
Abstract
Plant biosecurity is increasingly challenged by emerging crop pests. The spider mite Tetranychus evansi has recently emerged as a new threat to solanaceous crops in Africa and the Mediterranean basin, with invasions characterized by a high reproductive output and an ability to withstand a wide range of temperatures. Mitochondrial (868 bp of COI) and nuclear (1,137 bp of ITS) loci were analyzed in T. evansi samples spanning the current geographical distribution to study the earliest stages of the invasive process. The two sets of markers separate the samples into two main clades that are only present together in South America and Southern Europe. The highest COI diversity was found in South America, consistent with the hypothesis of a South American origin of T. evansi. Among the invaded areas, the Mediterranean region displayed a high level of genetic diversity similar to that present in South America, that is likely the result of multiple colonization events. The invasions of Africa and Asia by T. evansi are characterized by a low genetic variation associated with distinct introductions. Genetic data demonstrate two different patterns of invasions: (1) populations in the Mediterranean basin that are a result of multiple cryptic introductions and (2) emerging invasions of Africa and Asia, each likely the result of propagules from one or limited sources. The recent invasions of T. evansi illustrate not only the importance of human activities in the spread of agricultural pests, but also the limits of international quarantine procedures, particularly for cryptic invasions.
Similar content being viewed by others

Introduction
The increase in movement of both people and commodities internationally favours the spread of organisms outside their indigenous ranges. In the newly colonized areas, exotic species often become pests with dramatic consequences for biodiversity, agriculture, and human health. A recent survey of the causes of new plant disease emergence worldwide concluded that 56% of recent outbreaks result from introductions associated with trade of plants and plant products and the movement of people (Anderson et al. 2004). In the case of invasive arthropods, annual damages caused by exotic insects and mites in the United States alone have been estimated to be at least $16 billion (Pimentel et al. 2005). Climate change is likely to complicate further the impact of invasive plant pests. For example, a lengthening of the growing season in mid- and high latitudes of the northern hemisphere has been reported as a clear indicator of the responses by insect pests to a warmer climate (Menzel et al. 2008). As a result of both increased trade and climate change, emerging crop pests are likely to present new concerns for plant biosecurity, motivating research and new technologies to detect, identify and monitor pests and infectious diseases, and to modify strategies for plant crop protection (Rodoni 2009; Waage and Mumford 2008).
The red tomato spider mite Tetranychus evansi likely originated in South America and has emerged in only the last 10 years as a new destructive pest of solanaceous crops in many parts of the world. First reported from North-East Brazil in 1952 (Silva 1954), the mite was not considered a harmful pest in South America, except for a few outbreaks discovered in limited areas of Brazil (Humber et al. 1981). In the past half century the mite has since been collected from other continents including North America, many countries in sub-Saharan Africa, some Indian and Atlantic Ocean Islands, the Mediterranean basin, as well as from several parts in the Pacific Ocean (Hawaii, Taiwan, Japan and the south-eastern coast of China; see Migeon and Dorkeld 2006; Migeon et al. 2009 for a complete and chronological list). In the past decade, the pest status of T. evansi has changed and it is now regarded as a harmful invasive species in several parts in Africa and in the Mediterranean basin. In Africa T. evansi has become one of the most important dry season pests of tomatoes, causing yield losses of up to 90% in South-East Africa (Sibanda et al. 2000) and West Africa (Duverney and Ngueye-Ndiaye 2005). In the Mediterranean region, where several solanaceous field and glasshouse crops are economically important (tomato, potato, eggplant, etc.), the high invasive potential of T. evansi has prompted the recent addition of the species to the European and Mediterranean Plant Protection Organization (EPPO) alert list (EPPO 2007).
Invasion of new areas by T. evansi is favored by its high intrinsic rate of increase within a broad range of temperatures (Bonato 1999). With an arrhenotokous mode of reproduction (females being diploid and haploid males developing from unfertilized eggs), a small number of founding individuals or even a single female can initiate new mite colonies that can build up rapidly as a result of several typical ‘r-selected’ traits including short generation time (Sabelis 1985) and high dispersal ability (Kennedy and Smitley 1985). In addition, because of its small size (0.3–0.5 mm depending on the development stage), T. evansi can be difficult to detect on plant shipments and may remain undetected in new localities until its presence is revealed by outbreaks and plant damage. These factors, often compounded by the fact that historical records for the species are incomplete, inaccurate or nonexistent, make it difficult to reconstruct reliably the invasion history. Information on population histories gained by genetic approaches is particularly valuable for species such as T. evansi, that are small or inconspicuous (Navia et al. 2005; Scheffer and Grissell 2003) and that resemble morphologically resident species or previous invaders (Müller 2001; Reitzel et al. 2008; Stepien and Tumeo 2006). Such undetected cryptic invasions are not uncommon for spider mites, eventually leading to considerable economic consequences (e.g. Knapp et al. 2003; Navajas et al. 2001).
This paper presents a genetic analysis of the emerging pest T. evansi, using two DNA sequence based markers, a fragment of the Cytochrome Oxidase subunit I (COI) of mitochondrial DNA (mtDNA) and the internal transcribed spacer (ITS) region of nuclear ribosomal DNA (rDNA). We include samples from the known geographical distribution of the species in South America, Africa, Asia and Southern Europe. Special effort in sampling the Mediterranean basin was made to understand the invasion process in this area, where T. evansi is currently rising as a significant pest. As a result of the coupling of: (a) information about the complex patterns of movement and historical demography obtained by genetic analysis, (b) knowledge of biological features associated with rapid population growth, and (c) the existence of reference collections that allow the predictions of potential distributions in the face of climate change (see Migeon et al. 2009), studies of T. evansi can be a model for understanding invasions of cryptic emerging invasive species.
