrival to a site, a new population may be established if the conditions are suitable. However, successful arrival is not sufficient, as there are several factors that determine establishment success. The main ones being how well the species’ biology fits the new environment, its competitive ability with other species, and propagule size (e.g. Berggren et al. 2001, 2002; Blackburn and Duncan 2001; Cohen 2002; Mächler and Altermatt 2012).
The likelihood of establishment is expected to increase if the species possesses traits that enable it to cope with novel environments and to overcome negative effects arising from low number of individuals at the initial stage (Puillandre et al. 2008; Blackburn et al. 2009). Such traits may, for example, be the ability of individuals to adjust their behaviour by increasing their movement to compensate for the effects of few close-mating opportunities (Kindvall et al. 1998; Wissmann et al. 2009). However, in spite of a suitable environment and biological traits, establishment may still fail due to genetic factors such as lack of adaptive genetic variation in the arriving individuals (Lee 2002; Puillandre et al. 2008; Lawson Handley et al. 2011), or fitness consequences from inbreeding depression (Fowler and Whitlock 1999; Ayroles et al. 2009).
It is challenging to study these genetic processes and disentangle them from other biological factors during the establishment phase in natural settings, and therefore little is known in spite this being one of the fundamental questions in invasion biology (e.g. Lawson Handley et al. 2011; Banks et al. 2015). The major limitations for gaining knowledge are a lack of spatiotemporal population data and information on the populations’ colonization history. It is, for example, extremely rare that we know how many individuals have founded a natural population or how the genetic variation has changed over time at early stages of the colonization process. Additionally, we often do not know if colonization originated from one single or multiple introduction events, from one or several population sources and the timing of the introduction event(s). Most introductions (up to 95%) will instead remain unnoticed until the species is naturalized (di Castri 1989). Thus, in these situations, analyses of the dynamics of successful establishment are impossible and controlled experiments are needed. However, these are very time and resource consuming and therefore rare.
We have access to such a completely unique dataset. It originates from a landscape-scale introduction experiment with the Roesel’s bush-cricket, Metrioptera roeselii that was initiated as far back as 25 years ago in central Sweden. Seventy groups of founders were introduced into 70 different patches within an area of ca. 2300 km2 and the study was designed to examine questions on invasion and conservation ecology. All individuals originated from one large and homogeneous source population but the groups varied in size (Berggren 2001). Thus, all propagules originated from the same well-known source and experienced the same local climate. This highly controlled, long-term and detailed study setup gave us a unique opportunity to address questions on genetic processes in this non-native species and investigate mechanisms behind these. In combination with knowledge about the species’ colonization history (Kaňuch et al. 2013; Preuss et al. 2015), dispersal behaviour (Berggren et al. 2001, 2002; Hochkirch and Damerau 2009; Eriksson et al. 2013; Preuss et al. 2014) and adaptations (Kaňuch et al. 2014, 2020), this creates opportunities that, to our knowledge, previously have not existed in any other system. The introduction sites have been censused regularly in order to gather information on the species establishment success, population dynamics and local dispersal, and this allowed us to obtain fundamental information about genetic processes and patterns associated with early establishment success.
The aim of this study was to examine how the number of founders and time since establishment affect genetic variation in introduced populations of M. roeselii over time. We compared population genetic diversity and differentiation at two different occasions, 7 and 15 years since time of introduction and this data also allowed us to estimate effective population sizes. We expected establishment success as well as genetic diversity to be positively correlated with the number of founders. Thus populations founded by small propagule sizes would exhibit stronger signatures of demographic bottlenecks and stronger signs of differentiation from the founder population compared to populations founded by large propagules. We also expected increased genetic differentiation between founded populations over the study period of 15 generations, reduced levels of genetic variation due to genetic drift and possibly inbreeding depression. Because dispersal of individuals may counteract the above effects, we controlled for the potential effects of gene flow among local introduction events through observational and simulation-based approaches.
Material and methods
Species
Roesel’s bush-cricket, M. roeselii (Orthoptera: Tettigoniidae), is a small (body length 14–18 mm) and common European insect that inhabits a wide range of grassland habitats in most parts of the continent (Ingrisch and Köhler 1998; Hochkirch et al. 2016). However, via unintentional human-mediated transport and due to recent changes of global climate the species’ range has increased during the last century into previously uninhabited areas in western and northern Europe (e.g. Hochkirch and Damerau 2009; Wissmann et al. 2009; Kaňuch et al. 2013; Preuss et al. 2014; Hochkirch et al. 2016). In northern Europe, individuals complete one generation per year and reach maturity in summer after six nymphal stages. Stridulating adult males can be easily heard during this time. During the mating period the females lay eggs into hollow grass stems, other plant substrates or soil. The nymphs hatch the following or next spring after one or two winter diapauses depending on climatic conditions (Ingrisch and Köhler 1998).
