
Abstract
The tent-making bat hepatitis B virus (TBHBV) is a hepadnavirus closely related to human hepatitis B virus. The ecology of TBHBV is unclear. We show that it is widespread and highly diversified in Peters’ tent-making bats (Uroderma bilobatum) within Panama, while local prevalence varied significantly between sample sites, ranging from 0 to 14.3%. Females showed significantly higher prevalence than males, and pregnant females were more often acutely infected than non-reproductive ones. The distribution of TBHBV in bats was significantly affected by forest cover, with higher infection rates in areas with lower forest cover. Our data indicate that loss of natural habitat may lead to positive feedback on the biotic factors driving infection possibility. These results underline the necessity of multidisciplinary studies for a better understanding of mechanisms in pathogen–host relationships and for predictions in disease ecology.
Similar content being viewed by others


Introduction
Over the last century, the majority of the world’s biosphere has been vastly transformed. Many forests are lost or fragmented due to plantations and farmland and traversed by road networks (Ellis et al. 2010; Ellis 2011). The loss of natural habitat directly results in loss of biodiversity and drives many specialist species to extinction. In contrast, some generalist species can persist and occasionally even profit from changes in the environment, with some showing inflated population densities in disturbed areas (Fahrig 2003; Suzán et al. 2008). Fragmentation of habitats has been shown to decrease health and fitness of animals and to promote diseases, parasitism, and virus occurrence (Keesing et al. 2006; Acevedo-Whitehouse and Duffus 2009; Cottontail et al. 2009; Brearley et al. 2013).
Highly diverse tropical forests, in particular, are especially affected by habitat loss, as the functioning of these ecosystems depends on intact animal assemblages (Laurance et al. 2011; Aide et al. 2013). Bats are a good indicator group for habitat health as they are highly diverse and play major roles in pollination, pest control and seed dispersal (Jones et al. 2009; Cunto and Bernard 2012). In addition to their importance for ecosystem functioning, bats also are long-lived (Seim et al. 2013) and can have large group sizes and high mobility (Luis et al. 2013), all of which make them suitable model organisms for epidemiological studies (Drexler et al. 2012; Brook and Dobson 2015). Bats were identified as hosts to a great variety of viruses, some of them causing zoonotic diseases (e.g., Ebola, SARS, rabies), but research has focused mainly on RNA viruses, leaving DNA viruses largely unexplored (Calisher et al. 2006; Jones et al. 2008; Brook and Dobson 2015).
Of particular interest among the DNA viruses are orthohepadnaviruses that infect mammals (Rasche et al. 2016). Hepatitis B, a representative of these orthohepadnaviruses, is one of the most common and serious viral infectious diseases in humans, causing acute and chronic infections of the liver, resulting in an estimated 900,000 deaths each year (WHO Global Hepatitis Report 2017). Transmission occurs chiefly by direct contact with infected blood (WHO fact sheet #204, updated July 2016). In humans, the course of infection depends on the time of virus acquisition. Infection with hepatitis B virus (HBV) during birth results in 90% of chronic courses, while infection during early childhood leads to chronicity in 30% of all cases. In contrast, over 95% of infections acquired by immune-competent adults result in acute and self-limiting course (Seeger and Mason 2000; Gish et al. 2015). Chronic courses of infection may be a common feature of orthohepadnaviruses. In woodchucks, an animal model of HBV, perinatal infection with the woodchuck hepatitis virus (WHV) results in 100% chronic courses infection (Seeger and Mason 2000).
Orthohepadnaviruses are known from North American rodents, non-human apes, and humans (Locarnini et al. 2013) but were recently also found in bats, both in the Old World and in the New World (Drexler et al. 2013; He et al. 2013, 2015). The tent-making bat Hepatitis B virus (TBHBV), isolated from Peters’ tent-making bats (Uroderma bilobatum) in Panama, is antigenically closely related to primate HBV (Drexler et al. 2013).