Materials and methods
Sample collection
This study includes specimens of T. evansi from most parts of the world where it is currently known, including South America (Brazil and Argentina)—the assumed native area of the species—invaded areas on three continents (Africa, Europe and East Asia), where T. evansi has previously been reported, as well as additional new records (supplementary Table 1). Mites were collected between 2004 and 2008 from plants in the family Solanaceae and stored in 95% ethanol at −20°C for DNA analysis. Some individuals from each sample were preserved in 70% ethanol for further morphological verification of species identity involving observations of the shape of the male aedeagus under microscope preparations.
DNA extraction, amplification and sequencing
Total genomic DNA was isolated from individual adult females using the DNeasy tissue Kit (Qiagen, USA), following the protocol described in (Tsagkarakou et al. 2007a). Two target DNA fragments were PCR-amplified and sequenced: a fragment of the COI mitochondrial gene (COI) and the internal transcribed spacer region (ITS: ITS1-5.8sRNA-ITS2). Several mites (1–9) per locality were sequenced for the COI (supplementary Table 1). Given the striking lack of variation of the ITS sequences with only two majority types detected worldwide (see results below), only 1–2 mites per locality were sequenced for this fragment. PCR conditions were as described previously (Tsagkarakou et al. 2007a) with modifications of annealing temperatures and primers used to amplify and sequence the ITS 1,137 base pairs (bp) and the COI 868 (bp) as described by Gotoh et al. (2009). PCR amplifications were carried out in an Eppendorf Mastercycler. PCR products were commercially sequenced (either by Cogenics, Meylan, France or Macrogen, Seoul, South Korea). GenBank accession numbers are included in Table 1.
DNA sequences alignment and analyses
Sequences were edited in the Seqscape package (SeqScape Software v.2.5, Applied Biosystems SeqScape® Software), and aligned using the default parameters of CLUSTAL W (Thompson et al. 1994) and refined by eye. There were no insertions/deletions in either COI or ITS regions. The boundaries of genes in the ITS regions were delimited by comparison to the ITS sequences in Hurtado et al. (2008), GenBank accession no. AM408033. Pairwise nucleotide divergence of COI and ITS sequences using the Kimura 2-parameter (K2P) model were calculated using the program Molecular Evolutionary Genetic Analysis (MEGA v.4, Tamura et al. 2007) and phylogenetic trees were constructed using the neighbour joining method (Saitou and Nei 1987). For comparison, we also generated a maximum likelihood tree [implemented in PhyML v.2. 4.4 (Guindon and Gascuel 2003)] employing the GTR + I model, as determined by Modeltest v.3.7 (Posada and Crandall 1998). The COI sequence of the closely related Tetranychus urticae (obtained in this study) was used as outgroup. Samples were pooled by country to calculate haplotype frequencies (supplementary Tables 1 and 2). When haplotype diversity within a country was high (i.e., presence of two haplotype clades, see below), samples were divided into regions (i.e., within Brazil, France, Spain and Portugal). In some cases, localities of several countries were pooled by geographical affinities based on sample size and sequence homogeneity (i.e., Kenya and Tanzania were pooled in the East Africa sample and the Argentinean sample from Corrientes was included in the Brazil-South West sample).
Indices of sequence diversity, including number of mtDNA haplotypes (Nh), haplotype diversity (h), nucleotide diversity (π) and average number of pairwise nucleotide differences (K), were estimated using DnaSP v.4.20.2 (Rozas et al. 2003). These parameters were calculated for samples grouped into four geographical regions: (1) South America; (2) Mediterranean region (including Portugal, the Canary and the Madeira Islands); (3) Africa (including also Reunion Island), and (4) Asia; as well as for the two detected clades. Molecular analysis of variance (AMOVA; Excoffier et al. 1992) as implemented in Arlequin v.3.1 (Excoffier et al. 2006) was used to assess the genetic differentiation among the four geographical regions (South American, Mediterranean region, Africa and Asia). Genealogical relationships among haplotypes of COI were reconstructed by a haplotype network analysis obtained by statistical parsimony method (default parsimony connection limit of 95%) implemented in the TCS v.1.21 software (Clement et al. 2000).
Results
Mitochondrial COI haplotyes
Analysis of 868 (bp) of mitochondrial COI DNA sequence data from 298 individuals revealed ten unique haplotypes. Among the sequences analysed, 29 segregating (polymorphic) sites were detected (Table 1a), 27 of which were parsimony-informative and two were singleton sites. Ten different haplotypes were identified representing 0.697 ± 0.021 and 0.0123 ± 0.0053 (mean ± SD) haplotype and nucleotide diversity, respectively.
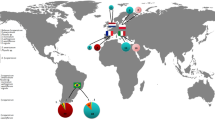
The distribution of the T. evansi mtDNA haplotypes across the sampled continents is presented in Fig. 1. Several haplotypes were found in a single locality (H2, H3, H5, H6, H8, H9 and H10) while others were widely distributed (H1, H4 and H7). The most widely distributed haplotype (H1) was found with a global frequency of 48.7% in Africa, Europe and South America (Brazil-South West) (supplementary Table 2; Fig. 1). The two other most frequent haplotypes were H4 and H7 with global frequencies of 16.1 and 17.1%, respectively. Whereas H4 occurred widely (Africa, Asia and Europe), H7 was found in only three localities in Europe (France-South, Spain-North East, and Portugal). In most localities a single haplotype was detected, but in others either two (Brazil-South West, Portugal Center, and Reunion Is.) or three (Spain-East) haplotypes were found (supplementary Tables 1 and 2).

