
Abstract
Eighty-three wild and domestic carnivores of nine species from Janos Biosphere Reserve (JBR), Mexico, were tested by serologic and molecular assays to determine exposure and infection rates of carnivore protoparvovirus 1. Overall, 50.8% (33/65) of the wild carnivores and 100% (18/18) of the domestic dogs tested were seropositive for Canine protoparvovirus 1 (CPV), while 23% (15/65) of the wild carnivores and 22.2% (4/18) of the domestic dogs were PCR positive for CPV. Phylogenetic analysis confirmed circulation of CVP-2 with residues 426 Asn (CPV2a = 1/19) and 426 Glu (CPV-2c = 18/19) among carnivores in JBR. The prevalence of both PCR positivity and antibodies to CPV varied significantly among wild host species. Of the six identified haplotypes, three were unique to kit foxes (Vulpes macrotis) (the species with higher haplotype richness) and two to striped skunks (Mephitis mephitis). The remaining haplotype was shared among all carnivore species including dogs suggesting non-host specificity and bidirectional and continuous viral transmission cycle in the JBR. The phylogenetic similarity of CPV strains from dogs and wild carnivores and the higher prevalence of CPV in wild carnivores captured near towns relative to those captured far from towns suggest that dogs might be an important source of CPV infection for wild carnivores in the JBR. We provide evidence that cross-species transmission occurs at the domestic–wildlife interface in JBR.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Infectious diseases can cause population declines in wildlife and are widely recognized as a threat to biodiversity conservation (Smith et al. 2009). Often, diseases affecting wildlife are associated with profound ecological changes in the ecosystem, shifting host–pathogen interactions (Daszak et al. 2000). The probabilities of contact and rates of transmission of pathogens among domestic and wild animals are increased by anthropogenic influences including habitat loss, land-use change, and human settlements (Fiorello et al. 2004; Belsare et al. 2014). Wild carnivores are at high risk of pathogen transmission due to their close phylogenetic relationships with domestic dogs and cats (Pedersen et al. 2007), which can be sources for canine distemper virus (CDV), canine parvovirus (CPV), feline immunodeficiency virus, feline leukemia virus, rabies, and others (Murray et al. 1999; Steinel et al. 2001; Fiorello et al. 2004; Pedersen et al. 2007; Belsare et al. 2014). Previous studies have showed that landscape features influence the spatial spread of viruses within wild carnivore populations. For instance, suburban red foxes (Vulpes vulpes) had higher seroprevalence of CPV than rural foxes (Truyen et al. 1998), and exposure to CDV in two south American foxes (Pseudalopex griseus and P. culpaeus) was more likely in areas closer to human settlements (Acosta-Jamett et al. 2011).
The subfamily Parvovirinae consists of non-enveloped, single-stranded DNA viruses that infect a wide range of mammalian hosts including members of the order Carnivora (Cotmore et al. 2014). Infections are primarily transmitted through fecal–oral contact, but may also be spread during predation, scavenging carcasses, or oronasal exposure (McCaw and Hoskins 2006). Carnivore protoparvovirus 1 comprises the most important parvoviruses that infect carnivores, such as canine parvovirus (CPV), feline panleukopenia virus (FPV), and mink enteritis virus (Steinel et al. 2001). Canine parvovirus type 2 (CPV-2) first emerged in the mid-1970s, apparently from an FPV-like virus (Parrish et al. 1991), and spread globally in 1978 to cause a pandemic in dogs, eventually becoming endemic worldwide (Hoelzer and Parrish 2010). After its adaptation to dogs, three new antigenic variants emerged: CPV-2a, CPV-2b, and CPV2c (Parrish et al. 1991; Buonavoglia et al. 2001), although there is not consensus among researchers regarding best nomenclature (Miranda and Thompson 2016; Li et al. 2017; Zhou et al. 2017). CPV may cause disease in several species of Felidae, Canidae, Procyonidae, Mustelidae, Ursidae, and Viverridae and can cause mortality primarily among young animals, which may result in a threat to carnivore conservation (Steinel et al. 2001). For instance, CPV has been implicated as preventing the recovery of a small population of the gray wolves (Canis lupus) in Minnesota (Mech and Goyal 1995).