Drexler et al. (2013) showed that TBHBV surface proteins allowed specific viral binding and in vitro entry into human hepatocytes via the human HBV high-affinity receptor NTCP (natrium taurocholate co-transporting protein), providing an interesting viral model organism. Uroderma bilobatum is a widespread, highly mobile frugivorous bat in the Neotropics that is specialized on eating figs. It commonly occurs in secondary forests and constructs its own roosts by modifying leaves of seven different plant families, often palms, into tent-like shelters (Baker and Clark 1987; Rodríguez-Herrera et al. 2007). It adapts well to anthropogenically modified habitats and readily roosts in the eaves of human houses, as well as in the leaves of non-native palms such as Cocos nucifera, Coccothrinax barbadensis, or Livistona chinensis (Timm and Lewis 1991; Rodríguez-Herrera et al. 2007) that are often planted as garden ornamentals (Sagot et al. 2013). Its potentially close contact with humans, the relatedness of TBHBV to primate HBV, and the ability of TBHBV to use the human NTCP are factors supporting the importance of virus ecology studies in U. bilobatum. We investigated environmental and host traits favoring TBHBV infection as well as patterns of transmission and distribution in U. bilobatum.
Materials and Methods
Between September 2012 and August 2015, we collected blood samples of Peters’ tent-making bat, U. bilobatum (Phyllostomidae: Stenodermatinae) in Panama. The samples were mainly obtained in the Panama Canal Zone (around Barro Colorado Island (BCI) and Gamboa) as well as from the regions Bocas del Toro, Los Santos, and Veraguas (Fig. 1), representing a total of 46 capture locations. Capturing and handling of bats were approved by the Smithsonian Tropical Research Institute (IACUC protocols 100316-1001-18 and 2013-0401-2016) and by the Panamanian Government (Ministerio de Ambiente, research permits: SE/A-68-11, SE/A-75-13, SE/A-69-14; export permits: SEX/A-37-14, SEX/A-22-15, and SEX/A-60-15). Bats were captured using mist nets (Ecotone, 6 × 2.5 m, 16 mm mesh and 2 × 70 denier) set in the forest or with hand nets directly at the roost. Individual bats were kept separately in freshly cleaned, soft cloth bags until processing. Sex, age [juvenile, subadult, and adult; based on grade of ossification of the epiphyseal plates (Sharifi et al. 2008)], and reproductive status (scrotal and non-reproductive in males; pregnant, lactating, post lactating and non-reproductive in females) were determined, and adult bats were individually marked with necklaces following Handley and Gardner (1991) or PIT tags (ID100; Euro ID, Weilerswist, Germany). Up to 70 µl of blood was drawn with EDTA coated capillaries (75 µl, Kabe, Germany) by puncturing the cephalic vein with a 27 G needle (Sterican, Braun, Germany) (Cottontail et al. 2009). After processing, all bats were offered sugar water and released at the capture site. Blood samples were stored on ice in the field and thereon at − 80°C until processing in the laboratory.

Sampling regions and prevalence of PCR-positive TBHBV infections in Uroderma bilobatum in Panama.
DNA was purified from blood specimens using the MagNA Pure 96 DNA and Viral NA Small Volume Kit (Roche, Mannheim, Germany). Following protocols described in Drexler et al. (2013), purified samples were screened for hepadnavirus DNA using a broadly reactive nested-PCR leading to amplification of a 354 bp fragment encoding the surface and polymerase proteins. Near-full genome amplifications were conducted using strain-specific primers (primer sequences available upon request). Viral loads were quantified by specific real-time PCR as described before (Drexler et al. 2013). To detect specific antibodies against TBHBV proteins in bat serum, immunofluorescence assays were conducted using HuH7 cells transfected with 1.1 overlength expression plasmids of TBHBV as described (Drexler et al. 2013).
