
Abstract
Flagella and invasin play important roles during the early stages of infection by the enteric pathogen Yersinia enterocolitica. Our previous study demonstrated that OmpR negatively regulates invasin gene expression at the transcriptional level. The present study focused on the role of OmpR in the regulation of flagella expression. Motility assays and microscopic observations revealed that an ompR mutant strain exhibits a non-motile phenotype due to the lack of flagella. An analysis of flhDC::lacZYA chromosomal fusions demonstrated a decrease in flhDC expression in ompR mutant cells, suggesting a role for OmpR in the positive control of flagellar master operon flhDC, which is in contrast to the negative role it plays in Escherichia coli. Moreover, high temperature or osmolarity and low pH decreased flhDC expression and OmpR was not required for the response to these factors. Evidence from an examination of the DNA binding properties of OmpR in vitro indicated that the mechanism by which OmpR regulates flhDC is direct. Electrophoretic mobility shift assays confirmed that OmpR binds specifically to the flhDC promoter region and suggested the presence of more than one OmpR-binding site. In addition, phosphorylation of OmpR by acetyl-P appeared to stimulate the binding abilities of OmpR. Together with the results of our previous studies revealing the negative role of OmpR in the regulation of invasin expression, these findings support a model in which invasion and motility might be reciprocally regulated by OmpR.
Similar content being viewed by others
Introduction
In Gram-negative bacteria of the Enterobacteriaceae family, the response regulator OmpR, together with histidine kinase EnvZ, constitutes the EnvZ/OmpR signal transduction pathway that regulates the transcription of several genes involved in adaptive responses to environmental signals (Egger et al. 1997; Stock et al. 1989). EnvZ of Escherichia coli senses changes in the osmolarity of the environment (Russo and Silhavy 1991). This transmembrane histidine kinase is autophosphorylated using ATP and the phosphate group of EnvZ-P is subsequently transferred to OmpR to form phosphorylated OmpR (OmpR-P). EnvZ also possesses phosphatase activity that removes the phosphoryl group from the phosphorylated/activated form of OmpR. The relative activities of the kinase and phosphatase functions of EnvZ regulate the cellular level of OmpR-P in response to environmental changes. OmpR-P is a transcription factor that binds within promoter regions and controls the expression of genes involved in the regulation of numerous functions in bacterial cells, including outer membrane permeability (Russo and Silhavy 1991), biofilm formation (Vidal et al. 1998), flagella synthesis (Shin and Park 1995), low pH tolerance (Bang et al. 2000), fatty acid transport (Higashitani et al. 1993), cell division (Yamamoto et al. 2000) and curli fiber formation (Jubelin et al. 2005). A role for OmpR in controlling the virulence properties of pathogenic bacteria has also been determined (Bernardini et al. 1990; Dorman et al. 1989; Lee et al. 2000). For example, a correlation between the functioning of OmpR and pathogenicity of Yersinia enterocolitica was demonstrated (Brzostek et al. 2003; Dorrell et al. 1998). OmpR was identified as the response regulator for osmolarity-regulated porins and Yop proteins in Y. enterocolitica (Brzostek et al. 2003). OmpR-dependent osmoregulation of Yops has also been recently confirmed in Yersinia pseudotuberculosis (Flamez et al. 2008). Lastly, we have demonstrated that OmpR negatively regulates invasin gene expression in Y. enterocolitica (Brzostek et al. 2007). It appears that OmpR operates as a global regulatory protein in Yersinia cells.
Flagella biogenesis of Gram-negative bacteria is subject to complex control in response to environmental stimuli, which involves a large variety of regulators. The human enteropathogen Y. enterocolitica is a peritrichously flagellated bacterium that becomes motile at temperatures below 30°C (Kapatral and Minnich 1995). Motility allows bacterial cells to locate the most favorable environment and is essential for Y. enterocolitica to contact host cells and invade the host organism (Young et al. 2000). The genes of the Y. enterocolitica flagellar regulon are organized into one large cluster expressed in a cascade that parallels the stepwise assembly of the flagellum (Horne and Pruss 2006). The flagellar transcriptional hierarchy is remarkably similar to that of E. coli and consists of three major flagellar gene classes: I, II and III. The flagellar regulon is modulated by key regulatory elements, particularly a master regulator FlhDC and the alternative sigma factor, FliA (σ28) (Iriarte et al. 1995). FlhDC, which is structurally and functionally conserved in Gram-negative bacteria (Young et al. 1999b), is encoded by the flhDC operon (class I). FlhDC is a heterotetrameric transcriptional activator at the top of the hierarchical cascade, which is required for the expression of all flagellar genes, while sigma factor FliA has a positive effect upon the expression of eight class III flagellar operons (Horne and Pruss 2006; Kapatral et al. 1996). In addition, several studies have shown that master regulator FlhDC affects the expression level of non-flagellar genes of Y. enterocolitica, including 21 genes that are involved in central metabolism (Bleves et al. 2002; Kapatral et al. 2004; Townsend et al. 2008; Young et al. 1999a).
Detailed inspection of the Y. enterocolitica flagellar gene cluster has revealed some differences in the organization of these genes compared with E. coli. The most intriguing is the presence of the inv gene coding for invasin—the main invasion and adhesion factor of this enteropathogen—within the flagellar unit. In addition, Y. enterocolitica has three separate flagellin genes (fleA, fleB, fleC), whereas only one gene (fliC) is present in E. coli.
Studies in E. coli and Salmonella have established that the expression of the master flagellar control operon flhDC is positively regulated at the transcriptional level by factors such as cAMP-CAP (Soutourina et al. 1999; Yokota and Gots 1970), H-NS (Soutourina et al. 1999, 2002) and the two-component system QseBC (Sperandio et al. 2002). Other regulators have been shown to negatively affect flhDC expression, including LrhA (Lehnen et al. 2002), RcsAB (Francez-Charlot et al. 2003) and OmpR (Shin and Park 1995). The transcriptional modulators of flhDC in Y. enterocolitica and their impact upon the regulation of flagellar genes have not yet been studied. In this study we show that OmpR controls the motility of Y. enterocolitica by positively regulating flhDC expression at the transcriptional level. The rationale for reciprocal regulation of motility and invasin production in Y. enterocolitica by OmpR is discussed.