a Distribution of mitochondrial DNA haplotypes of Tetranychus evansi across the geographical areas analyzed in this study. Nearby populations are pooled with the size of the symbols proportional to the number of individuals sampled: b in South America and c in the Mediterranean Basin. Pie charts illustrate the proportion haplotypes at each sampled locality
The highest COI diversity was found in South America with six out of ten haplotypes present (h = 0.685 ± 0.031, π = 0.0137 ± 0.0004, K = 11.8) (Table 2). Among the invaded areas, the Mediterranean region where four haplotypes were detected, displayed the highest genetic diversity (h = 0.642 ± 0.020, π = 0.0122 ± 0.0008, K = 10.6), which was similar to the level of genetic diversity in South American samples (Table 2a).
The phylogenetic reconstruction based on COI sequences separates the samples into two clades (99% of support through bootstrap analysis, Fig. 2a). The maximum likelihood tree (not shown) generated using GTR+I model was not different from the NJ topology shown and also supported the two major clades with high bootstrap values (Fig. 2a). Haplotypes in each clade were closely related and separated from each other by only one, and in a few cases two, nucleotide difference/s. Clade I showed a lower diversity (h = 0.435 ± 0.033, π = 0.0006 ± 0.0001, K = 0.5) than Clade II (h = 0.595 ± 0.030, π = 0.0014 ± 0.0002, K = 1.2 Table 2b), included the two most common haplotypes worldwide (H1 and H4) and was represented in samples from all continents. Clade II was represented in South American and Mediterranean samples only. These patterns of geographical differentiation support those from the TCS network, in which the two clades are separated from each other by 20 mutational steps (Fig. 2b). COI sequence divergence between the two clades averaged 2.72% in contrast to intra-clade divergences, which ranged from 0.12 to 0.35% and from 0.12 to 0.46% for clades I and II, respectively (Table 1a). Analysis by AMOVA shows that the genetic differentiation within each geographical region explains most (79%) of the total variance (Table 3), which results from the presence of sequences of the two clades in South America and the Mediterranean region.

a Phylogenetic reconstruction inferred through Neighbor-Joining based on mitochondrial COI (left) and ribosomal ITS sequences (right) from individual Tetranychus evansi.Numbers on branches indicate in the order bootstrap values for Neighbor-Joining and Maximum likelihood trees. Tetranychus urticae was used as outgroup. b A mitochondrial haplotype network constructed from samples of T. evansi using statistical parsimony. The areas of the circles are proportional to the number of samples sharing each haplotype. Lines represent a single mutational step and small black circles represent an unobserved haplotype
Internal Transcribed Spacer (rDNA ITS) variation
The complete ITS region (1,137 bp) was sequenced for a total of 96 individuals. Two major types of ITS sequences were found, named here T1 and T2, which differed from each other by two point mutations in the ITS2 region (positions 723 and 1,045). Variation among the two ITS types were consistent with the two COI clades revealed by the mtDNA sequence data. The single exception was ITS sequences (T3) found in the Argentina sample, which differed from T1 by a single point mutation (Table 2b). Four heterozygotes (T1/T3) were detected in this Argentinean sample of five individuals. Otherwise, ITS heterozygotes were rare: one individual from Portugal Center was a T1/T2 heterozygote. The most frequent (70.83%) and widely distributed ITS type was T1, which was found in most of the invaded areas as well as South West Brazil and Argentina (supplementary Table 2). The T2 type (22.92%) was found in some parts of the West Mediterranean region (France-South, Spain-North East and Portugal) and in the Atlantic Brazilian Coast which includes the North East, South East and South Brazil samples.
Discussion
Origins of invasive Tetranychus evansi
A common assumption, though not always correct, is that the geographical natal range of a species contains the oldest populations which will therefore have the greatest genetic diversity (Lozier et al. 2009; Nardi et al. 2005; Roderick 2004). In this study, patterns of genetic variation of T. evansi are consistent with the hypothesis of a South American origin of the species. Populations in South America were found to contain six out of the ten identified mitochondrial haplotypes and the single variant (T3) of the most common ITS sequence (T1), despite a relatively small sample size (the cumulative number of individuals analyzed in the other continents was approximately three times as great). Although the measured genetic diversity of T. evansi would certainly increase with a greater number of populations examined (as modeled for several invasive species, Puillandre et al. 2008), the sample from South America comprises collections from Recife (Brazil) to Tucuman (Northern Argentina), covering more than 3,800 km, spanning the distribution area of the pest. Tetranychus evansi is not evenly distributed in Brazil and geographical predictions based on climatic modelling pinpointed the Atlantic coast and South Brazil as the most suitable geographical areas for the species (Migeon et al. 2009). More generally, based on predictions modelled by CLIMEX by these authors, the potential distribution of T. evansi worldwide appears limited by cold stress together with the impact of other climatic parameters, mainly dryness and humidity. The restricted geographical distribution of T. evansi in South America, which has been confirmed by several intensive surveys (Da Silva et al. 2008; Furtado et al. 2005; Furtado et al. 2006) might be explained by climatic conditions, but could also reflect other environmental or ecological factors. Low densities of T. evansi were generally observed in South America compared to most of the newly invaded continents where the mite undergoes heavy outbreaks, such as in Africa (Saunyama and Knapp 2003) and in Southern Europe along the Spanish Mediterranean coast (Ferragut and Escudero 1999).