Introductions and sampling of data
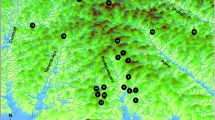
A number of 70 introductions of M. roeselii propagules were carried out in May 1994 and 1995 (70% and 30% of the introduction events respectively) in the intensively managed farmland landscapes of Uppland and Stockholm, central Sweden (Berggren 2001). The population source of the founding individuals was the oldest naturalized population in the Scandinavian Peninsula and is found close to the harbour of Västerås in Sweden (59°35′N, 16°29′E). Here the species was first observed in 1882 and nowadays M. roeselii covers an area of several thousands of square kilometres around Västerås (https://www.artportalen.se/). Five levels of propagule sizes were used for the introduction experiment; 2, 4, 8, 16, or 32 individuals of equal sex ratio and in the last virginal nymphal stage (i.e. no females carried fertilised eggs). The individuals were introduced on habitat islands within crop fields. The introduction sites were previously uninhabited by the species and situated at a distance of 17–82 km from the edge of the species’ current distribution area (Fig. 1). The average distance between two neighbouring introductions calculated by Delaunay triangulation was 6.3 km (25–75% = 3.4–11.5 km; Fig. S2 of Supplementary material). This was, at that time, estimated to be sufficient to prevent gene flow from natural dispersal (Berggren et al. 2001). There were no differences between the five propagule sizes regarding size of the habitat patch they were introduced in, and its degree of isolation, landscape composition or amount of ungrazed semi-natural grasslands in the landscape (see more in Berggren 2001).

© OpenTopoMap
Locations of 70 M. roeselii populations in Sweden that were experimentally introduced using different propagule sizes in 1994 and 1995. Some of the populations have been successfully established, while others were unsuccessful. A subset of locations was re-sampled after 7 (n = 29) and 15 (n = 17) years since the introduction. Landscapes of four areas (dashed circles) where inter-distances between introduction sites were ≤ 5 km apart and individual movement between neighbouring populations was expected with time, are enlarged to show details (white = grass or farmland, green = forests, blue = water, grey and yellow = roads). For IDs of all introductions see Fig. S1 of Supplementary material. The violet polygon shows the range of spread of the founder population, isolated from the introduction sites, until 2009 (data from https://www.artportalen.se/ by courtesy of Oskar Kindvall). Map source
From the successfully established populations individuals were collected for genetic analyses on two separate occasions. Because of technical limitations a reduced set of established introductions was used in the study. The first sampling was done during July and August 2001 in 29 of the introduction sites. In 2009 another sampling was carried out in 17 of the sites that previously had been sampled (Fig. 1). The number of individuals that we were able to collect was 9–38 (median 20) per population (n = 591 and 348 individuals in total in 2001 and 2009, respectively). In 2001 we also collected material from the source population for genetic analyses (n = 25 individuals). Since this sample did not differentiate significantly from the sample collected in 2008 (Kaňuch et al. 2013) we were confident that it was representative of the well established population. Although the initial introduction of the populations was not done in the same year, we expect that the difference of 1 year since introduction would not significantly affect any results. Therefore, the sampling occasions were named 7 or 15 years, respectively, since introduction. Population size estimates at each site were obtained using a bioacoustic census method and using the number of singing males as a proxy of population size. The census area around each introduction site was initially a minimum of 30 ha around the introduction patch. Each census was carried out using a standard technique; listening for stridulating males, mainly focusing on suitable habitat (details in Berggren 2001). To control for individuals moving away from the introduction location during the years of the experiment, the censused area was increased up to 225 ha around the introduction patch. Previous studies show an equal sex ratio in bush-crickets (e.g. Ingrisch and Köhler 1998) and therefore the abundance was estimated as two times the number of calling males.
Sampled individuals were stored in 96% ethanol at − 80 °C until analysed and measurements of genetic characteristics were based on microsatellite data. We isolated DNA from the muscle tissue of the hind-femur using the Chelex 100 (Bio-Rad) extraction method (Walsh et al. 1991) and amplified nine polymorphic microsatellite markers (MR2-16, 2-42, 3-24, 3-34; Holzhauer and Wolff 2005 and Metroe08, 19, 20, 24, 27; Kaňuch et al. 2010) according to adapted PCR protocol described by Preuss et al. (2015). All microsatellite loci were tested for the presence of null alleles, effects of stuttering, and large allele dropout using MicroChecker 2.2.3 (van Oosterhout et al. 2004). The Fisher’s combined probability test found evidence for a null allele in two loci (MR3-34 and Metroe20) of 2009 sampling only.
Estimation of genetic diversity, effective size and differentiation
We used a set of different indices of genetic diversity. Allelic richness (AR) was calculated as the mean number of alleles per locus rarefied on seven diploid individuals in the software HP-Rare 1.1 (Kalinowski 2005). Observed (HO) and expected (HE) heterozygosity and unbiased HE averaged over loci and population were calculated in Genetix 4.05 (Belkhir et al. 2004). When estimating the inbreeding coefficient (FIS), we used a Bayesian approach of individual inbreeding model (IIM) in the software INEST 2.2 (Chybicki and Burczyk 2009; Chybicki 2017) which takes into account that both null alleles and inbreeding can produce an excess of homozygotes. Based on initial data exploration, we omitted HO and HE from further analysis as they were strongly positively correlated (r = 0.84–1.00, p < 0.001) with an unbiased derivate of this index.
To estimate the effective size (Ne) of each population 15 years since the introduction we used a temporal method, thus we employed here only populations that were re-sampled in both sampling periods. This method makes use of the standardized variance in allele frequency (Waples 1989) and also accounts for estimates of the census population sizes at the time of the initial sampling as implemented in NeEstimator 2.1 (Do et al. 2014). We employed Fs estimator, which yields unbiased estimates also when based on a modest number of loci and alleles that may occur at low frequencies, thus it eliminates the bias associated with other estimators (Jorde and Ryman 2007). The contemporary effective sizes of introduced populations were compared with the Ne of the population used as source for the founding individuals. Ne of the founder population was calculated by the linkage disequilibrium method, which is the most commonly used genetic estimator of contemporary Ne if only one sample is available, as it computes estimates comparable to more unbiased temporal methods (Waples and Do 2010).