Materials and methods
Bacterial strains, plasmids and growth conditions
The strains and plasmids used in this study are listed in Table 1. Tryptone broth (TB; 1% tryptone, 0.25% NaCl) and minimal medium A (MMA) were used as growth media (Miller 1972). To examine the influence of the osmolarity and pH of the growth medium, an overnight culture was grown at 25°C in 5 ml of TB. This culture was diluted 1:100 in 10 ml of TB0, i.e. TB without NaCl, and the osmolarity was adjusted by adding NaCl to final concentrations of 0.1, 0.2, 0.3 or 0.4 M. In some experiments the osmolarity of the growth medium was raised by adding sorbitol or KCl instead of NaCl. Variation of the pH was achieved by buffering TB with MOPS [3-(N-morpholino) propanesulfonic acid—pH 7.0, 8.0], MES [2-(N-morpholino)ethanesulfonic acid—pH 5.5, 6.5], or homoPIPES [homopiperazine-N,N′-bis(2-ethanesulfonic acid)—pH 5.0] at 0.1 M. To study the effect of different carbon sources, MMA medium was supplemented with glycerol, glucose, pyruvate or acetate to 0.2%. For environmental assays bacteria were grown to exponential phase OD560 of ~0.4). Where necessary, growth media were supplemented with antibiotics: kanamycin (Km)—50 μg/ml, chloramphenicol (Cm)—12.5 or 25 μg/ml, nalidixic acid (Nal)—15 μg/ml, tetracycline (Tc)—12.5 μg/ml.
DNA techniques
Plasmid and chromosomal DNA was purified using Invitrogen kits. DNA fragments were amplified by PCR using Taq or Pfu DNA polymerase (Invitrogen) with oligonucleotide primers. Restriction endonuclease analysis, ligation and transformation were carried out using standard methods (Sambrook et al. 1989). To characterize the flhDC promoter region of strain Ye9 (serotype O:9), three PCR-amplified fragments were applied: a 448-bp fragment amplified using the forward FlhBa1 (5′-CGCTCTTTTCTGACTTCTGG-3′) and reverse FlhBa448 (5′-TCATTTTATACATCCCGACTGA-3′) primers. The 196- and 249-bp fragments of flhDC were amplified using primers: BFlh1 (5′-CCAACTGCCAGATAGACGAC-3′) and BFlh196 (5′-CTCATTTTATACATCCCGACTGA-3′) and OFlh1 (5′-GGGCCTGGATCCTTAGTTTT-3′) and OFlh249 (5′-TTGCATCTTCGACATGAAAC-3′), respectively. All primers were designed based on the flhDC sequence of Y. enterocolitica W1024 serotype O:9 (GenBank accession no. Z48169). The nucleotide sequence of the region upstream of flhDC was examined for potential OmpR binding sites by comparison with the consensus sequence (Egger et al. 1997). Alignments were carried out using NCBI BLAST software.
Motility assays
Swimming assays were performed on TB0 plates containing 0.3% agar. Strains were grown overnight in TB medium at 25°C and a 4 μl aliquot was spotted onto the plates, which were then incubated at 25 and 37°C. After overnight incubation, the plates were photographed and the swimming zones evaluated.
Transmission electron microscopy (TEM)
For the examination of flagella by transmission electron microscopy, bacteria grown on swimming plates (0.3% agar, 25°C, 16 h) were applied to formvar-coated grids (10 min) and then fixed by adding a drop of 0.5% glutaraldehyde for 2 min. The grids were washed three times for 10 s in water and negatively stained with 0.5% phosphotungstic acid for 10 s. Flagella were visualized using a LEO 912AB electron microscope (Zeiss) (Biology Faculty, Warsaw University).
Construction of envZ insertion mutant
The inactivation of envZ in Y. enterocolitica strain Ye9 was performed by plasmid insertion via homologous recombination using the conjugative suicide vector pEP185.2 (Kinder et al. 1993). A 673-bp intragenic fragment of envZ was amplified by PCR using primers EnvZ1 (5′-GAAAGTGGCCTGCGTTGG-3′) and EnvZ673 (5′-TCAATCACCCGCTCATAGC-3′). This DNA fragment was cloned into the pDrive cloning vector (Qiagen) to generate plasmid pDE673, then an XbaI/BamHI fragment of envZ was prepared from this construct and ligated with XbaI/BamHI-digested pEP185.2. The resulting plasmid pEZ1 was transferred from E. coli S17-1 λpir to Y. enterocolitica strain Ye9 NalR (Ye9N) by biparental conjugation. Strains harboring the plasmid integrated into the chromosome were recovered by selecting for Cm and Nal resistance. The insertional mutant obtained by this strategy was designated EZ10 (envZ). Correct integration at the envZ locus was confirmed by PCR with primers UPEnvZ (5′-TGAGAGCTCGTGAAGAAGTTGACCGTATCG-3′), located upstream of the homologous region used for recombination, and pCYC2 (5′-ACTCATCGCAGTACTGTTGTAA-3′), within the chloramphenicol resistance cassette of the suicide vector (data not shown).
Construction of reporter strains carrying a flhDC::lacZYA fusion
To generate a chromosomal flhDC::lacZYA operon fusion, a 519-bp fragment of the flhC gene was first amplified by PCR using Ye9 chromosomal DNA as the template with primers flhDC1 (5′-TGTCTAGAAGATGTTAGAAAGCGAGACG-3′) and flhDC519 (5′-TGCCCGGGTCAAACTGCGCGTCTAACC-3′). The DNA fragment was cloned into the pDrive vector to produce plasmid pD519, then an XbaI/SmaI flhC fragment was subcloned into the corresponding sites of the suicide vector pFUSE (Baumler et al. 1996) to give plasmid pF519. Integration of pF519 into the Y. enterocolitica chromosome was achieved by homologous recombination during the conjugation step, as described previously (Brzostek et al. 2007). The presence of the flhDC::lacZYA fusion in chromosomal DNA was confirmed by PCR with primers flhDRT1 (5′-CTCAGCGATGTTTCGTCTCG-3′), located upstream of the homologous region used for recombination, and lacZ991 (5′-CATCGCAGGCTTCTGCTTC-3′), within the lacZ sequence of pFUSE (data not shown).
β-Galactosidase assays
Y. enterocolitica strains containing the flhDC::lacZYA fusion were grown in MMA, TB and TB0 media under different environmental conditions (temperature, pH, osmolarity). The β-galactosidase activity was measured in bacterial cells permeabilized with SDS and CHCl3 using ONPG as the substrate, according to a standard method (Minnich and Rohde 2007). Most assays were performed at least three times and the data were expressed as the mean ± standard deviation.