Among the factors that might account for different mite densities in the native and invaded areas is competition with other tetranychid species, which are abundant on solanaceous plants in Brazil [32 and 17 tetranychid species have been reported from solanaceous plants in South America and Europe, respectively (Migeon and Dorkeld 2006)]. In addition, effective native natural enemies in South America maintain T. evansi populations at low densities (Furtado et al. 2005; Furtado et al. 2006; Rosa et al. 2005). For example the neotropical predatory mite Phytoseiulus longipes has been described as successfully developing on T. evansi (Maxime Ferrero, personal communication; Furtado et al. 2007), and several other mite predators of the family Phytoseiidae have been reported associated with T. evansi on tomato and wild solanaceous plants in Brazil (Fiaboe et al. 2006; Furtado et al. 2006; Rosa et al. 2005). A fungal pathogen was also suggested as the causative agent of the decline of T. evansi populations in northeastern Brazil, when in 1979 occasional outbreaks in tomato field crops were reported (Humber et al. 1981). However, outside of South America, several attempts to control T. evansi with different mite predators that have been used successfully on other Tetranychus species, have not shown promising results (Escudero and Ferragut 2005; De Moraes and McMurtry 1985). These findings are consistent with the enemy release hypothesis, which predicts less predatory pressure in introduced populations (Keane and Crawley 2002). Knowledge of a South American origin of T. evansi should help to target regions for additional exploration of biocontrol candidates (see Roderick and Navajas 2003).
Species-wide genetic homogeneity and invasion events
Assuming a South American origin of T. evansi, the invasion of new areas by this mite sampled in this study, except Mediterranean region, was characterized either by a very low genetic diversity (e.g., Africa) or no genetic variation (e.g., East Asia). A paradox often encountered in genetic studies of invasive species, is a colonization success of the introduced populations despite a strong reduction of genetic diversity (Frankham 2005; see also reviews by Dlugosch and Parker 2008 and Roman and Darling 2007). Several studies to date have shown that founder effects and bottlenecks are not an obstacle for invasion success (Ahern et al. 2009; Puillandre et al. 2008; Solignac et al. 2005), whereas plasticity in life history traits seems to be important for the successful expansion of an invasive species (Chen et al. 2006; Valiente et al. 2010; Wang et al. 2005). Mites belonging to clade I have been found across a wide range of habitats and regions beyond the predicted geographical distribution as modeled based on climatic suitability T. evansi (Migeon et al. 2009). Although well established in the Mediterranean basin where climatic conditions are favorable to the species, haplotype H4 also occurs beyond the modeled climatic borders for the species, such as near the Southern Sahelian border, where according to modeling predictions (Migeon et al. 2009) dryness should limits the distribution. Likewise, in Asia the same haplotype was detected in areas predicted to experience stress through humidity (in Taiwan) or cold (in Japan). With an unexpected wide range of climatic conditions being tolerated by one of the T. evansi haplotypes, its increasing distribution is not likely to be well predicted by existing climate-based modeling, and special attention should be given to its expansion worldwide.
Colonization from multiple sources may mitigate the loss of genetic diversity needed to face novel selective challenges as encountered in the new invaded areas (Facon et al. 2006; Kolbe et al. 2004; Roman 2006). The genetic variability of mites from the western Mediterranean region was the highest among the colonized areas. Given that records of T. evansi in the colonized areas date from less than 50 years ago, and considering a mutation rate of mitochondrial genes typically estimated at 1.4–2.3% per million years in Arthropods (Xu et al. 2006), it seems unlikely that the haplotype diversity originated in the new region, but rather represents the combined effects of ancestral diversity and multiple introduction events. Although the level of divergence (ranging from 2.35 to 3.08%), between the two mtDNA clades detected might suggest the presence of two distinct taxa within a species complex, evidence of a single species exists. First, a comparable amount of intraspecific genetic diversity of COI sequences has been found within other Tetranychidae mite species (ranging from 1 to 6%) (Navajas and Boursot 2003; Navajas et al. 1994; Navajas et al. 1999; Navajas et al. 1998). Second, we have detected ITS sequence hybrids (each of the ITS types corresponds to one of the two COI clades) in the same sample (from Portugal), thus showing that crosses between mites stemming from the two clades are not only possible, but exist in nature. Additional evidence of conspecificity for individuals of the two clades comes from cross breeding experiments, which demonstrated fertile F1 females, although with some incompatibility (Gotoh et al. 2009).
Invasion pathways
In Africa, where the oldest records of T. evansi outside the Americas have been reported [in 1952 from the Mauritius Is. (Moutia 1958) and in 1979 from Zimbabwe (Blair 1983)], the colonization of wide areas by T. evansi results from unique introductions (for example, a single haplotype, H1, was detected in 13 samples from Kenya, Tanzania and South Africa). With H1 also encountered in Southwest Brazil, this region might be a potential source of the invasion of the African continent. Haplotype H4 from clade I, found in the Maghreb countries and Niger, is largely distributed in the Mediterranean basin and is the only one found in Japan and Taiwan. The virtual lack of genetic diversity in Asian samples supports a single and recent introduction event. The first documented record in Asia is from Taiwan in 2001 (Ho et al. 2004), although older reports of misidentified T. evansi from Taiwan exist back to 1992 (Ho and Wang 2007) with subsequent reports from Japan in 2002. All current mite records of T. evansi in Asian continent were from regions surrounding port cities such as Kagoshima, Tokyo, Osaka, Kyoto, Fukuoka and Okinawa in Japan (T. Gotoh, unpublished data) and from Taitung in Taiwan, suggesting the central role of human commodities in the invasion of Asia by T. evansi. It remains however, unclear whether a single introduction event or several from the same source are responsible for colonization of this region. It cannot be ruled out that the Mediterranean region or the Western Africa, where mites bear the same haplotype, might have played a role as a secondary source in the colonization of T. evansi of the Asian continent.