In Mexico, CPV is a common pathogen in dogs (Pedroza-Roldán et al. 2015; Ortega et al. 2017), but little is known about CPV prevalence, genetic diversity, and the impact on wild carnivores. To our knowledge, only one study has been published on CPV in Mexican wild carnivores, showing that ringtail cats (Bassariscus astutus), along with other medium-sized mammals, were positive to antibodies against CPV in two protected areas in central Mexico (Suzán and Ceballos 2005). In this study, the high CPV seroprevalence in wild animals and free-roaming dogs suggested common cross-species transmission and spillover events. Therefore, there is a need to investigate the eco-epidemiology of CPV at the interface of wild carnivores and domestic dogs in different areas of Mexico, particularly evaluating risk to wild carnivores in areas impacted by anthropogenic disturbance.
A natural area considered a conservation priority for North American biodiversity is the Janos Biosphere Reserve (JBR) located in the northwestern part of Mexico (Ceballos et al. 2005; List et al. 2010). Several wild carnivores inhabit this reserve, and also reintroduction programs of endangered carnivores within the reserve or nearby locations have been conducted, such as the black-footed ferret (Mustela nigripes) and the Mexican gray wolf (Canis lupus baileyi) (List et al. 2010; Lara-Díaz et al. 2015). However, human settlements and farming activities are increasing in JBR and nearby areas (Ceballos et al. 2010), likely increasing contacts between wildlife and domestic animals. High densities (0.5–1.8 dogs/ha: Almuna 2016) of free-roaming dogs in this area may increase encounters with wild carnivores.
The objective of this study was to investigate the exposure and the genetic diversity of CPV in wild carnivores and the potential cross-species transmission risk from domestic dogs from urban settlements to wild carnivores at the JBR. We hypothesized that: (1) there is at least one CPV genotype circulating among domestic and wild carnivores and (2) there is a higher risk of CPV infection in wild carnivores from nearby areas of settlements in JBR.
Materials and Methods
Study Area and Animal Sampling
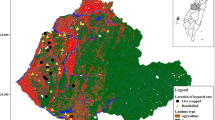
The JBR is located within a transitional zone between the Sierra Madre Occidental and the Chihuahuan Desert, which comprise a mosaic of grasslands, mesquite shrubland, and oak forests. The climate varies with elevation, from arid to temperate, with mean annual temperatures from 15.7°C in the plains at 1200 m above sea level to 11.8°C in mountains at 2700 m above sea level. A rainfall gradient occurs from a mean annual precipitation of 381–581 mm, with 77% of the precipitation falling from April through August (García 1981). Wild carnivores were sampled during fall 2013 (October–November) and spring 2014 (April–May) at five locations (El Cuervo, La Bascula, Monte Verde, Ojitos, and Rancho San Pedro) of the Janos Biosphere Reserve (JBR), Chihuahua, Mexico (30°51′50″N, 108°30′09″W) in northwestern Mexico (Fig. 1). Animals were captured at each of the five locations with 16 trapping sets placed at intervals of 500–800 meters along a 10 km transect (Fig. 1). Each trapping set contained one box trap (Tomahawk Live Trap Inc. WI) and one leg-hold trap (Victor Coil Soft Catch™). The traps were kept active for nine consecutive days and baited with sardine, chicken, and commercial lures, such as Coon Digger I, Coyote Urine, and Bobcat Gland (Kishel’s®). Each captured animal was chemically immobilized with a mixture of ketamine hydrochloride (Anesket®, Pisa, Atitalaquia, Hidalgo, Mexico) and xylazine hydrochloride (Procint, Pisa®) using doses reported by Kreeger and Arnemo (2012). Animals were weighed, sexed, identified to species, and examined for any sign of disease. We collected blood samples from cephalic or femoral veins: 1 ml was separated in Microtainers® with EDTA, and the rest of the blood was centrifuged at 1400 X G for 5 min to obtain serum that was stored in liquid nitrogen in the field. Whole blood and serum samples were transferred to a − 70°C freezer until laboratory testing. All individuals were marked with numbered metal ear-tags and were released at the site of capture.

Shaded polygons indicate trapping sets where wild carnivores were captured and sampled to test for antibodies and molecular detection of Canine protoparvovirus 1 in the Janos Biosphere Reserve, Mexico.
In order to evaluate the presence of CPV in domestic dogs and the genotypes circulating in JBR, blood and sera samples were collected from dogs with prior authorization of the owners. We performed a physical examination for clinical signs of disease, recorded data on age, vaccination, and clinical history of each dog and obtained blood from the cephalic vein.
All procedures for trapping and handling carnivores followed the guidelines of the American Society of Mammalogists (Sikes and Gannon 2011) and were approved by the Mexican Secretary of Environment and Natural Resources (Permit FAUT-0250).