Separation of OmpR from OmpR-P by reversed phase HPLC
Full length OmpR was overproduced and purified as described previously (Brzostek et al. 2007). The recombinant OmpR protein migrated as a single band at around 27 kDa on a 12% SDS-polyacrylamide gel. 20 μg of purified OmpR was phosphorylated by treatment with acetyl-P (20 mM) for 1 h at room temperature.
Separation of OmpR from OmpR-P was performed by HPLC according to the method of Head et al. (1998) with slight modifications. Protein samples were injected onto a Symmetry C8 reversed phase column (2.1 mm × 150 mm, particle 5 μm, Waters, USA) and separated using an acetonitrile gradient with solvent A that varied from 80 to 0% over 60 min and solvent B that went from 20 to 100% over the same period, with a constant flow rate of 0.2 ml/min. Between 60 to 135 min, the samples were chromatographed with 100% solvent B. Solvent A was composed of 20% acetonitrile, 0.1% trifluoroacetic acid; solvent B was 60% acetonitrile, 0.1% trifluoroacetic acid. An HPLC system (Waters Alliance 2695) with a PDA detector (2996 PDA, Waters, USA) was used. The eluent was monitored at 254 nm and the peaks were recorded using Empower software (Waters).
Electrophoretic mobility shift assays (EMSAs)
OmpR-binding studies were performed using an N-terminal His-tagged OmpR protein and fragments of the flhDC promoter region containing the putative OmpR binding sites. OmpR-His6 synthesized in E. coli M-15 was purified as described previously (Brzostek et al. 2007). Fragments of flhDC promoter region were amplified by PCR using Pfu polymerase (EURx) with Y. enterocolitica Ye9 chromosomal DNA as the template. The purified fragments (1–3 pmol in 20 μl) were incubated with varying concentrations of OmpR-His6 (0.2–4.0 μM) in the presence of binding buffer P [40 mM Tris–HCl (pH 8.0), 100 mM KCl, 10 mM MgCl2, 5 mM dithiothreitol (DTT), 5% glycerol] at room temperature for 30 min and the reactions were then analyzed by electrophoresis on 5% native polyacrylamide gels (29:1 acrylamide/bis acrylamide) in 0.5× Tris–borate–EDTA buffer for 3 h at 120 V at 4°C. For phosphorylation of OmpR, 20 mM acetyl phosphate (acetyl-P, Sigma) was added to the reaction mixture. As a negative control, a 211-bp fragment of Y. enterocolitica 16S rRNA gene amplified by PCR with primers S1 (5′-TACGCATTTCACCGCTACAC-3′) and S211 (5′-CAGAAGAAGCACCGGCTAAC-3′) was included in the binding assays. The DNA bands were visualized by staining the gels with ethidium bromide. To show the binding of different amounts of OmpR, to the purified 448-bp fragment, the gels were stained with Coomassie Brilliant Blue R-250. To study the influence of phosphorylation time on OmpR binding ability, OmpR-His6 was pre-incubated in buffer P containing 20 mM acetyl-P and used in binding reactions after the indicated times. The nucleoprotein complexes in these reactions were then analyzed by gel electrophoresis as described above.
Results
The ompR mutant is non-motile and non-flagellated
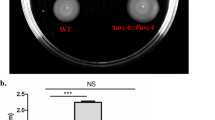
Swimming motility assays were performed with the wild-type Ye9 and Ye9N and the AR4 and EZ10 mutant strains of Y. enterocolitica at 25 and 37°C. Mutant AR4 (ΔompR::Km), lacking OmpR protein, was created as described previously (Brzostek et al. 2007). Mutant EZ10 (envZ::pEP185.2) was constructed in this study by insertional mutagenesis. A motility assay at 37°C revealed a non-motile phenotype for all strains, as expected (data not shown). The analysis of motility of strains Ye9, AR4 and EZ10 incubated at 25°C (Fig. 1) revealed that the ompR mutant had failed to swim when compared with the strain Ye9 or the envZ mutant. However, the envZ mutant was less motile than the wild-type strain Ye9. In addition, the motility of Ye9N strain, a NalR derivative of Ye9, was unchanged (data not shown). Complementation of the ompR mutation was achieved using the plasmid pHR4 (Brzostek et al. 2003), carrying the gene coding for OmpR. The introduction of this plasmid into mutant AR4 restored swimming motility, although the motility of the AR4/pHR4 strain was slightly weaker than that of wild-type cells. These results indicated that OmpR is required for the expression of motility. To determine whether the loss of motility of strain AR4 was due to the absence of flagella, cells of Y. enterocolitica strains Ye9, ompR mutant AR4, envZ mutant EZ10 and AR4/pHR4 were examined by electron microscopy (Fig. 2). Electron micrographs showed that all the cells are rod-shaped and those of the parent strain Ye9 and the envZ mutant strain EZ10 are flagellated, but the ompR mutant cells lack flagella. The flagellated phenotype was restored to strain AR4 by the introduction of an active copy of ompR on plasmid pHR4.

OmpR is required for Y. enterocolitica swimming motility. Swimming zones were observed on 0.3% TB0 agar plates after 16 h incubation at 25°C. Ye9, wild-type strain; AR4, ompR mutant; AR4/pHR4, ompR mutant with plasmid pHR4 expressing OmpR; EZ10, envZ mutant

Transmission electron microscopy of Y. enterocolitica strains. Bacteria grown for 16 h at 25°C on 0.3% TB0 agar plates were negatively stained with phosphotungstic acid. Ye9, wild-type strain; AR4, ompR mutant; AR4/pHR4, ompR mutant with plasmid pHR4 expressing OmpR; EZ10, envZ mutant. The scale bar represents 1 μm
Effect of growth conditions and OmpR on flhDC promoter function
To investigate OmpR-dependent activity of the flhDC promoter, an operon fusion of flhDC with the lacZ reporter gene was constructed in the wild-type strain Ye9 and in the ompR mutant AR4 to produce strains Ye11 and AR6, respectively. The reporter fusion was created by plasmid insertion via homologous recombination. The levels of expression of flhDC::lacZYA, as measured by β-galactosidase assay, were compared for strains Ye11 and AR6 under different growth conditions (Fig. 3). Complementation analyses were performed using strain AR6 carrying plasmid pBR3 encoding His-tagged OmpR (Brzostek et al. 2007).