Emerging pest: lessons learned by Tetranychus evansi
Among the different haplotypes present in the Mediterranean region, H1 and H4, both from clade I, are the most widely spread, whereas a single haplotype of clade II (H7) was found in a restricted area of the Mediterranean region. Worth noting is that H1 and H4 are in areas where heavy mite outbreaks have been reported, e.g. in South-East Africa (Saunyama and Knapp 2003; Sibanda et al. 2000), West Africa (Duverney and Ngueye-Ndiaye 2005), Spain (Ferragut and Escudero 1999), and where we have also observed the mite in the field on tomato and eggplant greenhouses in France and in the Canary Is. and on pepper and potatoes in Algeria (Yamina Guenaoui, personal communication). By contrast, high mite densities in regions where H7 and other haplotypes belonging to the clade II are present have never been reported and mite populations have been observed associated with wild solanaceous plants such as nightshade, Solanum nigrum. In addition to differences in host associations, the two clades also appear to differ in invasive potential, with mites from clade I having a higher invasive potential having colonized the entire Mediterranean basin, together with Africa and East Asia. Demographic parameters and adaptive performance underlying the differential invasive success of mites of clade I and II, are currently under investigation. The existence of two biologically distinct clades that differ in invasive potential is similar to what is known about the recent invasive biology of the tropical whitefly with Bemisia tabaci, which there exist at least two worldwide invasive biotypes, B and Q, (Boykin et al. 2007). Biotype Q is dominant in the Mediterranean basin and shows performance differences related to extreme temperature (Bonato et al. 2007) and to insecticide tolerance (Horowitz et al. 2005; Tsagkarakou et al. 2007b).
The threat that T. evansi represents for local agriculture in the Mediterranean basin is increased by the sympatric presence of distantly related mites. In the case of multiple introductions which might have unequal potential for becoming invasive (Allendorf and Lundquist 2003; Jousson et al. 2000; Sakai et al. 2001), crosses among individuals from distant populations might generate novel genetic combinations leading to new traits which eventually results in increased invasiveness (Allendorf and Lundquist 2003; Ellstrand and Schierenbeck 2000; Facon et al. 2006; Lee 2002). Hybrid vigour has been invoked to explain why some introduced populations may overcome their parents’ fitness in new environments (Facon et al. 2005) and the reproductive compatibility between mites from the two clades has recently been demonstrated (Gotoh et al. 2009).
Tetranychus evansi is regarded as an important agricultural pest in Southern Europe but also in South East Africa and the detection of different lineages with different invasive potential is crucial to help in designing pest management strategies involving quarantine measures and the search for biocontrol candidates. The assumption that a local adaptation between biological control agents and the invasive genotypes of a pest exists (see Hufbauer and Roderick 2005), implies a targeted search of natural enemies in the area of provenance of the particular invasive genotypes. Among several mite predators reported associated with T. evansi in Brazil, only Brazilian (Furtado et al. 2007) and Argentine (Maxime Ferrero, personal communication) P. longipes populations were the most promising to control T. evansi populations in introduced areas. By contrast, the Chilean (Maxime Ferrero, personal communication) and South African populations (De Moraes and McMurtry 1985) of this mite predator are inefficient to control T. evansi.
This study illustrates the extent to which molecular markers can aid in the study of an emerging pest in the earliest stages of the invasion process and thus help to address questions related to management and biosecurity. Unfortunately, regulation generally does not take into account the fact that some genotypes of an invasive species may differ in invasive potential (Allendorf and Lundquist 2003). Human activities have played a significant role in explaining the present distribution of T. evansi, and thus monitoring pathways will be critical for its control. The spread of T. evansi throughout the world is testimony of the limits of international quarantine measures, particularly important in the case of inconspicuous pests, which are prone to cryptic invasions. Integrating international cooperation will be critical to identify sources and pathways of invasion as well as build and/or maintain resilience to invasion to enhance plant biosecurity for emerging arthropod pests.
References
Ahern R, Hawthorne D, Raupp M (2009) Founder effects and phenotypic variation in Adelges cooleyi, an insect pest introduced to the eastern United States. Biol Invasions 11:959–971
Allendorf FW, Lundquist LL (2003) Introduction: population biology, evolution, and control of invasive species. Conserv Biol 17:24–30
Anderson PK, Cunningham AA, Patel NG, Morales FJ, Epstein PR, Daszak P (2004) Emerging infectious diseases of plants: pathogen pollution, climate change and agrotechnology drivers. Trends Ecol Evol 19:535–544
Blair BW (1983) Tetranychus evansi Baker & Pritchard (Acari: Tetranychidae): a new pest of tobacco in Zimbabwe. In: Corest phytopathology and agronomy study group, Bergerac