Serological test: inhibition of hemagglutination (HI)
The hemagglutination inhibition (HI) assay was performed to detect antibodies against CPV according the description by Coligan et al. (2005). Non-specific inhibitors were removed by mixing 20 µL of serum samples with 80 µL of 25% kaolin in phosphate-buffered saline [PBS] (1:5 final dilution) and incubating at room temperature (RT) for 20 min. Kaolin was removed by centrifuge at 10,000 g at 4°C for 15 min. For the HI test, 50 µL of 4 hemagglutination units of CPV2a (Strain L-85) were added to 50 µL of the treated serum. After incubation for 1 h at RT, 50 µL of 0.8% guinea pig erythrocytes was added, and microplates were incubated at RT for 1 h. Antibody titers were defined as the reciprocal of the highest serum dilution that inhibited hemagglutination. Serum samples showing HI titers equal or up to 1:60 were considered positive.
DNA Extraction and Polymerase Chain Reaction (PCR)
To detect CPV-2 DNA, a highly conserved portion of the parvovirus VP2 gene was amplified. Total genomic DNA was extracted from blood samples, using the DNeasy Blood and Tissue Kit® (Qiagen, Hilden, Germany) according to manufacturer’s instructions, 0.5–3 µg of sample DNA were used as template. The PerfeCTa SYBR® Green FastMix kit was used (Quantabio Scientific, Beverly, MA, USA) with the primers 3778f (GACCAGCTGAGGTTGGTTATAG) and 4243r (GGTGCATTTACATGAAGTCTTGG), which amplified a 466 base pairs (bp) fragment of VP2 residues 333–487. Amplification conditions were 95°C 5 min, 94°C 30 s, 57°C 30 s, and 72°C 1 min, for 35 cycles and an extension of 72°C 5 min. The amplification of partial VP2 gene and subsequent sequencing of the PCR products, where many important informative amino acids reside, would give us definitive antigenic and genetic difference between the original CPV-2, its variants. As positive controls, DNA from commercial vaccines with modified live CPV-2 strains was used. Molecular biology grade water was used as negative controls. PCR products were visualized on a 2% agarose gel. Amplicons of the desired size were then purified and sequenced at the Instituto de Investigaciones Biomédicas, Universidad Nacional Autónoma de México. The obtained sequences were compared to homologous sequences in GenBank using the BLASTn tool.
Phylogenetic Analysis and Genetic Diversity
Nucleotide sequences were analyzed using MEGA6 (Tamura et al. 2013) to determine the consensus of sequences for the amplified region of the VP2 gene. The sequences of this study and the known Protoparvovirus 1 variants retrieved from GenBank were aligned using Muscle. A phylogenetic tree was constructed using the Maximum Likelihood method by Hasegawa–Kishino–Yano model with invariant sites and 1000 bootstrap replicates. The nucleotide substitution model for each alignment was selected based on the lowest AIC (Akaike information criterion) using JModelTest (Darriba et al. 2012).
A haplotype network based on independent median-joining algorithm was used to evaluate the relationship among CPV haplotypes circulating in JBR, taking host species into account. All of these analyses were conducted using PopART software (Population Analysis with Reticulate Trees) version 1.7 (Leigh and Bryant 2015).
Anthropization Degree
The level of anthropization was measured as the distance to the nearest settlement area (urban distance). Spatial data on human settlements were obtained from satellite images available in Google Earth (GoogleEarth_7.1.8 2016). A total of five towns (El Cuervo, Ejido San Pedro, Casa de Janos, Ejido Pancho Villa and Ejido Monte Verde) were identified in the JBR (Fig. 1). We calculated the distance from the capture locations of each individual wild carnivore to the closest settlement. The resulting data were grouped into four categories based on the distances: (1) < 5; (2) 5–10; (3) 10–15; and (4) > 15 km.
Statistical Analysis
The average of PCR prevalence and seroprevalence was calculated for each species of wild carnivore, and 95% confidence intervals were calculated based on Wilson score interval (Newcombe 1998).
To assess possible associations between sex and distance to human settlements (as a proxy for domestic dog populations) and CPV seropositivity and PCR positivity, generalized linear mixed models (GLMM, “glmer” in R package “lme4”: R Core Team 2014) were fitted by Laplace approximation (Bolker et al. 2009), considering a binomial error distribution and logit link function. In order to account for non-independence due to phylogeny, species was considered as a random effect in the model, and sex and distance as fixed effects. All statistical tests were two-tailed with α = 0.05.