Effect of OmpR and different temperature, osmolarity and pH conditions on the expression of the flhDC operon fusions: a the strains Ye11 (wild-type), AR6 (ompR mutant) and AR6/pHR4 (ompR mutant with plasmid pHR4 expressing OmpR) were grown at different temperatures in TB0 medium (pH 7.0; low osmolarity), b and c the strains Ye11 (wild-type) and AR6 (ompR mutant) were grown at 25°C in TB0 medium, b different osmolarities (0–0.4 M NaCl), c different pH values (5.0–8.0). Values represent the mean β-galactosidase activities expressed in Miller units ± standard deviations from three independent experiments
To study the influence of OmpR and temperature on the expression of flhDC, the flhDC::lacZYA strains were grown in TB0 medium at 25, 31, 34 or 37°C (Fig. 3a). The lack of the OmpR protein in AR6 resulted in a ~3.5-fold decrease in the level of β-galactosidase activity at 25°C compared with the level obtained for strain Ye11 (524 ± 36 vs. 1762 ± 37 Miller units). This result implied a positive role for the OmpR protein in the regulation of flhDC expression. In addition, complementation of the ompR mutation in strain AR6 with plasmid pBR3 resulted in an increase in β-galactosidase activity (1303 ± 80 Miller units), indicating that the His-tagged OmpR protein, expressed from the gene introduced in trans, was able to positively regulate flhDC expression. Moreover, a ~6.5-fold reduction in β-galactosidase activity at 31°C compared with the level at 25°C (1762 ± 37 vs. 138 ± 18 Miller units) was observed for strain Ye11, and an increase in the temperature up to 37°C resulted in a further decrease in β-galactosidase activity. The same inverse relationship between temperature and the level of flhDC expression was also observed for strain AR6. These results showed that flhDC expression is repressed by high temperature and that OmpR is not involved in the observed thermoregulation.
To examine whether OmpR modulates the expression of flhDC in response to high osmolarity, flhDC promoter activity was measured in strains grown in TB0 medium or TB0 supplemented with NaCl (Fig. 3b). In the strain with active OmpR (Ye11), high osmolarity conditions caused a decrease in expression from the flhDC promoter. β-galactosidase activity was reduced ~10-fold from 2238 ± 263 Miller units at zero NaCl to 221 ± 48 Miller units at 0.3 M NaCl. Besides the normally decreased level of flhDC expression in the ompR mutant AR6, an inhibitory effect of high osmolarity on the β-galactosidase activity of this strain was also observed. These results imply that OmpR may not be involved in the osmoregulation of flhDC at higher osmolarity. To ensure that the observed effect of NaCl on flhDC expression was caused by an increase in osmolarity and not an increase in the NaCl concentration in the medium, NaCl was replaced by KCl or sorbitol. Similarly to NaCl, the expression of flhDC decreased with the increasing concentration of the added osmolytes (data not shown).
To study the influence of pH on the expression of flhDC in the strains Ye11 and AR6, the cells were grown in TB0 medium at different pH (Fig. 3c). The optimal pH for flhDC expression in Ye11 was found to be 7.0 (2255 ± 129 Miller units). Raising the pH to 8.0 resulted in a ~2-fold decrease in flhDC promoter activity (1044 ± 55 Miller units). Moreover, a shift to acidic pH values resulted in a dramatic fall in activity, with the greatest decrease of ~10-fold occurring when the pH was reduced to 5.0 (233 ± 47 Miller units). In strain AR6 (lacking OmpR protein) grown under different pH conditions, decreases in β-galactosidase activity were observed at pH values above and below 7.0. The level of flhDC promoter activity declined from 668 ± 130 Miller units at pH 7.0 to 294 ± 80 Miller units at pH 8.0, and to 175 ± 32 Miller units at pH 5.0. These results suggest that the control of flhDC expression in response to changes in pH is not mediated by the OmpR protein.
Effect of OmpR and carbon source on flhDC expression
The level of acetyl-phosphate (acetyl-P, the phosphodonor for OmpR phosphorylation) within the cell may vary considerably depending on the carbon source in the growth medium. To determine the effect of carbon source on OmpR-dependent flhDC expression, the β-galactosidase activity was measured for Y. enterocolitica strains Ye11 and AR6 (with and without OmpR, respectively) grown in MMA supplemented with 0.2% glycerol, glucose, pyruvate or acetate (Table 2). A higher level of β-galactosidase activity was obtained for the wild-type strain compared with the ompR mutant when grown on the different carbon sources. The activity in Ye11 was highest in cells grown on acetate (757 ± 48 Miller units) and slightly lower with pyruvate (662 ± 36 Miller units). In the presence of glycerol and glucose in the medium the flhDC expression was reduced to 505 ± 56 and 497 ± 57 Miller units, respectively. Interestingly, the pattern of β-galactosidase activity with the different carbon sources was different for the ompR mutant strain AR6. No significant differences in the activity of flhDC were observed in AR6 grown in the presence of pyruvate, acetate, glycerol or glucose.
Separation of OmpR from OmpR-P by reversed phase HPLC
It has previously been shown that response regulators of two component signal transduction systems react with acetyl-P to become phosphorylated (McCleary and Stock 1994). Phosphorylation of Y. enterocolitica OmpR in vitro was performed by treatment with 20 mM acetyl-P. After 60 min incubation, a sample of the reaction mixture was subjected to reversed phase HPLC to separate OmpR from OmpR-P. When OmpR was incubated with acetyl-P, two peaks were detected, eluting at 60 and 80 min (Fig. 4), in contrast to the single peak that eluted at approximately 80 min when OmpR without phosphorylation was analyzed (data not shown). It is assumed that these two peaks correspond to OmpR-P and OmpR, respectively.

Identification of two fractions of OmpR after phosphorylation by acetyl-P. Following treatment with acetyl-P (20 mM, 60 min), 2 μg of OmpR protein was subjected to reversed-phase HPLC using a symmetry C8 column as described in “Materials and methods”. The two peaks, I and II, correspond to two fractions of OmpR (OmpR and OmpR-P)
Binding of OmpR to the promoter region of flhDC examined using electrophoretic mobility shift assays (EMSAs)
Bioinformatic analysis of the flhDC promoter region of Y. enterocolitica Ye9 revealed the presence of two putative OmpR binding sites located at positions from −110 to −93 nt (B1) and from −44 to −27 nt (B2) relative to the AUG start codon (Fig. 5). These two sequences closely resemble the consensus OmpR binding site, both exhibiting 61% identity.