Bonato O (1999) The effect of temperature on life history parameters of Tetranychus evansi (Acari: Tetranychidae). Exp Appl Acarol 23:11–19
Bonato O, Lurette A, Vidal C, Fargues J (2007) Modelling temperature-dependent bionomics of Bemisia tabaci (Q-biotype). Physiol Entomol 32:50–55
Boykin LM, Shatters RG Jr, Rosell RC et al (2007) Global relationships of Bemisia tabaci (Hemiptera: Aleyrodidae) revealed using Bayesian analysis of mitochondrial COI DNA sequences. Mol Phylogenet Evol 44:1306–1319
Chen Y, Opp S, Berlocher S, Roderick G (2006) Are bottlenecks associated with colonization? genetic diversity and diapause variation of native and introduced Rhagoletis completa populations. Oecologia 149:656–667
Clement M, Posada D, Crandall KA (2000) TCS: a computer program to estimate gene genealogies. Mol Ecol 9:1657–1660
Da Silva F, De Moraes G, Knapp M (2008) Distribution of Tetranychus evansi and its predator Phytoseiulus longipes (Acari: Tetranychidae, Phytoseiidae) in southern Brazil. Exp Appl Acarol 45:137–145
De Moraes GJ, McMurtry JA (1985) Comparison of Tetranychus evansi and T. urticae (Acari: Tetranychidae) as prey for eight species of phytoseiid mites. Entomophaga 30:393–397
Dlugosch KM, Parker IM (2008) Founding events in species invasions: genetic variation, adaptive evolution, and the role of multiple introductions. Mol Ecol 17:431–449
Duverney C, Ngueye-Ndiaye A (2005) Essais préliminaires pour limiter les dégâts de Tetranychidae sur les cultures maraîchères dans le Sine-Saloum (Sénégal). In: Comptes rendus du deuxième colloque international sur les acariens des cultures. AFPP Paris, Montpellier
Ellstrand NC, Schierenbeck KA (2000) Hybridization as a stimulus for the evolution of invasiveness in plants? Proc Natl Acad Sci USA 97:7043–7050
EPPO (2007) European and mediterranean plant protection rganization. EPPO A2 List of pests recommended for regulation as quarantine pests. (http://www.eppo.org/QUARANTINE/listA2.htm). Accessed 25 Sep 2009
Escudero LA, Ferragut F (2005) Life-history of predatory mites Neoseiulus californicus and Phytoseiulus persimilis (Acari: Phytoseiidae) on four spider mite species as prey, with special reference to Tetranychus evansi (Acari: Tetranychidae). Biol Control 32:378–384
Excoffier L, Smouse P, Quattro J (1992) Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial-DNA restriction data. Genetics 131:479–491
Excoffier L, Laval G, Schneider S (2006) Arlequin version 3.01: an integrated software package for population genetic data analysis. Computational and Molecular Population Genetics Lab (CMPG). Institute of Zoology, University of Berne, Switzerland
Facon B, Jarne P, Pointier JP, David P (2005) Hybridization and invasiveness in the freshwater snail Melanoides tuberculata: hybrid vigour is more important than increase in genetic variance. J Evol Biol 18:524–535
Facon B, Genton BJ, Shykoff J, Jarne P, Estoup A, David P (2006) A general eco-evolutionary framework for understanding bioinvasions. Trends Ecol Evol 21:130–135
Ferragut F, Escudero LA (1999) Tetranychus evansi Baker & Pritchard (Acari, Tetranychidae), una nueva araña roja en los cultivos horticolas españoles. Boletin de Sanidad Vegetal, Plagas 25:157–164
Fiaboe KKM, Fonseca RL, De Moraes GJ, Ogol CKPO, Knapp M (2006) Identification of priority areas in South America for exploration of natural enemies for classical biological control of Tetranychus evansi (Acari: Tetranychidae) in Africa. Biol Control 38:373–379
Frankham R (2005) Resolving the genetic paradox in invasive species. Heredity 94:385–385
Furtado I, Kreiter S, De Moraes GJ, Tixier MS, Flechtmann C, Knapp M (2005) Plant mites (Acari) from Northeastern Brazil, with descriptions of two new species of the family Phytoseiidae (Mesostigmata). Acarologia 45:131–143
Furtado PI, De Moraes JG, Kreiter S, Knapp M (2006) Search for effective natural enemies of Tetranychus evansi in south and southeast Brazil. Exp Appl Acarol 40:157–174
Furtado IP, De Moraes GJ, Kreiter S, Tixier MS, Knapp M (2007) Potential of a Brazilian population of the predatory mite Phytoseiulus longipes as a biological control agent of Tetranychus evansi (Acari: Phytoselidae: Tetranychidae). Biol Control 42:139–147
Gotoh T, Araki R, Boubou A, Migeon A, Ferragut F, Navajas M (2009) Evidence of co-specificity between Tetranychus evansi and T. takafujii (Acari: Prostigmata, Tetranychidae) suggested by molecular, morphological and compatibility data: comments on taxonomical and agricultural aspects. Int J Acarol 35:485–501
Guindon S, Gascuel O (2003) A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol 52:696–704
Ho CC, Wang SC (2007) Addendum on the records of Tetranychus evansi in Taiwan. Plant Prot Bull 49:167–168
Ho CC, Wang SC, Chien YL (2004) Field observation on two newly recorded spider mites in Taiwan. Plant Prot Bull 47:391–402
Horowitz AR, Kontsedalov S, Khasdan V, Ishaaya I (2005) Biotypes B and Q of Bemisia tabaci and their relevance to neonicotinoid and pyriproxyfen resistance. Arch Insect Biochem Physiol 58:216–225
Hufbauer RA, Roderick GK (2005) Microevolution in biological control: mechanisms, patterns, and process. Biol Control 35:227–239
Humber R, De Moraes G, dos Santos J (1981) Natural infection of Tetranychus evansi (Acarina: Tetranychidae) by a Triplosporium sp (Zygomycetes: Entomophthorales) in northeastern Brazil. Biocontrol 26:421–425