Results
During the fall of 2013 (October–November) and the spring of 2014 (April–May), a total of 65 wild carnivores belonging to eight species were captured and sampled, including coyotes (Canis latrans), kit foxes (Vulpes macrotis), gray foxes (Urocyon cinereoargenteus), bobcats (Lynx rufus), raccoons (Procyon lotor), striped skunks (Mephitis mephitis), hooded skunks (Mephitis maroura), and badgers (Taxidea taxus) (Table 1). Of these 65 wild carnivores, 63 were adults and two were juveniles (one coyote and one kit fox). Additionally, eighteen dogs were sampled from two settlements (Monte Verde [3] and Ejido San Pedro [5]), and four ranches (Rancho El Uno [3], El Cuervo [1], Ojitos [3], and Rancho San Pedro [3]) within the JBR (Fig. 1).
Prevalence of Antibodies Against CPV
We detected antibodies against CPV in all species sampled. All domestic dogs (18/18; 95% C.I. = 82.4–100%) and 50.8% (33/65; 95% C.I. = 38–63%) of wild carnivores were seropositive (Table 1). The GLMM analysis showed no statistical association between sex, distance of wild carnivores to human settlements, and CPV seroprevalence in wild carnivores.
Molecular Identification of CPV
Overall, 23% (15/65: 95% CI 0.14–0.35) of the wild carnivores were PCR positive for canine parvovirus (CPV). The prevalence of CPV in wild carnivores varied significantly among the eight-host species (Table 1). Eight (53.3%) of the fifteen PCR-positive wild carnivores were kit foxes, while the rest included three coyotes, three striped skunks, and one raccoon (Table 1).
As there were no statistical differences between distance and distance + sex (as fixed factors) in the models (Table 2), we concluded that there was no statistical association between sex and the prevalence of CPV in wild carnivores. GLMM analysis showed that there was a significant negative relationship between the distance to human settlements and PCR prevalence in wild carnivores from the JBR (Table 3).
Phylogenetic Analysis and Genetic Diversity
Phylogenetic analysis of the VP2 sequences of the CPV showed a total of six genetic variants of 19 sequences obtained from wild carnivores and domestic dogs with 97.9–99.8% similarity among them (Fig. 2). All sequences were submitted to GenBank with accession numbers MG425948–MG425966. Among these, five sequences were identified as novel genetic variants. All genotypes clustered into two distinct antigenic variants, the first consisting of five genetic variants of 18 sequences obtained from coyotes [3], kit foxes [7], raccoons [1], striped skunks [3], and dogs [4], all of which associated with Canine Parvovirus type 2 with VP2 residue 426 Glu (CPV-2c) (98.5–99.8% similarity). Fifteen sequences obtained from wild carnivore species and domestic dogs were identical to a strain previously reported in a dog from the USA (GenBank acc. no. FJ005236). The other four genetic variants of five sequences were novel and were assigned with GenBank accession numbers MG425956, MG425961, MG425962, and MG425964. The second group contained one genetic variant with one sequence obtained from a kit fox (GenBank acc. no. MG425951) which had VP2 residue 426 Asn and clustered with CPV2-a. This sequence had 99.8% similarity to a strain previously described in a dog from Italy (GenBank acc. no. FJ222824) (Decaro et al. 2009). In addition, we found other mutations in the parvovirus VP2, such as N375D and T440A among others, in CPV strains obtained from kit foxes (Supplementary table S1).

Maximum likelihood phylogenetic tree based on variants of VP2 gene of Canine protoparvovirus 1. The analysis involved 32 nucleotide sequences. Each genotype is indicated by its GenBank accession number, with novel genetic variants in boldface. Abbreviations used include Feline panleukopenia virus (FPV), Blue fox virus (BFP), Mink enteritis virus (MEV), Raccoon parvovirus (RPV), Argentina (ARG), Brazil (BRA), Italy (ITA), Germany (GER), Mexico (Mx), United States of America (US), El Cuervo (EC), Monte Verde (MV), Ojitos (OJ), La báscula (LB), Ejido San Pedro (ESP), Rancho San Pedro (RS), Rancho El Uno (REU).
The haplotype network showed a total of six different haplotypes circulating in JBR: kit foxes harbored the greatest diversity with three unique haplotypes, followed by striped skunks with two. One haplotype shared by all carnivores including dogs was central to the network. All unique haplotypes spread from this haplotype, except for one found in a kit fox that derived from a striped skunk (Fig. 3). Overall, the nucleotide diversity was π = 0.002 with 13 segregating sites and 5 parsimony-informative sites.