Bioinformatic analysis of the flhDC regulatory region. Two putative OmpR binding sites (B1, B2) are shown in bars within the flhDC promoter sequence. Two nucleotide sequences P1 and P2 similar to the O1 and O2 binding sites for OmpR identified within the Y. pseudotuberculosis flhDC promoter region (Hu et al. 2009) are underlined. These four sequences (B1, B2, P1, P2) are shown separately in alignments with the consensus OmpR binding sequence; the conserved C residues are in bold. The ATG start codon is marked in bold and M indicates methionine
To determine whether OmpR binds to the flhDC promoter region containing these two putative binding sites, EMSAs were performed. Shifted OmpR-P/DNA complexes were clearly produced when the 448-bp flhDC fragment (433 bp from the start codon) interacted with the protein at concentrations of 0.8, 1.2, 1.85 or 3.0 μM (Fig. 6a). DNA binding was specific for the flhDC promoter fragment because no shifting of an unrelated control DNA fragment (211-bp 16S rDNA) was observed. Furthermore, a stepwise shifting of the OmpR-P/DNA complexes was evident, which suggests the presence of more than one OmpR binding site. This result indicated that OmpR protein interacts directly with the flhDC promoter to activate transcription.

Interactions between OmpR and the flhDC promoter region examined by EMSA. A 448-bp flhDC promoter fragment and 211-bp 16S rDNA control fragment were incubated together in different binding reactions: a with increasing concentrations of OmpR-P (0.2, 0.4, 0.8, 1.2, 1.85, 3.0 μM; lanes 2–7), or without OmpR (lane 1), b with non-phosphorylated (OmpR) (1.2 μM) or phosphorylated (OmpR-P) (1.2 μM) protein (lanes 1 and 2), or without OmpR (lane 3), c with 1.2 μM of OmpR phosphorylated by acetyl-P for different times (90, 60, 45, 30, 15 min; lanes 2–6) or without OmpR (lane 1). The upper and lower panels in parts a and b show the 5% native polyacrylamide gels stained with ethidium bromide and Coomassie Brilliant Blue R-250, respectively
To determine the effect of phosphorylation of OmpR, EMSAs were performed with OmpR (1.2 μM—sufficient for flhDC promoter shifting) phosphorylated by incubation with 20 mM acetyl-P for 60 min and OmpR without phosphorylation (Fig. 6b). OmpR applied without phosphorylation was found to bind the flhDC fragment with lower affinity than the phosphorylated form. To examine the effect of the time of phosphorylation on OmpR binding ability, EMSAs were carried out with the OmpR protein treated with acetyl-P for 15, 30, 45, 60 or 90 min. The results shown in Fig. 6c revealed similar OmpR-P binding affinities regardless of the time of phosphorylation from 15 up to 60 min: the same amount of shifted, specific OmpR-P/DNA complexes was seen in all cases. However, when the time of phosphorylation was increased to 90 min, a reduction in the amount of specific OmpR/DNA shifted complexes was observed (Fig. 6c, lane 2). It may be speculated that the formation of higher order structures of OmpR during the 90 min incubation might decrease the level of phosphorylation and thus reduce DNA binding.
During our studies of OmpR binding abilities to the flhDC promoter region in Y. enterocolitica, the involvement of OmpR in the regulation of flhDC in Y. pseudotuberculosis was reported and two binding sites for OmpR in the promoter region (O1 and O2) were identified (Hu et al. 2009). In silico examination of the nucleotide sequence, within the 448-bp flhDC promoter region of Y. enterocolitica, corresponding to the putative OmpR binding sites O1 and O2 of Y. pseudotuberculosis, led to the identification of these sequences (P1 and P2) with 66 and 50% identity to the OmpR consensus, respectively (Fig. 5). Bioinformatic analysis also indicated that these putative P1 and P2 Y. enterocolitica binding sites for OmpR do not overlap with B1 and B2 sites revealed at the beginning of our analysis. Moreover, the O1 and O2 sites of Y. pseudotuberculosis exhibited 64 and 94% identity to the P1 and P2 binding sites of Y. enterocolitica. These two putative OmpR binding sites P1 and P2 were recognized in the Y. enterocolitica flhDC promoter region, located between −425 and −409 nt (P1) and 246 and −228 nt (P2), relative to the translational start site. To determine whether OmpR actually binds to these sequences EMSAs were performed with a PCR-amplified 196-bp fragment comprising the B1 and B2 binding sites and with a 249-bp fragment of the flhDC comprising both P1 and P2 binding sites. Different concentrations of the purified OmpR were incubated with these fragments and the binding reactions were analyzed by electrophoresis in 6% native polyacrylamide gels. The results presented in Fig. 7 demonstrate a stepwise shifting profile of OmpR-DNA complexes when the 196-bp flhDC and 249-bp fragment interacts with 1.6, 2.8 and 4.0 μM of the protein. DNA binding was specific for these both flhDC fragments because the mobility of 16S rDNA was unaltered in the presence of different amounts of purified OmpR.

Interactions between OmpR and two different fragments of the flhDC promoter region examined by EMSA: a a 211-bp 16S rDNA control fragment, b a 196-bp fragment comprising the B1 and B2 binding sites, c a 249-bp fragment comprising both P1 and P2 binding sites of the flhDC promoter region. DNA fragments were incubated independently in binding reactions with increasing concentrations of OmpR-P (1.6, 2.8, 4.0 μM; lanes 2–4, 6–8, 10–12), or without OmpR (lane 1, 5, 9)
Discussion
Flagella and invasin play important roles during the early stages of infection by the enteric pathogen Y. enterocolitica (Pepe and Miller 1993; Young et al. 2000). Our previous study demonstrated that OmpR, the response regulator of the two-component signal transduction system EnvZ/OmpR, negatively regulates invasin gene expression at the transcriptional level (Brzostek et al. 2007).
The present study focused on the role of OmpR in the regulation of flagella expression. Motility assays on swimming plates at 25°C showed that an ompR mutant strain exhibits a non-motile phenotype. Microscopic observation revealed the lack of flagella on the ompR cells, confirming that OmpR is required for flagella synthesis. The motility of all Y. enterocolitica strains tested was abolished at high temperatures (37°C). This phenomenon is characteristic for both Y. enterocolitica and Y. pseudotuberculosis. The sole mediator of the thermoregulation of flagella synthesis is FliA (σF), a flagellum-specific sigma factor, required for the transcription of class III genes of the flagellar regulon (Horne and Pruss 2006; Kapatral and Minnich 1995; Kapatral et al. 1996).