For genetic differentiation between the introduced populations and the founder population we calculated pairwise FST estimates, corrected for null alleles by the ENA method using FreeNA software (Chapuis and Estoup 2007). We also estimated genetic differentiation by Dest values, which account for different alleles being fixed in different populations (Jost 2008), as opposed to FST where only levels of heterozygosity in different samples are considered. The Dest was calculated as a harmonic mean across all loci in SMOGD 1.2.5 (Crawford 2010).
To model the effect of propagule size and time since introduction on genetic diversity, effective population size and level of differentiation from the founder population, we constructed regression models with a Gamma distribution, log-link function and type II SS using ‘car’ package (Fox and Weisberg 2011) of the R 3.4.4 software (R Core Team 2018). For all indices except Ne we constructed multiple models with propagule size (five values) and year (two levels) as factors including their interaction. The effective size of each population was modelled by propagule size in a one-factor model (Ne values were calculated for 15 years since the introduction only).
Bottleneck analyses
For testing demographic bottleneck effects we defined the null hypothesis as being presence of mutation-drift equilibrium under a two-phased mutation model (TMM). The selected TMM model should have the best fit for most microsatellite data as it combines the stepwise mutation model and the infinite allele model (Piry et al. 1999). To test for a recent reduction in effective size (excess in heterozygosity), we implemented a procedure where both proportion of multi-step mutations (pg) and average multi-step mutation size (δg) varied randomly during simulations. In addition to equilibrium heterozygosity values, we controlled for signatures of a bottleneck by the deficiency in M-ratio (Garza and Williamson 2001). The total number of alleles (k) divided by overall range in allele size (R) produces the ratio (M), and this is expected to be smaller in recently reduced populations compared to populations in mutation-drift equilibrium (it is supposed that k decreases faster than R when the population size is reduced). Thus, the heterozygosity test assumes that a bottleneck eliminates rare alleles, whereas the M-ratio test is based on the idea that drift removes alleles randomly with respect to allele size (Williamson-Natesan 2005). Both the excess of heterozygotes and the deficiency in M-ratio were tested by the Z-test based on combined Z scores for particular loci and also by the Wilcoxon signed-rank test in the software INEST 2.2 (Chybicki 2017). In the case of the Wilcoxon test, p values were determined after 1,000,000 permutations to approximate the exact value (normality of data distribution not assumed here).
Inference of gene flow between adjacent introductions
To explore to what extent gene flow had had any effect on the genetic variation in the populations, we first computed directional relative migration values among populations for each sampling period using the divMigrate-online tool (Sundqvist et al. 2016). Assuming an island model of population structure, migration among populations was estimated by D measure (Jost 2008), which is independent of gene diversity. However, a preliminary analysis revealed relatively high genetic similarity between founder population and introductions (mean 25%, range 19–71%). This may increase the risk of overestimating migration values among pairs of introduced populations. Therefore, we abandoned this measure. Instead, to analyse gene flow among introductions we used a more conservative screening of allelic variation. First, we explored allele frequency distributions and estimated private allelic richness in relation to time since the introduction. Next, we used simple linear regressions to analyse the relation between the distance to the nearest other introduction and genetic diversity as well as differentiation estimates (we expected either positive or negative relationships, respectively). To further analyse potential effects of unintentional gene flow we employed computer-simulated genotypes produced in EASYPOP software (Balloux 2001). Simulations were done to assess the theoretically expected values of genetic variability with known levels of gene flow (e.g. none, medium, high) among our study populations. The other settings of the simulations were: random mating system, island model of migration and mixed mutation model of loci. The number of loci, possible number of alleles per each locus and number of generations was equivalent to the sampled introductions. We compared our field data with simulated genotypes using four different indices of individual heterozygosity; all relevant when analysing genetic variability either in populations with high inbreeding or in populations with migration and admixture of founders (Aparicio et al. 2006). The measures were (1) proportion of heterozygous loci (PHt), (2) standardized heterozygosity based on the mean expected heterozygosity (Hs_exp), (3) internal relatedness (IR) and (4) homozygosity by locus (HL) calculated by the R function GENHET (Coulon 2010). Differences among groups were determined by the non-parametric Kruskal–Wallis ANOVA with post-hoc Dunn test for multiple comparisons in the R-package ‘FSA’ 0.8.30 (Ogle et al. 2020).
Finally, in order to test for unintended exchange of individuals among introductions we selected four groups of introduction sites where the individual sites were found no more than 5 km from each other and where all had been sampled in both sampling periods. This distance was chosen as a more recent study had shown that exchange of short-winged (flightless) individuals could potentially occur due to natural dispersal (Berggren et al. 2001). In these population groups we searched for individual traces of gene flow and its effects on the population genetic structure. We examined this by a Principal Coordinates Analysis (PCoA) of the R-package ‘adegenet’ 2.1.1 (Jombart 2008). This multivariate method considers the best approximation of a dissimilarity matrix of Euclidean and short distances while its principal components optimize the representation of the squared pairwise distances between individuals. Additionally, a Bayesian clustering using tessellations and spatial Markov models implemented in TESS 2.3.1 software (Chen et al. 2007) assigned each individual into distinct genetic clusters. In the model with admixture (Durand et al. 2009), we assumed the number of clusters (K) to equal the number of introduced populations and computed the proportion of individuals that could be assigned to each cluster. We ran the simulations for 50,000 sweeps from which the first 10,000 sweeps were discarded as burn-in. Because TESS analysis requires unique coordinates for each individual, a measure that we had not collected, we created random individual coordinates within an area of the mean size of introduction patch (ca. 1 ha) prior to the run.