Phylogenetic Analyses
Sequences were aligned with Geneious 9.1.3 using the MAFFT package (version 7.222). DnaSP 5.10.01 was used to detect point mutations within a near-complete genome alignment of 15 novel TBHBV sequences and 4 published sequences (NC_024445, KC790379, KC790380, KC790381) (Drexler et al. 2013). The antigenic loop within the S-ORF was predicted in TMHMM (v.2.0).
Neighbor-joining phylogenetic analysis of a 2503 nucleotide fragment was conducted in MEGA7 (Kumar et al. 2016) using the complete deletion option and a percentage distance substitution model including a bootstrap analysis with 1000 replicates.
Statistical Analyses
All statistics were performed in R v3.3.1 (R Development Core Team 2016). We tested for virus genetic isolation by geographic distance using a Mantel test (mantel(), R package vegan, Oksanen et al. 2016). Tip-randomization tests for capture year and capture region were conducted using BaTS (Parker et al. 2008). Therefore, we used the PCR screening fragments available from all positive samples, including a total of 24 sequences and a sequence length of 354 nucleotides. We used MCMC trees created in BEAST v.1.8.3 (Drummond and Rambaut 2007) using a HKY substitution model under a strict clock and 10 million generations (burn-in 1 million).
As a basis for further analyses, we extracted data for forest cover and forest cover loss using ArcMap (ESRI 2011) and maps based on Landsat images provided by the University of Maryland (Hansen et al. 2013, http://earthenginepartners.appspot.com/science-2013-global-forest). We used a 1 km radius around each capture site (resulting in 314 ha), based on recent telemetry studies estimating the home range of U. bilobatum at around 23 ha (Villalobos-Chaves et al. 2016). Forest cover was calculated based on a map of tree canopy cover density (0–100%) from 2000, setting the threshold at 85% density, which resulted in 99% of the tree canopy cover density values for each of the protected study sites (Bohio, Gigante, and Peña Blanca). Forest cover loss was extracted from a map representing the loss of forested areas during the period 2000–2014, encoded as percental loss of forested area in a 1 km radius around each capture site.
Using generalized linear mixed effects models (glmer(), R package lme4, Bates et al. 2007), we identified the variables best predicting TBHBV infections. Initial data exploration was conducted following Zuur et al. (2010). Our full model consisted of the infection status (infected vs. non-infected, binomial distribution) as a response variable and the variables sex, reproductive status, percent forest cover, and percent forest cover loss in a 1 km radius around each capture site as fixed effects. We also included the year of capture as well as the capture location nested in capture region as random effects. We scaled the fixed variables and tested for multi colinearity using variance inflation factors (VIFs) implemented in the R library AED (Zuur 2010). Further, we tested for spatial autocorrelation of infected bats in our data set as well as in model residuals of our candidate models using Moran’s I test (Moran.I(), R package ape, Paradis et al. 2004).
The best models were selected based on corrected Akaike’s information criterion (AICc) and the ‘all-subset approach’ as described in (Symonds and Moussalli 2011) (dredge(), R package MuMIn, Barton 2016). Parameter estimates were obtained using model averaging of set of top models, based on Δ2 AICc as cutoff (Burnham and Anderson 2003; Symonds and Moussalli 2011). For all models included in the top-model set, we calculated pseudo-R2 values to estimate model fit by accounting for the variation explained by both fixed and random effects (rsquared.glmm(), R packaged MuMIn).
Pregnant and lactating females were grouped together with scrotal males as reproductive, to obtain a single reproductive status variable for both females and males. To identify biotic and abiotic factors influencing TBHBV infections, we included in our statistical analysis 54 U. bilobatum blood samples from a former study (Drexler et al. 2013), as well as 7 additional individuals of the same sampling period that had not been previously analyzed, all collected in the BCI canal zone.