Median-joining haplotype network reconstructed with the VP2 gene sequences of Canine protoparvovirus 1 obtained from wild carnivores in the Janos Biosphere Reserve, Mexico.
Discussion
This study represents the first molecular characterization of Protoparvovirus genotypes at the interface of domestic and wild carnivores in Mexico. As far as we know, this represents the first evidence of CPV2 with the VP2 residue 426 Asn in Mexico and the first report of CPV variants in kit foxes (Vulpes macrotis). The finding of several viral sequences with VP2 residue 426 as acid glutamic (so-called CPV-2c) is consistent with the fact that it is the predominant variant in Latin America (Zhou et al. 2017) including Mexico (Pedroza-Roldán et al. 2015). However, data for Mexico are sparse, consisting only of a local survey on dogs.
As we expected, PCR prevalence was lower in most species than seroprevalence. Once CPV infection occurs, the quantity of virus in the blood and tissues decreases rapidly and typically is undetectable by days 6–8 (Murphy et al. 1999). Carnivores develop antibodies against CPV that are detectable by days 5–7 (Murphy et al. 1999) and have antibody protection for life (Maclachlan et al. 2017). However, for kit foxes, the PCR-positive prevalence was higher than seroprevalence. This could be consistent with an outbreak of CPV in kit foxes during the fall of 2013 in the JBR, particularly given that the PCR-positive samples were collected from kit foxes that were almost all negative to antibodies against CPV, indicating that there were exposed recently to the virus. Although several studies have reported CPV outbreaks and/or mortalities in captive (Steinel et al. 2001) and free ranging wild carnivores (Williams and Barker 2001; Mech et al. 2008; Mech and Goyal 2011), our results suggest that subclinical disease is likely, because none of the animals captured in JBR showed clinical signs of parvovirus infection.
In this study, we provide evidence of cross-species transmission in the JBR, including at the domestic–wildlife interface. Similar to other studies (Allison et al. 2014; Miranda et al. 2017), we found a high prevalence and a close genetic similarity of CPV amplicons among carnivores. Most sequences obtained from carnivores (including dog, kit fox, skunk, coyote, and raccoon) were clustered in a monophyletic branch suggesting non-host specificity and bidirectional and continuous viral transmission cycle in the JBR. However, our finding of mutations in four parvovirus strains obtained from kit foxes and skunks suggests the evolution and presence of other sylvatic cycles involving specific wild carnivore species in the region. The haplotype network was consistent with this concept, showing that kit foxes and skunks harbored the greatest diversity of parvoviruses, with one haplotype that was shared by all carnivores including dogs. In addition, the analysis suggests that all unique haplotypes evolve from this haplotype, except for one found in a kit fox that derived from a striped skunk. It has been theorized that wildlife hosts might play an important role in the evolution of CPV, which in turn might explain the emergence of new variants in dogs (Truyen et al. 1998; Allison et al. 2013). To date, there are no findings of an evolutionary link between FPV and CPV-2 in dogs, whereas two studies have found viruses genetically intermediate between FPV-like and CPV-2 (Truyen et al. 1998), and CPV-2 and CPV-2a (Allison et al. 2012), in wild carnivores. Nevertheless, further studies of parvovirus evolution and host specificity should be based on an analysis of complete VP2 gene, where more mutations (e.g., residue 300) have been reported as determining host range (Allison et al. 2016).
Despite there being evidence of CPV circulation at the domestic–wildlife interface worldwide, the role of domestic dogs on maintenance and transmission of CPV to wild carnivores has not been well explored (Knobel et al. 2014). For instance, Truyen et al. (1998) found a higher but not statistically significant seroprevalence in red foxes (Vulpes vulpes) in suburban areas (15.2%) compared with rural areas (10.8%). In contrast, Nelson et al. (2012) did not evidence that prevalence of CPV in wolves from Canada differed near towns where there were more domestic dogs. According to the reservoir-target system described by Viana et al. (2014), the combination of high percentage of seropositive dogs, the genetic similarity of CPV among wild and domestic carnivores, and the higher prevalence of CPV in wild carnivores captured near towns suggests that even our phylogenetic analysis suggests that bidirectional transmission among carnivores, dogs might be the main source of CPV infection for wild carnivores. CPV is profusely shed by infected dogs (McCaw and Hoskins 2006) and has a high stability and persistence in the environment (Cotmore and Tattersall 2007). Thus, the high densities of free-roaming dogs ranged from 0.5 to 1.8 dogs/ha in human settlements within the JBR with very low vaccination rates (< 10% of dogs) (Almuna 2016) may increase the risk of cross-species transmission and potential negative impacts to wild carnivores. A vaccination campaign of dogs could reduce risk to dogs and wild carnivores and serve as an experiment to test the hypothesis of dogs as a reservoir of CPV at the domestic and wild carnivore interface in JBR (Viana et al. 2014).