Results
Genetic diversity
Seven years after introductions, 71% of the populations had successfully established. Higher establishment success was found in introductions with larger propagule sizes (propagule size 2 – 43%, 4 – 64%, 8 – 64%, 16 – 86%, 32 – 100%; R2 = 0.87, p = 0.021, n = 14 introductions per category) and there were no systematic differences across the study area (Fig. 1). We found lower genetic diversity (AR and unbiased HE) in most examined introductions 7 years after introduction compared to the founder population (Fig. 2a, b). This changed as populations reached 15 years after introduction, when allelic richness and heterozygosity were regained in all populations regardless of propagule size (Table 1). The values of AR even exceeded those in the founder population (Fig. 2a). However, genetic diversity indices did not relate to propagule size (Table 1), but there was a tendency of a positive relationship during the first sampling period in AR and FIS (Fig. 2a, c). The estimated genetic effective population size 15 years after the introduction was significantly larger in introductions with larger propagule sizes compared to the estimation from the founder population (Fig. 2d, Table 1).

a Allelic richness (AR, number of alleles per locus rarefied on seven diploid individuals); b unbiased expected heterozygosity (HE); c inbreeding coefficient (FIS) and d effective population size (Ne) in relation to propagule size in populations after 7 (diamonds) and 15 (circles) years since introduction (points with vertical segments show means and standard deviations). Ne was calculated using a temporal method that accounts for both sampling periods. Trends of the effect of propagule size are derived from linear regression models (solid line significant p < 0.05; dashed line non-significant). Horizontal lines denote the values of the founder population
Population differentiation and bottleneck
We found that propagule size had significant effect on how much the introduced population of M. roeselii resembled their founder population as measured by two different estimators of genetic differentiation (FST and Dest). As expected, introduced populations were more similar to their founder the larger the propagule size was. The effect of propagule size on level of differentiation was lower in the second compared to the first sampling occasion (Fig. 3, Table 1). There were signatures of demographic bottleneck in almost all populations and in both sampling occasions manifested as a deficiency in M-ratio. However, we found excess in heterozygosity only in the first sampling period and, similar to M-ratio, the presence of a bottleneck was independent of the propagule size (Table 2).

Genetic differentiations calculated for two alternative types of pairwise estimators a FST and b Dest, between each introduced population of M. roeselii and the founder population in relation to propagule size after 7 (diamonds) and 15 (circles) years since introduction (points with vertical segments show means and standard deviations). Trends for the effect of propagule size are derived from linear regression models (solid line significant p < 0.05; dashed line non-significant)
Counteracting effect of gene flow
Allele sizes found after 15 generations were clustered around the alleles that were present 7 years since the introduction (Fig. 4). However, the number of private alleles did not increase in the later sampling period (paired t = −0.55, df = 15, p = 0.59; Fig. S3 of Supplementary material). An increase of private alleles should be expected if populations are isolated but we could not rule out that observed pattern was a result of limited set of polymorphic loci. A linear regression showed no relationship between estimates of genetic diversity or differentiation and geographical distance to the nearest other introduction (adj. R2 = −0.74–0.45, p = 0.22–0.98; Fig. S4 of Supplementary material). Similarly, genetic variability in introduced populations after 15 generations was akin to simulated genotypes of theoretically expected values with no gene flow level among populations (PHt, HL) or ranged between simulated values of no and medium level of gene flow (Hs_exp, IR). In all these indices of individual heterozygosity the observed values were significantly different (p < 0.05) from the simulated populations having medium or high level of gene flow (Fig. 5).

Allele frequency distribution for 9 microsatellite loci in a set of individuals sampled in the founder population (n = 25 individuals) and in introduced populations as sampled after 7 (n = 591 individuals) and 15 years (n = 348 individuals) since the introduction. The x-axis represents the allele size (bp) and the y-axis represents the observed allele frequencies

Estimates of individual heteorzygosity (a proportion of heterozygous loci, b standardized heterozygosity, c internal relatedness, d homozygosity by locus) in introductions (n = 17 populations, 348 individuals), and in simulated multilocus genotypes of theoretically expected values according to different gene flow level among populations (n = 10 populations, 200 individuals in each level). Values shown are after 15 generations both in introductions and simulations. Violin plots show medians and 25–75% quartiles in box with rotated kernel density estimation. Groups sharing a letter are not significantly different (α = 0.05)
Still, we found some signs of exchange of individuals among introductions in one of the groups within a radius of 5 km. Genetic structures of populations founded by two or eight individuals became more similar in the later sampling occasion (Fig. 6a). This temporal change was evident with both methods applied. In the other three groups where the pattern did not change significantly between two sampling periods (Fig. 6b–d), we could not distinguish whether individuals that appeared as intermediates between two genetic clusters resulted from a very high genetic similarity of founding individuals (see Methods) or from migration. If the latter was true, one would expect homogenization of the structure in the second sampling.