It is known that regulation of the flagellar regulon occurs at the level of the FlhDC master regulator (Young et al. 1999b). Therefore, the present study investigated the mechanism by which OmpR regulates flhDC expression using flhDC::lacZYA operon fusion analysis and in vitro biochemical approaches. Our results revealed the involvement of OmpR in positive regulation of flhDC transcription. However, contrary to these findings, a negative regulatory function of OmpR in flhDC expression has previously been reported in E. coli (Shin and Park 1995). In addition, it has been suggested that OmpR negatively regulates flhDC in Xenorhabdus nematophila (Park and Forst 2006). On the other hand, the ompR mutation had no effect on either motility or flhDC expression in Salmonella enterica sv. Typhimurium (Kutsukake 1997). Thus, it seems that the pattern of regulation of flhDC expression by OmpR identified here is specific to Yersinia strains.
To clarify the role of OmpR in the regulation of Y. enterocolitica flhDC, the influence of different environmental stimuli on the activity of the flhDC promoter was measured. High temperature (31°C and above) appears to be the environmental factor that decreases flhDC expression in the wild-type strain and in the ompR strain, suggesting that temperature-mediated repression of flhDC is OmpR-independent. The thermoregulation of flhDC transcription was previously demonstrated in the wild-type Y. enterocolitica strain 8081v, serotype O:8 (Minnich and Rohde 2007) However, the degree of temperature-dependent repression was smaller than found in our study which was probably the result of differences in the flhDC regions between Y. enterocolitica serotypes O:8 and O:9. It has been suggested that sigma factor FliA and not the FlhDC regulator is directly responsible for the temperature regulation of flagellar genes (Horne and Pruss 2006; Kapatral et al. 1996, 2004).
As the EnvZ/OmpR signal transduction system plays a well characterized role in the osmoregulation of several genes in E. coli (Hall and Silhavy 1979; Higashitani et al. 1993) and Y. enterocolitica (Brzostek et al. 2003, 2007), we examined the effect of osmotic conditions on the flhDC expression level in strains with and without OmpR protein. An in-depth analysis of flhDC expression in the presence of osmolytes in the growth medium, demonstrated that flhDC promoter activity in the wild-type strain decreases at high osmolarity. However, this effect was also observed in the absence of OmpR, indicating that under these conditions flhDC expression might be OmpR-independent. The present studies were extended by investigating the effect of pH on OmpR-dependent flhDC expression. These data imply that the pH regulation of flhDC in Y. enterocolitica, like osmolarity is not mediated by the OmpR protein. It may be speculated that changes in DNA supercoiling or the activity of other regulatory factors or mechanisms might contribute to the modulation of flhDC transcription in response to particular osmolarity or pH conditions, as has been reported for some enterobacterial genes (Higgins et al. 1988).
The regulation of the flhDC operon has been intensively studied in E. coli and several factors have been found to negatively or positively regulate the transcription of flhDC in response to different environmental cues. The E. coli OmpR has been shown to negatively regulate flhDC expression in response to changes in osmolarity (Shin and Park 1995), and the function of H-NS in the modulation of flhDC transcription as well as motility under low-pH conditions has been demonstrated (Soutourina et al. 2002).
Thus, apart from the contrary nature of the regulation (i.e. the positive role of OmpR in the regulation of flhDC expression in Y. enterocolitica revealed in the present study), the osmolarity control of the Y. enterocolitica flagellar regulon differs from that of E. coli. However, we do not rule out the possibility that the degree of OmpR phosphorylation triggered by particular, possibly (combined) osmotic and pH conditions, could influence its regulatory properties.
Several mechanisms could account for the enhanced expression of flhDC in the ompR strain. One hypothesis is that OmpR directly activates the expression of the master regulator FlhDC by binding within its promoter. To investigate this possibility, the DNA binding properties of OmpR were studied in vitro with an EMSA.
Our data revealed that OmpR binds specifically to the 448-bp fragment of flhDC promoter region and the stepwise shifting profile of nucleoprotein complexes suggested the presence of more than one binding site. These data were in agreement with a bioinformatic analysis of the region upstream of the start codon of flhDC which identified two sequences with high similarity to the consensus binding site for OmpR (Egger et al. 1997). Both sites (B1 and B2) exhibit 61% identity to the OmpR consensus binding sequence and contain two conserved cytosine residues, critical for OmpR binding, located at 8- and 10-nt intervals, respectively.
During these studies, the involvement of OmpR in the positive regulation of flhDC in Y. pseudotuberculosis was reported and two binding sites for OmpR in the promoter region (O1 and O2) were identified by Hu et al. (2009). However, alignment of the flhDC regulatory regions of Y. enterocolitica and Y. pseudotuberculosis shows high nucleotide sequence divergence, including the P1 and P2 sites (corresponding to O1 and O2 sites of Y. pseudotuberculosis) identified in promoter region of Y. enterocolitica. EMSAs performed with fragments comprising both P1 and P2 or B1 and B2 OmpR binding sites demonstrate that OmpR may bind independently to these fragments, however, the binding affinity of OmpR for these specific sequences will be investigated in future studies. The presence of high-affinity sites and low-affinity sites for the OmpR has been suggested for the regulation of the ompC, ompF genes of E. coli and the ssrA gene of S. enterica sv Typhimurium (Feng et al. 2003; Rampersaud et al. 1994).
It has previously been shown that phosphorylation of OmpR activates this regulatory protein. OmpR phosphorylation by EnvZ in response to high osmolarity has been well studied in E. coli (Igo et al. 1989; Lan and Igo 1998; Russo and Silhavy 1991). Our experiments indicate that an envZ mutant strain of Y. enterocolitica is motile and synthesizes flagella, suggesting no influence of EnvZ on the OmpR-dependent regulation of flhDC under the conditions used.
Thus, OmpR is likely to be available for phosphorylation by other phospho-donors besides its partner kinase EnvZ, which might result in its activation. In E. coli, some portion of OmpR seems to be phosphorylated by acetyl-P (Head et al. 1998; McCleary and Stock 1994; Pruss 1998; Shin and Park 1995) and the occurrence of this reaction has been suggested for S. enterica sv. Typhimurium (Bang et al. 2000). Here, reversed phase HPLC was used to confirm the ability of acetyl-P to phosphorylate Y. enterocolitica OmpR in vitro. In addition, EMSAs indicated that phosphorylation of OmpR slightly enhances its binding abilities. It has previously been suggested that the intracellular level of acetyl-P may vary greatly according to the growth conditions, such as the nature of the carbon source (McCleary and Stock 1994; Wanner and Wilmes-Riesenberg 1992). The use of acetate or pyruvate as the carbon source in the growth medium was found to increase flhDC transcription in the wild-type Y. enterocolitica strain compared with glycerol or glucose, but this effect was not observed in the ompR strain. Raised intracellular concentrations of pyruvate or acetate might result in a higher level of acetyl-P which would increase the phosphorylation state of OmpR.