Despite study limitations, such as the relatively small sample size and the fact that each of the eight wild carnivore species captured in this study has an unequal capture probability (non-random sampling), we provide useful elements to the understanding of the carnivore parvovirus ecology and epidemiology in this geographic area. The presence of CPV in JBR and the potential risk of CPV infection for wild carnivores highlight the importance of implementing strategies for wildlife conservation programs in northwestern Mexico, where reintroduction programs of the Mexican gray wolf are carried out (Lara-Díaz et al. 2015), an endangered wild carnivore that is lethally susceptible to parvovirus infection (Hedrick et al. 2003). Nevertheless, more studies should be conducted to further understand the role of domestic dogs and wild carnivores in the epidemiology of CPV in this region.
References
Acosta-Jamett G, Chalmers WSK, Cunningham AA, Cleaveland S, Handel IG, Bronsvoortet BMC (2011) Urban domestic dog populations as a source of canine distemper virus for wild carnivores in the Coquimbo region of Chile. Veterinary Microbiology 152: 247-257
Allison A, Kohler DJ, Fox KA, Brown JD, Gerhold RW, Shearn-Bochsler VI, Dubovi EJ, Parrish CR, Holmes EC (2013) Frequent cross-species transmission of parvoviruses among diverse carnivore hosts. Journal of Virology 87: 2342–2347
Allison A, Kohler DJ, Ortega A, Hoover EA, Grove DM, Holmes EC, Parrish CR (2014) Host-specific parvovirus evolution in nature is recapitulated by in vitro adaptation to different carnivore species. PLoS Pathogen 10: e1004475
Allison AB, Harbison CE, Pagan I, Stucker KM, Kaelber JT, Brown JD, Ruder MG, Keel MK, Dubovi EJ, Holmes EC, Parrish CR (2012) Role of multiple hosts in the cross-species transmission and emergence of a pandemic Parvovirus. Journal of Virology 86: 865–872
Allison A, Organtini L, Zhang S, Hafenstein S, Holmes EC, Parrish CR (2016) Single mutations in the VP2 300 loop region of the three-fold spike of the carnivore parvovirus capsid can determine host range. Journal of Virology 90:753–767
Almuna MR (2016) Factores de riesgo asociados a tasas de infección de distemper canino en perro doméstico (Canis familiaris) y carnívoros silvestres en la Reserva de la Biósfera de Janos, Chihuahua, México. Bachelor´s Thesis, Universidad de Chile
Belsare AV, Vanak AT, Gompper ME (2014) Epidemiology of viral pathogens of free-ranging dogs and Indian foxes in a human-dominated landscape in central India. Transboundary and Emerging Diseases 61:78–86
Bolker BM, Brooks ME, Clark CJ, Geange SW, Poulsen JR, Stevens MHH, White JSS (2009) Generalized linear mixed models: a practical guide for ecology and evolution. Trends in Ecology and Evolution 24:127–135
Buonavoglia C, Martella V, Pratella A, Tempesta M, Cavalli A, Buonavoglia D, Bozzo G, Elia G, Decaro N, Carmichael L (2001) Evidence for evolution of Canine Parvovirus type 2 in Italy. Journal of General Virology 82: 3021–3025
Ceballos G, Davidson A, List R, Pacheco J, Manzano-Fischer P, Santos-Barrera G, Cruzado J (2010) Rapid decline of a grassland system and its ecological and conservation implications. PLoS One 5: e8562