Discussion
In this controlled natural experiment we show that an introduced species can cope well in a new environment in the early colonisation stage despite a severe bottleneck. We argue that this fact plays a large role in the invasion process for an exotic species (e.g. Forsyth and Duncan 2001; Memmott et al. 2005; Blackburn et al. 2009). Instead of an expected continuous loss of genetic variation at the establishment stage (due to random genetic drift), we observed a surprisingly fast resurgence of genetic diversity only 15 generations after the bottleneck. From previous knowledge on the ecology of establishment (both from empirical studies and theoretical simulations) we expected a significant and continued negative effect of small propagule sizes on genetic diversity (Lockwood et al. 2005, 2009; Simberloff 2009). Instead, populations founded by only a handful of individuals, regained genetic diversity shortly after introduction, to a level comparable to a large population with random mating. Only the results from the smallest propagule population followed our predictions, but this population had only reduced variation during the first sampling event (Fig. 2). The overall distribution of alleles in the introductions over time and the theoretical estimates of simulated multilocus genotypes supported our expectations that the majority of the introductions remained isolated within the study period, at least until the first sampling 7 years after introduction. The fact that populations founded by few individuals were genetically differentiated from the founding population was likely due to genetic drift (Lockwood et al. 2005; Schulte et al. 2013; Bock et al. 2015). This was expected as single individuals only carry a small fraction of the genetic diversity from the founding population. Individual heterozygosity simulated for conditions similar to the introductions, also suggested no or low levels of gene flow among populations. The mean inbreeding coefficient of introduced populations was similar to the founder population regardless of propagule size and sampling period, irrespective of the presence of a significant bottleneck or not (as results of bottleneck analyses were not consistent). Even if the study setup did not fully prevent the introductions from gene flow (Figs. 5b, c and 6a), the fact that the reduced variability as well as differentiation did not last but were regained at an unusually rapid rate was highly interesting.

Genetic distances between individuals using a Principal Coordinates Analysis (scatter plots) and their assignment into assumed genetic clusters by the Bayesian clustering using tessellations and Markov models (bar plots) in nearby populations of four selected areas (locations of populations groups a–d and their propagule sizes see Fig. 1). Ellipses indicate credible distribution of the individuals in to different clusters according to the first and second principal components of the PCoA. The bar plots show the estimated assignment probabilities of individuals to different clusters
Potential gene flow is improbable by regular migration of mostly flightless individuals between distant populations (Berggren et al. 2001), whereas some long-winged individuals, with capacity to disperse longer distances, are expected to be vectors of limited gene flow as this morphological modification is important in natural dispersal events (Wissmann et al. 2009). However, increased incidence of this phenomenon is known to occur when population densities are high or in very warm years (Hochkirch and Damerau 2009). We did not measure the frequency of macropterous individuals every year since introduction but in 2008, at the end of our sampling period, only 0.5% of long-winged individuals were found in isolated populations in Northern Europe, including Sweden (n = 203 ind. from 10 pops; Kaňuch et al. 2013). Based on this, we find that influence of dispersing long-winged individuals likely is of low importance for our findings, as also the densities in the experimental introductions were low and no observations of increased proportions (< 1%) of long-winged morphs were seen in sampled individuals. In another study in 2019 we found a much higher proportion (14%) of long-winged adults in 10 year old introduction sites in central Sweden (n = 251 ind. from 14 pops; unpublished data). Potentially, an increase in temperature the last years (public information from the Swedish Meteorological and Hydrological Institute; https://www.smhi.se/) may have an effect on increased dispersal in this species, but this is less likely for our study two decades ago (e.g. in 2001). Another potential source of gene flow that we could not control for is passive transport via farming products, something that has been seen in other studies of the species (e.g. Kindvall et al. 1998; Berggren et al. 2001; Wagner 2004). Although a single successfully migrating individual may provide enough gene flow to decrease differentiation substantially, we believe it unlikely that long-distance migrants affected a large part of our introductions. Data from Artportalen (https://www.artportalen.se/) show that first observation of the species in the region was 2004; still in 2010 no individuals were reported close (< 5 km) to the study sites. This is supported by the finding that migrants from a neighbouring population were likely only one single case. The isolation of populations and the lack of gene flow did not seem to be important for the establishment success of the species, as even marginal and/or more distant populations established well (Fig. 1, Fig. S4 of Supplementary material).
Our study suggests that M. roeselii possesses yet to be explored important reproductive and/or genetic traits associated with an invasive species; traits that in their interaction with the environment affect establishment success (e.g. Fowler and Whitlock 1999; Hochkirch and Damerau 2009; Lawson Handley et al. 2011). When effective population sizes are extremely small and isolated, we suggest that behavioural adaptations can be a key element that determine the outcome for the establishment. Presence of a negative assortative mating that reduces inbreeding level is unlikely in our study as nuptial gift-giving bush-crickets prefer to mate with similar phenotypes (Dorková et al. 2019), and assortative mating is more common under high population densities and strong competition (Jiang et al. 2013). Rather, the polyandrous system provides a mechanism that can reduce inbreeding in small populations. During early stages of colonization, a female can have offspring that are half siblings rather than full siblings; thereby the degree of inbreeding in her grandchildren is reduced. Recovery of genetic variation in a population would therefore be faster with an increased level of polyandry (Cornell and Tregenza 2007). Although such behavioural mechanism can increase heterozygosity, it does not explain an increase in number of alleles.