Hurtado MA, Ansaloni T, Cros-Arteil S, Jacas JA, Navajas M (2008) Sequence analysis of the ribosomal internal transcribed spacers region in spider mites (Prostigmata: Tetranychidae) occurring in citrus orchards in Eastern Spain: use for species discrimination. Ann Appl Biol 153:167–174
Jousson O, Pawlowski J, Zaninetti L, Zechman FW, Dini F, Di Guiseppe G, Woodfield R, Millar A, Meinesz A (2000) Invasive alga reaches California. Nature 408:157–158
Keane RM, Crawley MJ (2002) Exotic plant invasions and the enemy release hypothesis. Trends Ecol Evol 17:164–170
Kennedy G, Smitley D (1985) Dispersal. In: Helle W, Sabelis MW (eds) Spider mites, their biology, natural enemies and control. Elsevier, Amsterdam, pp 233–242
Knapp M, Wagener B, Navajas M (2003) Molecular discrimination between the spider mites Tetranychus evansi Baker & Pritchard, an important pest of tomatoes in southern Africa, and the closely related species T. urticae Koch (Acarina: Tetranychidae). Afr Entomol 11:300–304
Kolbe JJ, Glor RE, Schettino LRG, Lara AC, Larson A, Losos JB (2004) Genetic variation increases during biological invasion by a Cuban lizard. Nature 431:177–181
Lee CE (2002) Evolutionary genetics of invasive species. Trends Ecol Evol 17:386–391
Lozier J, Roderick G, Mills N (2009) Tracing the invasion history of mealy plum aphid, Hyalopterus pruni (Hemiptera: Aphididae), in North America: a population genetics approach. Biol Invasions 11:299–314
Menzel A, Estrella N, Heitland W, Susnik A, Schleip C, Dose V (2008) Bayesian analysis of the species-specific lengthening of the growing season in two European countries and the influence of an insect pest. Int J Biometeorol 52:209–218
Migeon A, Dorkeld F (2006) Spider Mites Web: a comprehensive database for the Tetranychidae. http://www.montpellier.inra.fr/CBGP/spmweb/. Last update 15 June 2009. Accessed 25 Sep 2009
Migeon A, Ferragut F, Escudero-Colomar L, Fiaboe K, Knapp M, de Moraes G, Ueckermann E, Navajas M (2009) Modelling the potential distribution of the invasive tomato red spider mite, Tetranychus evansi (Acari: Tetranychidae). Exp Appl Acarol 48:199–212
Moutia LA (1958) Contribution to the study of some phytophagous acarina and their predators in Mauritius. Bull Entomol Res 49:59–75
Müller J (2001) Invasion history and genetic population structure of riverine macroinvertebrates. Zoology 104:346–355
Nardi F, Carapelli A, Dallai R, Roderick GK, Frati F (2005) Population structure and colonization history of the olive fly, Bactrocera oleae (Diptera, Tephritidae). Mol Ecol 14:2729–2738
Navajas M, Boursot P (2003) Nuclear ribosomal DNA monophyly versus mitochondrial DNA polyphyly in two closely related mite species: the influence of life history and molecular drive. Proc R Soc Lond B 270:124–127
Navajas M, Gutierrez J, Bonato O, Bolland HR, Mapangou-Divassa S (1994) Intraspecific diversity of the Cassava Green Mite Mononychellus progresivus (Acari: Tetranychidae) using comparisons of mitochondrial and nuclear ribosomal DNA sequences and cross-breeding. Exp Appl Acarol 18:351–360
Navajas M, Lagnel J, Gutierrez J, Boursot P (1998) Species-wide homogeneity of nuclear ribosomal ITS2 sequences in the spider mite Tetranychus urticae contrasts with extensive mitochondrial COI polymorphism. Heredity 80:742–752
Navajas M, Gutierrez J, Lagnel J, Fauvel G, Gotoh T (1999) DNA sequences and cross-breeding experiments in the hawthorn spider mite Amphitetranychus viennensis reveal high genetic differentiation between Japanese and French populations. Entomol Exp Appl 90:113–122
Navajas M, Gutierrez J, Williams M, Gotoh T (2001) Synonymy between two spider mite species, Tetranychus kanzawai and T. hydrangeae (Acari: Tetranychidae), shown by ribosomal ITS2 sequences and cross-breeding experiments. Bull Entomol Res 91:117–123
Navia D, De Moraes GJ, Roderick G, Navajas M (2005) The invasive coconut mite Aceria guerreronis (Acari: Eriophyidae): origin and invasion sources inferred from mitochondrial (16S) and nuclear (ITS) sequences. Bull Entomol Res 95:505–516
Pimentel D, Zuniga R, Morrison D (2005) Update on the environmental and economic costs associated with alien-invasive species in the United States. Ecolog Econ 52:273–288
Posada D, Crandall KA (1998) Modeltest: testing the model of DNA substitution. Bioinformatics 14:817–818
Puillandre N, Dupas S, Dangles O, Zeddam JL, Capdevielle-Dulac C, Barbin K, Torres-Leguizamon M, Silvain JF (2008) Genetic bottleneck in invasive species: the potato tuber moth adds to the list. Biol Invasions 10:319–333
Reitzel A, Darling J, Sullivan J, Finnerty J (2008) Global population genetic structure of the starlet anemone Nematostella vectensis: multiple introductions and implications for conservation policy. Biol Invasions 10:1197–1213
Roderick GK (2004) Tracing the origin of pests and natural enemies: genetic and statistical approaches. In: Ehler LE, Sforza R, Mateille T (eds) Genetics, evolution, and biological control. CAB International, Wallingford, pp 97–112
Roderick GK, Navajas M (2003) Genes in new environments: genetics and evolution in biological control. Nature Rev Genet 4:889–899
Rodoni B (2009) The role of plant biosecurity in preventing and controlling emerging plant virus disease epidemics. Virus Res 141:150–157
Roman J (2006) Diluting the founder effect: cryptic invasions expand a marine invader’s range. Proc R Soc Lond B 273:2453–2459
Roman J, Darling JA (2007) Paradox lost: genetic diversity and the success of aquatic invasions. Trends Ecol Evol 22:454–464