Ceballos G, Pacheco J, List R, Manzano-Fischer P, Santos G, Royo M (2005) Prairie dogs, cattle, and crops: diversity and conservation of the grassland-shrubland habitat mosaics in northwestern Chihuahua, Mexico. In: Ecosystems, and Conservation in Northern Mexico. Cartron JLE, Ceballos G, Felger RS (editor) Biodiversity, New York: Oxford University Press, pp 425–438
Coligan J, Bierer B, Margulies D, Shevach E, Strober W (2005) Short protocols in immunology: a compendium of methods from current protocols in immunology. Wiley Blackwell Publishing, Hoboken, New Jersey
Cotmore SF, Agbandje-McKenna M, Chiorini JA, Mukha DV, Pintel DJ, Qiu J, Soderlund-Venermo M, Tattersall P, Tijssen P, Gatherer D, Davison AJ (2014) The family Parvoviridae. Archives of Virology 159: 1239–1247
Cotmore SF, Tattersall P (2007) Parvoviral host range and cell entry mechanisms. Advances in Virus Research 70: 183–232
Darriba D, Taboada GL, Doallo R, Posada D (2012) JModelTest 2: More models, new heuristics and parallel computing. Nature Methods 9: 772
Daszak P, Cunningham A, Hyatt D (2000) Emerging infectious diseases of wildlife threats to biodiversity and human health. Science 287: 443–9
Decaro N, Desario C, Parisi A, Martella V, Lorusso A, Miccolupo A, Mari V, Colaianni ML, Cavalli A, Trani LDi, Buonavoglia C (2009) Genetic analysis of Canine Parvovirus type 2c. Virology 385: 5–10
Fiorello CV, Deem SL, Gompper ME, Dubovi EJ (2004) Seroprevalence of pathogens in domestic carnivores on the border of Madidi National Park, Bolivia. Animal Conservation 7: 45–54
García E (1981) Modificaciones al Sistema de Clasificación Climática de Köeppen. Instituto de Geografía. Universidad Nacional Autónoma de México, D.F., México. p 246
GoogleEarth_7.1.8 (2016) DigitalGlobe 2016; INEGI 2016. http://www.earth.google.com [October 15, 2016]., (October 24, 2014). Janos, Mexico. 12R 730618.95 E, 3402834.78 N, 2.27 km. [Online]
Hedrick PW, Lee RN, Buchanan C (2003) Canine parvovirus enteritis, canine distemper, and major histocompatibility complex genetic variation in Mexican wolves. Journal of Wildlife Diseases 39: 909-913.
Hoelzer K, Parrish CR (2010) The emergence of parvoviruses of carnivores. Veterinary Research 41: 1-13
Knobel DL, Butler JR, Lembo T, Critchlow R, Gompper ME (2014) Dogs, disease, and wildlife. In: Free-ranging dogs and wildlife conservation, Gompper, ME (editor) Oxford Uni, Oxorfd, United Kingdom, pp. 144–164
Kreeger TJ, Arnemo JM (2012) Handbook of Wildlife Chemical Immobilization (4th ed). Sybille, Wyoming: Terry J Kreeger
Lara-Díaz NE, López-González CA, Coronel-Arellano H, Cruz-Romo JL (2015) Nacidos libres: en el camino a la recuperación del lobo mexicano. CONABIO Biodiversitas 119: 1–6.
Leigh JW, Bryant D (2015) POPART: full-feature software for haplotype network construction. Methods in Ecology and Evolution 6: 1110-1116.
Li G, Ji S, Zhai X, Zhang Y, Liu J, Zhu M, Zhou J, Su S (2017) Evolutionary and genetic analysis of the VP2 gene of canine parvovirus. BMC Genomics 18: 1-13
List R, Pacheco J, Ponce E, Sierra-Corona R, Ceballos G (2010) The Janos Biosphere Reserve, Northern Mexico. International Journal of Wilderness 16: 35–41
Maclachlan J, Dubovi EJ, Barthold SW, Swayne DF, Winton JR (2017) Fenner’s Veterinary Virology, Academic Press, San Diego, USA
McCaw DL, Hoskins JD (2006) Canine viral enteritis. In: Greene CE (editor) Infectious Diseases of the Dog and Cat, 3rd edn. St. Louis, MO: Saunders pp 63–73.