Therefore, a possible mechanism causing a regain of genetic variation when gene flow is lacking may be also an exceptional rapid genome wide mutation rate. Mutation rate is suggested as the most significant factor increasing genetic diversity in populations with small effective sizes (Talla et al. 2019a, b). In orthopteran species, the mean mutation rate in microsatellite loci may be one order of magnitude higher than in other insects (Chapuis et al. 2015), pointing towards this as a factor of interest. High mutation rate has been suggested in unintentionally introduced populations of M. roeselii, but in previous studies the number of founding individuals has been unknown and multiple introductions could not be excluded (see discussion in Kaňuch et al. 2014). Our current study may provide further support for this mechanism, which also can be present in other Orthoptera species. Due to mutation dependence on microsatellite length, increased level of genetic diversity observed in this group of insects is likely shaped by molecular mechanisms that determine very long microsatellite repeat arrays (Chapuis et al. 2012).
The severe bottlenecks in combination with limited gene flow were expected to result in reduced genetic variation (e.g. Simberloff 2009; Demandt 2010; Kaňuch et al. 2014). It was therefore surprising that we did not find support for the hypothesis that genetic diversity was related to the number of founders. An increase in allelic richness and/or heterozygosity with time has previously been observed in other invasive species, but this has required several dozens of generations (Dlugosch and Parker 2008). With no or very limited gene flow between small populations, our results clearly suggest that this species has traits that make it very capable of overcoming demographic bottlenecks. We suggest that a fast recovery of genetic diversity at reduced population sizes is an important factor for high establishment success in species and is a feature that is important when evaluating if a species is likely to become invasive in a new environment.
Conclusions
When the mechanisms that lie behind successful colonisations are studied, we know that the number of colonizing individuals is one major factor (Berggren 2001). The initial number of individuals determines not only the likelihood of extinction from stochastic effects, but also density-behaviourally linked negative consequences such as the Allee effect. A non-escapable fact is also that a small propagule will bring with it just a fraction of the source population’s genetic diversity. For the bush-cricket M. roeselii we found in this study mechanisms that may explain the species successful large-scale expansion over regions in Europe and North America. We conclude that successful establishment of populations of this species could be a result of synergetic effects of rare long-distance individual dispersal, behavioural adaptations and/or even increased mutation rate. The possibility for isolated populations to bounce back from low levels of genetic diversity would be a very valuable trait for any species and it likely determines establishment and long-term persistence of potential invaders of new areas. To further examine the importance of intra-population mutation rates for invasive species we suggest studies focused on the temporal change of genetic diversity using a genome-wide assessment. Experimental studies on invasion success using different species with similar ecology but with different mating systems would be especially helpful. Information that a species is less likely to suffer from severe bottlenecks will help the management of invasive species and should be incorporated in assessing the invasive risk of ‘door-knockers’ as well as the expansion potential for species already established.
Data availability
Data will be made available on reasonable request.
References
Aparicio JM, Ortego J, Cordero PJ (2006) What should we weigh to estimate heterozygosity, alleles or loci? Mol Ecol 15:4659–4665
Ayroles JF, Hughes KA, Rowe KC, Reedy MM, Rodriguez-Zas SL, Drnevich JM, Caceres CE, Paige KN (2009) A genomewide assessment of inbreeding depression: gene number, function, and mode of action. Conserv Biol 23:920–930
Balloux F (2001) EASYPOP (version 1.7): a computer program for population genetics simulations. J Hered 92:301–302
Banks NC, Paini DR, Bayliss KL, Hodda M (2015) The role of global trade and transport network topology in the human-mediated dispersal of alien species. Ecol Lett 18:188–199
Belkhir K, Borsa P, Chikhi L, Raufaste N, Bonhomme F (2004) GENETIX 4.05, logiciel sous WindowsTM pour la génétique des populations. Laboratoire Génome, Populations, Interactions, Université de Montpellier II, Montpellier
Berggren Å (2001) Colonization success in Roesel’s bush-cricket Metrioptera roeseli: the effects of propagule size. Ecology 82:274–280
Berggren Å, Carlson A, Kindvall O (2001) The effect of landscape composition on colonization success, growth rate and dispersal in introduced bush-crickets Metrioptera roeseli. J Anim Ecol 70:663–670
Berggren Å, Birath B, Kindvall O (2002) Effect of corridors and habitat edges on dispersal behavior, movement rates, and movement angles in Roesel’s bush-cricket (Metrioptera roeseli). Conserv Biol 16:1562–1569
Blackburn TM, Duncan RP (2001) Determinants of establishment success in introduced birds. Nature 414:195–197