Results
We collected a total of 474 individual blood samples, from which 20 (4.2%) tested positive for TBHBV DNA (Table 1, Supplementary Table 1). Immunofluorescence analyses revealed the presence of antibodies against TBHBV in samples from 12 additional TBHBV PCR-negative individuals and showed positive reactions in 4 of the PCR-positive samples. The detection rate of antibody-positive bats (3.4%) was comparable to PCR-positive bats (4.2%). Ratios of IFT-positive to PCR-positive varied between 0 and 4.5 and differed significantly between the sampling sites BCI and Gamboa (pairwise Fisher’s test with Bonferroni correction, p < 0.01). The viral loads were high with up to 3.20 × 1012 copies per ml serum and an average of 1.96 × 1011cp/ml serum (see Table 1 for details).
Near-complete TBHBV genomes (2503 nt, whole genome comprises 3149 nt (Drexler et al. 2013) were sequenced from 15 individual samples and shorter fragments for the five remaining samples. All sequences were uploaded to Genbank and are accessible under the following numbers: MG231914-MG231918 and MG252537-MG252551. To detect hints for immune pressure, non-synonymous substitutions were analyzed among the genome of 19 sequences, including 4 formerly published TBHBV sequences (Drexler et al. 2013) (Fig. 2). Accumulation of mutations was detected in presumably antigenically active sites within the surface protein-coding open reading frame (ORF), including c-terminal amino acids within the preS1 domain and the predicted antigenic loop.

Analysis of non-synonymous nucleotide exchanges in 19 near-complete TBHBV genomes. The alignment comprises 2503 nucleotides of 15 novel TBHBV sequences and published sequences NC_024445, KC790379, KC790380, KC790381. For details on ORF position of amino acid exchanges, see Supplementary 1.
In a phylogenetic analysis, viruses clustered with previously described TBHBV strains from Panama (Drexler et al. 2013) (Fig. 3). Virus strains did not show any clustering according to year of sampling using a tip-randomization test including all available TBHBV fragments (354 nt). Further, clustering according to sampling sites was only significant for two regions, namely Gamboa and Veraguas (Table 2). Some virus strains sampled among large geographic ranges showed low genetic distance (Fig. 3), for example, B404 from Los Santos, which is closely related to sequences from the BCI canal zone, approximately 200 km away. Indeed, using a Mantel test, no support for isolation by distance was detected (Mantel test, R = 0.023, p = 0.166).

Neighbor-joining phylogenetic tree of near-complete TBHBV genomes. A total of 2503 nucleotides were used for the analysis conducted in MEGA7. Bootstrap values (%) larger than 75 are shown. Novel sequences are indicated in red (Color figure online).
Combining results of PCR- and IFT-screening females exhibited a higher prevalence (9.7%) than males (3.8%) (Chi-squared, X2 = 5.55, df = 1, p = 0.019). Local prevalence of PCR-positive bats varied between 0 and 14.3%.
All variables used in our mixed models to predict TBHBV infections had variance inflation factors (VIFs) below 3, indicating the absence of colinearity among those variables (Zuur et al. 2010). We further did not detect spatial colinearity between our observations (Moran’s I, I = 0.00, p = 0.24). To ascertain that no regional effect is driving our modeling outcome, we excluded during our preliminary model selection process individual sample regions, however, we obtained throughout consistent results. Of the 16 models created by the all-subset model approach (Supplementary Table 3), only two entered in the Δ2 AICc model set (Table 3). Both top models shared three variables, the extrinsic factor forest cover as well as the intrinsic variables sex and reproductive status, indicating a high importance of these variables. For decreasing forest cover the local TBHBV prevalence was increasing; additionally, females as well as reproductive active individuals tended to have a higher prevalence compared to males or non-reproductive active individuals, respectively (Table 4). The variable forest cover loss was slightly positively correlated with higher TBHBV prevalence, but it was weighted fairly low when averaged over all subset models (Table 4). Model residuals of both top models did not show spatial autocorrelation (Moran’s I, I = − 0.00, p = 0.91; I = − 0.00, p = 0.99). The conditional pseudo-R2 values estimating model fit for the two top models were 0.33 and 0.34, respectively (Table 3).