Mech L, Goyal S (2011) Parsing demographic effects of Canine Parvovirus on a Minnesota wolf population. Journal of Veterinary Medicine and Animal Health 3: 27–30
Mech, L, Goyal S, Paul W, Newton W (2008) Demographic effects of Canine Parvovirus on a free-ranging wolf population over 30 years. Journal of Wildlife Disease 44: 824–836
Mech LD, Goyal SM (1995) Effects of Canine Parvovirus on gray wolves in Minnesota. The Journal of Wildlife Management 59: 565–570
Miranda C, Santos N, Parrish C, Thompson G (2017) Genetic characterization of Canine Parvovirus in sympatric free-ranging wild carnivores in Portugal. Journal of Wildlife Disease 53: 824–831
Miranda C, Thompson G, (2016) Canine parvovirus: the worldwide occurrence of antigenic variants. Journal of General Virology 97: 2043-2057
Murphy FA, Gibbs EPJ, Horzinek MC, Studdert MJ (1999) Veterinary Virology. Academic Press San Diego, USA
Murray DL, Kapke CA, Evermann JF, Fuller TK (1999) Infectious disease and the conservation of free ranging large carnivores. Animal Conservation 2: 241–254
Nelson B, Hebblewhite M, Ezenwa V, Shury T, Merrill EH, Paquet PC, Schm F, Skinner G, Webb N (2012) Prevalence of antibodies to Canine Parvovirus and Distemper Virus in wolves in the Canadian Rocky Mountains. Journal of Wildlife Diseases 8: 68–76
Newcombe RG (1998) Two-sided confidence intervals for the single proportion: comparison of seven methods. Statistics in Medicine 17: 857–872
Ortega AF, Martínez-Castañeda JS, Bautista-Gómez LG, Muñoz RF, Hernández IQ (2017) Identification of co-infection by rotavirus and parvovirus in dogs with gastroenteritis in Mexico. Brazilian Journal of Microbiology 48: 769–773
Parrish CR, Aquadro CF, Strassheim ML, Evermann JF, Sgro JY, Mohammed HO (1991) Rapid antigenic-type replacement and DNA sequence evolution of Canine Parvovirus. Journal of Virology 65: 6544–6552
Pedersen AB, Jones KE, Nunn CL, Altizer S (2007) Infectious diseases and extinction risk in wild mammals. Conservation Biology 21: 1269–1279
Pedroza-Roldán C, Páez-Magallan V, Charles-Niño C, Elizondo-Quiroga D, Leonel De Cervantes-Mireles R, López-Amezcua MA (2015) Genotyping of Canine Parvovirus in western Mexico. Journal of Veterinary Diagnostic Investigation 27: 107–111
R Core Team (2014) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria
Sikes RS, Gannon WL (2011) Guidelines of the American Society of Mammalogists for the use of wild mammals in research. Journal of Mammalogy 92: 235–253
Smith KF, Acevedo-Whitehouse K, Pedersen AB (2009) The role of infectious diseases in biological conservation. Animal Conservation 12: 1–12
Steinel A, Parrish CR, Bloom ME, Truyen U (2001) Parvovirus infections in wild carnivores. Journal of Wildlife Disease 37: 594–607
Suzán G, Ceballos G (2005) The role of feral mammals on wildlife infectious disease prevalence in two nature reserves within Mexico City limits. Journal of Zoo and Wildlife Medicine 36: 479–484
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: Molecular evolutionary genetics analysis version 6.0. Molecular Biology and Evolution 30:2725–2729
Truyen U, Muller T, Heidrich R, Tackmann K, Carmichael LE (1998) Survey on viral pathogens in wild red foxes (Vulpes vulpes) in Germany with emphasis on parvoviruses and analysis of a DNA sequence from a red fox parvovirus. Epidemiology and Infection 121: 433–440
Viana M, Mancy R, Biek R, Cleaveland S, Cross PC, Lloyd-Smith JO, Haydon DT (2014) Assembling evidence for identifying reservoirs of infection. Trends in Ecology and Evolution 29:270–279
Williams ES, Barker IK (2001) Infectious diseases of wild mammals, 3rd edn. Wiley-Blackwell, Iowa
Zhou P, Zeng W, Zhang X, Li S (2017) The genetic evolution of Canine Parvovirus - A new perspective. PLoS One 12: 1–13
Acknowledgements
This study was supported by CONACyT Project No. 179482. We would like to thank A. Vigueras, H. Mendoza, J. Lopez, L. Aguilar, L. Orozco, and M. Moguel for helping during field sampling. We thank J. Diaz, E. Ponce, and R. Sierra (Janos Grassland Biological Station, IE-UNAM), and A. Esquer and L. Garcia (Rancho El Uno TNC) for logistical support in the field, and Laboratorios Tornel for helping with PCR diagnostic.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
López-Pérez, A.M., Moreno, K., Chaves, A. et al. Carnivore Protoparvovirus 1 at the Wild–Domestic Carnivore Interface in Northwestern Mexico. EcoHealth 16, 502–511 (2019). https://doi.org/10.1007/s10393-019-01436-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10393-019-01436-0