Blackburn TM, Cassey P, Lockwood JL (2009) The role of species traits in overcoming the small initial population sizes of exotic birds. Global Change Biol 15:2852–2860
Bock DG, Caseys C, Cousens RD, Hahn MA, Heredia SM, Hübner S, Turner KG, Whitney KD, Rieseberg LH (2015) What we still don’t know about invasion genetics. Mol Ecol 24:2277–2297
Chapuis M-P, Estoup A (2007) Microsatellite null alleles and estimation of population differentiation. Mol Biol Evol 24:621–631
Chapuis M-P, Streiff R, Sword GA (2012) Long microsatellites and unusually high levels of genetic diversity in the Orthoptera. Insect Mol Biol 21:181–186
Chapuis M-P, Plantamp C, Streiff R, Blondin L, Piou C (2015) Microsatellite evolutionary rate and pattern in Schistocerca gregaria inferred from direct observation of germline mutations. Mol Ecol 24:6107–6119
Chen C, Durand E, Forbes F, François O (2007) Bayesian clustering algorithms ascertaining spatial population structure: a new computer program and a comparison study. Mol Ecol Notes 7:747–756
Chybicki IJ (2017) INEST 2.2. http://www.ukw.edu.pl/pracownicy/strona/igor_chybicki/software_ukw/. Accessed 28 April 2019
Chybicki IJ, Burczyk J (2009) Simultaneous estimation of null alleles and inbreeding coefficients. J Hered 100:106–113
Cohen AN (2002) Success factors in the establishment of human-dispersed organisms. In: Bullock JM, Kenward RE, Hails RS (eds) Dispersal ecology. Blackwell Publishing, Oxford, pp 374–394
Cornell SJ, Tregenza T (2007) A new theory for the evolution of polyandry as a means of inbreeding avoidance. Proc R Soc B-Biol Sci 274:2873–2879
Coulon A (2010) GENHET: an easy-to-use R function to estimate individual heterozygosity. Mol Ecol Res 10:167–169
Crawford NG (2010) SMOGD: software for the measurement of genetic diversity. Mol Ecol Res 10:556–557
Demandt MH (2010) Temporal changes in genetic diversity of isolated populations of perch and roach. Conserv Genet 11:249–255
di Castri F (1989) History of biological invasions with emphasis on the Old World. In: Drake J, Mooney HA, di Castri F, Groves RH, Kruger FJ, Rejmánek M, Williamson M (eds) Biological invasions: a global perspective. Wiley, New York, pp 1–30
Dlugosch KM, Parker IM (2008) Founding events in species invasions: genetic variation, adaptive evolution, and the role of multiple introductions. Mol Ecol 17:431–449
Do C, Waples RS, Peel D, Macbeth GM, Tillett BJ, Ovenden JR (2014) NeEstimator V2: re-implementation of software for the estimation of contemporary effective population size (Ne) from genetic data. Mol Ecol Res 14:209–214
Dorková M, Naďo L, Jarčuška B, Kaňuch P (2019) Size-dependent mating pattern in a nuptial gift-giving insect. Ecol Evol 9:454–462
Douma JC, Pautasso M, Venette RC, Robinet C, Hemerik L, Mourits MCM, Schans J, van der Werf W (2016) Pathway models for analysing and managing the introduction of alien plant pests-an overview and categorization. Ecol Model 339:58–67
Durand E, Jay F, Gaggiotti OE, François O (2009) Spatial inference of admixture proportions and secondary contact zones. Mol Biol Evol 26:1963–1973
Eriksson A, Low M, Berggren Å (2013) Influence of linear versus network corridors on the movement and dispersal of the bush-cricket Metrioptera roeseli (Orthoptera: Tettigoniidae) in an experimental landscape. Eur J Entomol 110:81–86
Forsyth DM, Duncan RP (2001) Propagule size and the relative success of exotic ungulate and bird introductions to New Zealand. Am Nat 157:583–595
Fowler K, Whitlock MC (1999) The variance in inbreeding depression and the recovery of fitness in bottlenecked populations. Proc R Soc B-Biol Sci 266:2061–2066
Fox J, Weisberg S (2011) An R companion to applied regression, 2nd edn. Sage, Thousand Oaks
Garza JC, Williamson EG (2001) Detection of reduction in population size using data from microsatellite loci. Mol Ecol 10:305–318
Hochkirch A, Damerau M (2009) Rapid range expansion of a wing-dimorphic bush-cricket after the 2003 climatic anomaly. Biol J Linn Soc 97:118–127
Hochkirch A, Willemse LPM, Szovenyi G, Rutschmann F, Presa JJ, Krištín A, Kleukers R, Chobanov DP (2016) Roeseliana roeselii. The IUCN Red List of threatened species 2016, e.T68468465A74623999. Accessed 11 June 2018
Holzhauer SIJ, Wolff K (2005) Polymorphic microsatellite loci in the bush-cricket Metrioptera roeseli. Mol Ecol Notes 5:502–503
Ingrisch S, Köhler G (1998) Die heuschrecken mitteleuropas. Westarp Wissenschaften, Magdeburg
Jiang Y, Bolnick DI, Kirkpatrick M (2013) Assortative mating in animals. Am Nat 181:E125–E138
Jombart T (2008) adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24:1403–1405
Jorde PE, Ryman N (2007) Unbiased estimator for genetic drift and effective population size. Genetics 177:927–935
Jost L (2008) GST and its relatives do not measure differentiation. Mol Ecol 17:4015–4026
Kalinowski ST (2005) HP-rare: a computer program for performing rarefaction on measures of allelic diversity. Mol Ecol Notes 5:187–189
Kaňuch P, Pfunder M, Berggren Å, Cassel-Lundhagen A (2010) Description of nine new microsatellite loci in Metrioptera roeselii (Orthoptera: Tettigoniidae) and their multiplex PCR protocols. Mol Ecol Res 10:404–408
Kaňuch P, Berggren Å, Cassel-Lundhagen A (2013) Colonization history of Metrioptera roeselii in northern Europe indicates human-mediated dispersal. J Biogeogr 40:977–987
Kaňuch P, Berggren Å, Cassel-Lundhagen A (2014) Genetic diversity of a successful colonizer: isolated populations of Metrioptera roeselii regain variation at an unusually rapid rate. Ecol Evol 4:1117–1126