To summarize, the OmpR regulator—probably phosphorylated by acetyl-P—might, under certain environmental conditions, act directly on the flhDC promoter region to activate flhDC transcription and thus play a role in coordinating the motility of Y. enterocolitica.
These findings, together with the results of our previous studies indicating a negative role for OmpR in the regulation of invasin expression, appear to support a model in which motility and invasin production, the two major factors involved in the early stages of Y. enterocolitica pathogenesis, might be reciprocally regulated. It has previously been suggested that invasin and motility may be regulated in an opposing manner in Y. enterocolitica (Badger and Miller 1998). This regulation seems appropriate during the process of pathogenesis of Y. enterocolitica, where motility is the first critical feature during host cell invasion, followed by adhesion and invasion processes. Therefore, depending on the infection stage, OmpR could regulate invasin production and motility in an opposing manner, and this regulation may be fine-tuned in response to changing environmental conditions and by the activities of the multiple regulatory factors involved.
Since FlhDC, besides its regulatory function in motility, may also act as a global regulator of Y. enterocolitica metabolism (Kapatral et al. 2004) and affect the secretion of virulence factors by the flagellar export apparatus (Young et al. 1999a), the modulation of flhDC operon expression by OmpR is likely to have considerable implications for Y. enterocolitica physiology. In addition, it seems that the role of OmpR in the regulation of flhDC is specific to enteropathogenic Yersinia species.
References
Badger JL, Miller VL (1998) Expression of invasin and motility are coordinately regulated in Yersinia enterocolitica. J Bacteriol 180:793–800
Bang IS, Kim BH, Foster JW, Park YK (2000) OmpR regulates the stationary-phase acid tolerance response of Salmonella enterica serovar Typhimurium. J Bacteriol 182:2245–2252
Baumler AJ, Tsolis RM, van der Velden AWM, Stojiljkovic I, Anic S, Heffron F (1996) Identification of a new iron regulated locus of Salmonella typhi. Gene 183:207–213
Bernardini ML, Fontaine A, Sansonetti PJ (1990) The two component regulatory system OmpR-EnvZ controls the virulence of Shigella flexneri. J Bacteriol 172:6274–6281
Bleves S, Marenne MN, Detry G, Cornelis GR (2002) Up-regulation of the Yersinia enterocolitica yop regulon by deletion of the flagellum master operon flhDC. J Bacteriol 184:3214–3223
Brzostek K, Raczkowska A, Zasada A (2003) The osmotic regulator OmpR is involved in the response of Yersinia enterocolitica O:9 to environmental stresses and survival within macrophages. FEMS Microbiol Lett 228:265–271
Brzostek K, Brzóstkowska M, Bukowska I, Karwicka E, Raczkowska A (2007) OmpR negatively regulates expression of invasin in Yersinia enterocolitica. Microbiology 153:2416–2425
Dorman CJ, Chatfield S, Higgins CF, Hayward C, Dougan G (1989) Characterization of porin and ompR mutants of a virulent strain of Salmonella typhimurium: ompR mutants are attenuated in vivo. Infect Immun 57:2136–2140
Dorrell N, Li SR, Everest PH, Dougan G, Wren BW (1998) Construction and characterisation of a Yersinia enterocolitica O:8 ompR mutant. FEMS Microbiol Lett 165:145–151
Egger LA, Park H, Inouye M (1997) Signal transduction via the histidyl–aspartyl phosphorelay. Genes Cells 2:167–184
Feng X, Oropeza R, Kenney LJ (2003) Dual regulation by phospho-OmpR of ssrA/B gene expression in Salmonella pathogenicity island 2. Mol Microbiol 48:1131–1143
Flamez C, Ricard I, Arafah S, Simonet M, Marceau M (2008) Phenotypic analysis of Yersinia pseudotuberculosis 32777 response regulator mutants: new insights into two-component system regulon plasticity in bacteria. Int J Med Microbiol 298:193–207
Francez-Charlot A, Laugel B, Van Gemert A, Dubarry N, Wiorowski F, Castanie-Cornet MP, Gutierrez C, Cam K (2003) RcsCDB His-Asp phosphorelay system negatively regulates the flhDC operon in Escherichia coli. Mol Microbiol 49:823–832
Hall MN, Silhavy TJ (1979) Transcriptional regulation of Escherichia coli K-12 major outer membrane protein 1b. J Bacteriol 140:342–350
Head CG, Tardy A, Kenney LJ (1998) Relative binding affinities of OmpR and OmpR-phosphate at the ompF and ompC regulatory sites. J Mol Biol 281:857–870
Higashitani A, Nishimura Y, Hara H, Aiba H, Mizuno T, Horiuchi K (1993) Osmoregulation of the fatty acid receptor gene fadL in Escherichia coli. Mol Gen Genet 240:339–347
Higgins CF, Dorman CJ, Stirling DA, Waddell L, Booth IR, May G, Bremer E (1988) A physiological role for DNA supercoiling in the osmotic regulation of gene expression in S. typhimurium and E. coli. Cell 26:569–584
Horne SM, Pruss BM (2006) Global gene regulation in Yersinia enterocolitica: effect of FliA on the expression levels of flagellar and plasmid-encoded virulence genes. Arch Microbiol 185:115–126
Hu Y, Wang Y, Ding L, Lu P, Atkinson S, Chen S (2009) Positive regulation of flhDC expression by OmpR in Yersinia pseudotuberculosis. Microbiology 155:3622–3631
Igo MM, Ninfa AJ, Stock JB, Silhavy TJ (1989) Phosphorylation and dephosphorylation of a bacterial transcriptional activator by a transmembrane receptor. Genes Dev 3:1725–1734
Iriarte M, Stainier I, Mikulskis AV, Cornelis GR (1995) The fliA gene encoding sigma 28 in Yersinia enterocolitica. J Bacteriol 177:2299–2304
Jubelin G, Vianney A, Beloin C, Ghigo JM, Lazzaroni JC, Lejeune P, Dorel C (2005) CpxR/OmpR interplay regulates curli gene expression in response to osmolarity in Escherichia coli. J Bacteriol 187:2038–2049
Kapatral V, Minnich SA (1995) Co-ordinate, temperature-sensitive regulation of the three Yersinia enterocolitica flagellin genes. Mol Microbiol 17:49–56
Kapatral V, Olson JW, Pepe JC, Miller VL, Minnich SA (1996) Temperature-dependent regulation of Yersinia enterocolitica Class III flagellar genes. Mol Microbiol 19:1061–1071