Rosa AA, Gondim MGC, Fiaboe KKM, De Moraes GJ, Knapp M (2005) Predatory mites associated with Tetranychus evansi Baker & Pritchard (Acari: Tetranychidae) on native solanaceous plants of coastal Pernambuco State, Brazil. Neotrop Entomol 34:689–692
Rozas J, Sánchez-DelBarrio JC, Messeguer X, Rozas R (2003) DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics 19:2496–2497
Sabelis MW (1985) Reproductive strategies. In: Helle W, Sabelis MW (eds) Spider mites, their biology,natural enemies and control. Elsevier, Amsterdam, pp 265–278
Saitou N, Nei M (1987) The neighbour-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
Sakai AK, Allendorf FW, Holt JS, Lodge DM, Molofsky J, With KA, Baughman S, Cabin RJ, Cohen JE, Ellstrand NC, McCauley DE, O’Neil P, Parker IM, Thompson JN, Weller SG (2001) The population biology of invasive species. Ann Rev Ecol Syst 32:305–332
Saunyama IGM, Knapp M (2003) Effects of pruning and trellising of tomatoes on red spider mite incidence and crop yield in Zimbabwe. Afr Crop Sci J 11:269–277
Scheffer SJ, Grissell EE (2003) Tracing the geographical origin of Megastigmus transvaalensis (Hymenoptera: Torymidae): an African wasp feeding on a South American plant in North America. Mol Ecol 12:415–421
Sibanda T, Dobson HM, Cooper JF, Manyangarirwa W, Chiimba W (2000) Pest management challenges for smallholder vegetable farmers in Zimbabwe. Crop Prot 19:807–815
Silva P (1954) Um novo àcaro nocivo ao tomateiro na Bahia. Bol Inst Biol Bahia 1:1–20
Solignac M, Cornuet JM, Vautrin D et al (2005) The invasive Korea and Japan types of Varroa destructor, ectoparasitic mites of the Western honeybee Apis mellifera, are two partly isolated clones. Proc R Soc Lond B 272:411–419
Stepien CA, Tumeo MA (2006) Invasion genetics of Ponto-Caspian gobies in the Great Lakes: a cryptic species, absence of founder effects, and comparative risk analysis. Biol Invasions 8:61–78
Tamura K, Dudley J, Nei M, Kumar S (2007) Molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol Biol Evol 24:1596–1599
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, positions-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680
Toroitich F, Navajas M, Knapp M (2008) Molecular analysis of the genetic diversity of Tetranychus evansi Baker and Pritchard from Kenya and Tanzania. In: VIth European Congress of Acarology, Montpellier, Poster
Tsagkarakou A, Cros-Arteil S, Navajas M (2007a) First record of the invasive mite Tetranychus evansi in Greece. Phytoparasitica 35:519–522
Tsagkarakou A, Tsigenopoulos CS, Gorman K, Lagnel J, Bedford ID (2007b) Biotype status and genetic polymorphism of the whitefly Bemisia tabaci (Hemiptera: Aleyrodidae) in Greece: mitochondrial DNA and microsatellites. Bull Entomol Res 97:29–40
Valiente A, Juanes F, Nuñez P, Garcia-Vazquez E (2010) Brown trout (Salmo trutta) invasiveness: plasticity in life-history is more important than genetic variability. Biol Invasions 12:451–462
Waage JK, Mumford JD (2008) Agricultural biosecurity. Phil Trans R Soc B 363:863–876
Wang B, Li W, Wang J (2005) Genetic diversity of Alternanthera philoxeroides in China. Aquat Bot 81:277–283
Xu W, Jameson D, Tang B, Higgs P (2006) The relationship between the rate of molecular evolution and the rate of genome rearrangement in animal mitochondrial genomes. J Mol Evol 63:375–392
Acknowledgements
We are grateful to P. Auger, R. Hufbauer, P. Jarne and G. Kargoat for helpful discussions on this work. We thank T. Gotoh, Ibaraki University, Japan, for providing unpublished data of the distribution of T. evansi haplotypes in Japan. We are also grateful to all the persons who have collected and contributed mites for this project: T. Ben-David (The Hebrew University of Jerusalem, Israel), P. Caplong (FDGDON, La Réunion), M. Castagnoli (CRA, Firenze), G. Daubigny (IRD-CBGP, Montpellier), G. De Moraes (Universidade de São Paulo, Brazil), F. Ferragut (Universidad Politécnica, Valencia, Spain), M. Ferrero (SupAgro, Montpellier), M. G. C. Gondim (UFR, Recife), T. Gotoh (Ibaraki University, Japan), J. V. C. Guedes (UF, Santa Maria), Y. Guenaoui (Mostaganem, Algeria), E. Hernández-Suárez (ICIA, Canary Islands), K, Hima (AGRHYMET, Niger), M. Knapp (ICIPE, Nairobi), S. Kreiter (SupAgro, Montpellier), K. Lebdi Grissa (INAT, Tunis), S. Magalhães (Instituto Gulbenkian de Ciência, Portugal), P. Martini (IRF, Sanremo), D. Navia (EMBRAPA, Brasilia), J. P. Quéré (INRA-CBGP, Montpellier), S. Rapetti (IRF, Sanremo), R. Santos de Mendonca (EMBRAPA, Brasilia), S. Simoni (CRA, Firenze), S. Toledo (EEAOC, Tucuman), F. J. Toroitich (ICIPE, Nairobi), A. Tsagkarakou (Heraklion, Greece). This work benefited from a France/Berkeley Fund grant on “Invasive Insects and Mites of Mediterranean Climates” to MN and GKR and an EGIDE-Picasso 2008-17080QH grant from the French Ministère des Affaires Etrangères et Européennes to MN. Financial support for field trips in Portugal was provided by the PESSOA exchange programme accorded to Sara Magalhães, University of Lisbon and Isabelle Olivieri, University of Montpellier.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Boubou, A., Migeon, A., Roderick, G.K. et al. Recent emergence and worldwide spread of the red tomato spider mite, Tetranychus evansi: genetic variation and multiple cryptic invasions. Biol Invasions 13, 81–92 (2011). https://doi.org/10.1007/s10530-010-9791-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10530-010-9791-y