Kaňuch P, Kiehl B, Cassel-LundhagenA, Laugen, AT, Low M, Berggren Å (2020) Gene flow relates to evolutionary divergence among populations at the range margin. PeerJ 8:e10036
Kindvall O, Vessby K, Berggren Å, Hartman G (1998) Individual mobility prevents an Allee effect in sparse populations of the bush cricket Metrioptera roeseli: an experimental study. Oikos 81:449–457
Lawson Handley L-J, Estoup A, Evans DM, Thomas CE, Lombaert E, Facon B, Aebi A, Roy HE (2011) Ecological genetics of invasive alien species. Biocontrol 56:409–428
Lee CE (2002) Evolutionary genetics of invasive species. Trends Ecol Evol 17:386–391
Lockwood JL, Cassey P, Blackburn T (2005) The role of propagule pressure in explaining species invasions. Trends Ecol Evol 20:223–228
Lockwood JL, Cassey P, Blackburn TM (2009) The more you introduce the more you get: the role of colonization pressure and propagule pressure in invasion ecology. Divers Distrib 15:904–910
Mächler E, Altermatt F (2012) Interaction of species traits and environmental disturbance predicts invasion success of aquatic microorganisms. PLoS ONE 7:e45400
Memmott J, Craze PG, Harman HM, Syrett P, Fowler SV (2005) The effect of propagule size on the invasion of an alien insect. J Anim Ecol 74:50–62
Ogle DH, Wheeler P, Dinno A (2020) FSA: fisheries stock analysis. R package version 0.8.30. https://github.com/droglenc/FSA
Piry S, Luikart G, Cornuet JM (1999) BOTTLENECK: a computer program for detecting recent reductions in the effective population size using allele frequency data. J Hered 90:502–503
Preuss S, Low M, Cassel-Lundhagen A, Berggren Å (2014) Evaluating range-expansion models for calculating nonnative species’ expansion rate. Ecol Evol 4:2812–2822
Preuss S, Berggren Å, Cassel-Lundhagen A (2015) Genetic patterns reveal an old introduction event and dispersal limitations despite rapid distribution expansion. Biol Invasions 17:2851–2862
Puillandre N, Dupas S, Dangles O, Zeddam J-L, Capdevielle-Dulac C, Barbin K, Torres-Leguizamon M, Silvain J-F (2008) Genetic bottleneck in invasive species: the potato tuber moth adds to the list. Biol Invasions 10:319–333
R Core Team (2018) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna
Schulte U, Veith M, Mingo V, Modica C, Hochkirch A (2013) Strong genetic differentiation due to multiple founder events during a recent range expansion of an introduced wall lizard population. Biol Invasions 15:2639–2649
Simberloff D (2009) The role of propagule pressure in biological invasions. Ann Rev Ecol Evol Syst 40:81–102
Sundqvist L, Keenan K, Zackrisson M, Prodöhl P, Kleinhans D (2016) Directional genetic differentiation and relative migration. Ecol Evol 6:3461–3475
Talla V, Johansson A, Dincă V, Vila R, Friberg M, Wiklund C, Backström N (2019a) Lack of gene flow: narrow and dispersed differentiation islands in a triplet of Leptidea butterfly species. Mol Ecol 28:3756–3770
Talla V, Soler L, Kawakami T, Dincă V, Vila R, Friberg M, Wiklund C, Backström N (2019b) Dissecting the effects of selection and mutation on genetic diversity in three wood white (Leptidea) butterfly species. Genome Biol Evol 11:2875–2886
van Oosterhout C, Hutchinson W, Wills D, Shipley P (2004) Micro-checker: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4:535–538
Wagner C (2004) Passive dispersal of Metrioptera bicolour (Phillipi 1830) (Orthopteroidea: Ensifera: Tettigoniidae) by transfer of hay. J Insect Conserv 8:287–296
Walsh S, Metzger D, Higuchi R (1991) Chelex 100 as a medium for simple extraction of DNA for PCR-based typing from forensic material. Biotechniques 10:506–513
Waples RS (1989) A generalized approach for estimating effective population size from temporal changes in allele frequency. Genetics 121:379–391
Waples RS, Do C (2010) Linkage disequilibrium estimates of contemporary Ne using highly variable genetic markers: a largely untapped resource for applied conservation and evolution. Evol Appl 3:244–262
Westphal MI, Brown M, MacKinnon K, Noble I (2008) The link between international trade and the global distribution of invasive alien species. Biol Invasions 10:391–398
Williamson-Natesan EG (2005) Comparison of methods for detecting bottlenecks from microsatellite loci. Conserv Genet 6:551–562
Wissmann J, Schielzeth H, Fartmann T (2009) Landscape scale expansion of Roesel’s bush-cricket Metrioptera roeselii at the north-western range limit in Central Europe (Orthoptera: Tettigoniidae). Entomol Gen 31:317–326
Acknowledgements
We are grateful to Lina Ahlbäck Widenfalk and Emilia Broberg for help with field work and to Ludvig Ahrén and Johan Wallén for lab assistance. We are grateful to two anonymous reviewers for valuable comments and suggestions that helped to improve a previous version of the manuscript.
Funding
Open access funding provided by Swedish University of Agricultural Sciences. The research by AL and ÅB was funded by the Carl Tryggers Foundation (CTS 08:237), PK was partly supported by the Slovak Scientific Grant Agency VEGA (2/0076/19) and from the NJ-faculty at SLU.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kaňuch, P., Berggren, Å. & Cassel-Lundhagen, A. A clue to invasion success: genetic diversity quickly rebounds after introduction bottlenecks. Biol Invasions 23, 1141–1156 (2021). https://doi.org/10.1007/s10530-020-02426-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10530-020-02426-y