Kapatral V, Campbell JW, Minnich SA, Thomson NR, Matsumura P, Pruss BM (2004) Gene array analysis of Yersinia enterocolitica FlhD and FlhC: regulation of enzymes affecting synthesis and degradation of carbamoylphosphate. Microbiology 150:2289–2300
Kinder SA, Badger JL, Bryant GO, Pepe JC, Miller VL (1993) Cloning of the YenI restriction endonuclease and methyltransferase from Yersinia enterocolitica serotype O8 and construction of a transformable R−M+ mutant. Gene 136:271–275
Kutsukake K (1997) Autogenous and global control of the flagellar master operon, flhD, in Salmonella typhimurium. Mol Gen Genet 254:440–448
Lan CY, Igo MM (1998) Differential expression of the OmpF and OmpC porin proteins in Escherichia coli K-12 depends upon the level of active OmpR. J Bacteriol 180:171–174
Lee AK, Detweiler CS, Falkow S (2000) OmpR regulates the two-component system SsrA–SsrB in Salmonella pathogenicity island 2. J Bacteriol 182:771–781
Lehnen D, Blumer C, Polen T, Wackwitz B, Wendisch VF, Unden G (2002) LrhA as a new transcriptional key regulator of flagella, motility and chemotaxis genes in Escherichia coli. Mol Microbiol 45:521–532
McCleary WR, Stock JB (1994) Acetyl phosphate and the activation of two-component response regulators. J Biol Chem 269:31567–31572
Miller JH (1972) Experiments in molecular genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor
Minnich SA, Rohde HN (2007) A rationale for repression and/or loss of motility by pathogenic Yersinia in the mammalian host. Adv Exp Med Biol 603:298–310
Park D, Forst S (2006) Co-regulation of motility, exoenzyme and antibiotic production by the EnvZ-OmpR-FlhDC-FliA pathway in Xenorhabdus nematophila. Mol Microbiol 61:1397–1412
Pepe J, Miller VL (1993) Yersinia enterocolitica invasin: a primary role in the initiation of infection. Proc Natl Acad Sci USA 90:6473–6477
Pruss BM (1998) Acetyl phosphate and the phosphorylation of OmpR are involved in the regulation of the cell division rate in Escherichia coli. Arch Microbiol 170:141–146
Rampersaud A, Harlocker SL, Inouyer M (1994) The OmpR protein of Escherichia coli binds to sites in the ompR promoter region in a hierarchical manner determined by its degree of phosphorylation. J Biol Chem 269:12559–12566
Russo FD, Silhavy TJ (1991) EnvZ controls the concentration of phosphorylated OmpR to mediate osmoregulation of the porin genes. J Mol Biol 222:567–580
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual, 2nd edn. Cold Spring Harbor Laboratory, Cold Spring Harbor
Shin S, Park C (1995) Modulation of flagellar expression in Escherichia coli by acetyl phosphate and the osmoregulator OmpR. J Bacteriol 177:4696–4702
Simon R, Priefer U, Puhler A (1983) A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in gram negative bacteria. Biotechnology 1:784–791
Soutourina OA, Kolb A, Krin E, Laurent-Winter C, Rimsky S, Danchin A, Bertin P (1999) Multiple control of flagellum biosynthesis in Escherichia coli: role of H-NS protein and the cyclic AMP-catabolite activator protein complex in transcription of the flhDC master operon. J Bacteriol 181:7500–7508
Soutourina OA, Krin E, Laurent-Winter C, Hommais F, Danchin A, Bertin PN (2002) Regulation of bacterial motility in response to low pH in Escherichia coli: the role of H-NS protein. Microbiology 148:1543–1551
Sperandio V, Torres AG, Kaper JB (2002) Quorum sensing Escherichia coli regulators B and C (QseBC): a novel two-component regulatory system involved in the regulation of flagella and motility by quorum sensing in E. coli. Mol Microbiol 43:809–821
Stock JB, Ninfa AJ, Stock AM (1989) Protein phosphorylation and regulation of adaptive responses in bacteria. Microbiol Rev 53:450–490
Townsend MK, Carr NJ, Iyer JG, Horne SM, Gibbs PS, Pruss BM (2008) Pleiotropic phenotypes of a Yersinia enterocolitica flhD mutant include reduced lethality in a chicken embryo model. BMC Microbiol 23:8–12
Vidal O, Longin R, Prigent-Combaret C, Dorel C, Hooreman M, Lejeune P (1998) Isolation of an Escherichia coli K-12 mutant strain able to form biofilms on inert surfaces: involvement of a new ompR allele that increases curli expression. J Bacteriol 180:2442–2449
Wanner BL, Wilmes-Riesenberg MR (1992) Involvement of phosphotransacetylase, acetate kinase, and acetyl phosphate synthesis in control of the phosphate regulon in Escherichia coli. J Bacteriol 174:2124–2130
Yamamoto K, Nagura R, Tanabe H, Fujita N, Ishihama A, Utsumi R (2000) Negative regulation of the bolA1p of Escherichia coli K-12 by the transcription factor OmpR for osmolarity response genes. FEMS Microbiol Lett 186:257–262
Yokota T, Gots JS (1970) Requirement of adenosine 3′,5′-cyclic phosphate for flagella formation in Escherichia coli and Salmonella typhimurium. J Bacteriol 103:513–516
Young GM, Schmiel DH, Miller VL (1999a) A new pathway for the secretion of virulence factors by bacteria: the flagellar export apparatus functions as a protein-secretion system. Proc Natl Acad Sci USA 96:6456–6461
Young GM, Smith MJ, Minnich SA, Miller VL (1999b) The Yersinia enterocolitica motility master regulatory operon, flhDC, is required for flagellin production, swimming motility, and swarming motility. J Bacteriol 181:2823–2833
Young GM, Badger JL, Miller VL (2000) Motility is required to initiate host cell invasion by Yersinia enterocolitica. Infect Immun 68:4323–4326
Acknowledgments
We thank Prof. A. Sklodowska and D. Ruszkowski (Laboratory of Environmental Pollution Analysis, Faculty of Biology, University of Warsaw) for their assistance with HPLC analysis. This work was supported by Warsaw University (grant BW 2007) and by the Polish Ministry of Science and Higher Education (grant N303 009 32/0537).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Raczkowska, A., Skorek, K., Bielecki, J. et al. OmpR controls Yersinia enterocolitica motility by positive regulation of flhDC expression. Antonie van Leeuwenhoek 99, 381–394 (2011). https://doi.org/10.1007/s10482-010-9503-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10482-010-9503-8