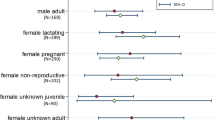
Based on these two models incorporating 535 individual observations, we plotted the predicted probabilities of a TBHBV infection showing all three significantly explaining variables, whereas forest cover loss was kept constant in the graph for the second model (ggeffect(), R package ggeffects, Lüdecke 2017). The graphs show probabilities at least ten times higher in areas without forest cover than in fully forested areas (Fig. 4) and fit well the distribution of the raw data used (Fig. 5). Our model implicates therefore a strong influence of the available forest cover throughout all combinations of sex and reproductive status on the infection probability with TBHBV.

Predicted probabilities of TBHBV infection based on the Δ2 AICc candidate model set for the effects of a the reproductive status, proportion of forest cover and sex; and b same as a but including the proportion of forest cover loss as estimated constant over the whole prediction range (see also Table 2).

Scatterplot of raw data for TBHBV infection status against percental forest cover.
Discussion
Despite the high similarity of TBHBV to human hepatitis B virus, no cross-species infections between the natural reservoir species U. bilobatum and humans are reported, nor expected (Drexler et al. 2013; Rasche et al. 2016). In fact, this close relatedness offers an exceptional possibility to study disease transmission among host bats in their natural environment, as mechanisms and restrictions might be directly adapted from well-studied host–virus interaction in humans and animals (Seeger and Mason 2000; Menne and Cote 2007; Wang et al. 2011).
Our study shows that TBHBV infections in U. bilobatum occur not only in the Panama Canal Zone where they were initially discovered (Drexler et al. 2013), but are actually widespread in Panama (more than 10.000 km2). Detected viral loads in infected bats were very high (mean 1.96 × 1011 ge/ml serum) and are comparable to highest levels reached in chronically infected woodchucks and untreated chronic human patients (Seeger and Mason 2000). Since high viral load is a predictor for HBV-related liver cirrhosis, hepatocellular carcinoma, and increased mortality in chronic human patients (Chen et al. 2009), similar consequences on the health of chronically TBHBV-infected bats are imaginable, but further studies are necessary for confirmation.
Near-complete genome sequencing of 15 new virus strains suggests that the currently observed TBHBV infections did not originate from a single outbreak in the Canal Zone, but are widely distributed across Panama. Since closely related TBHBV sequences are found in distant sample sites and the diversity at individual sample sites reflects the overall TBHBV diversity combined with the relatively slow evolution of hepadnaviruses, a long-term relationship between the bat host U. bilobatum and TBHBV in Panama can be assumed (Littlejohn et al. 2016). All samples cluster together with sequences previously published (Drexler et al. 2013), confirming the assignment as TBHBV. We did not find significant clustering of samples within years of collection, coinciding with the relatively slow virus evolution. However, samples collected in Veraguas as well as in Gamboa clustered more often together than expected by chance. These samples were frequently obtained from bats sharing the same roost or were roosting in close vicinity to each other, suggesting a largely unobstructed gene flow of virus strains within a given population. Clearly, we found no indication of genetic isolation by distance, suggesting a largely unobstructed gene flow of the virus strains within the country. This concurs with the wide distribution, high mobility, and abundance of this common bat species (Baker and Clark 1987; Meyer et al. 2009; Mantilla-Meluk 2014).
Notably, TBHBV strains from the Gamboa sampling site are dispersed among the phylogenetic tree, embracing the complete TBHBV diversity at this particular site. Here, in contrast to the other sampling sites, U. bilobatum were frequently observed roosting in the village of Gamboa closely together under overhanging roofs (O’Mara et al. 2017). Suitable roosting conditions are known to attract bats and favor the formation of larger groups (Campbell et al. 2006). A recent study has shown that resident bats are attracted to unknown, new individuals, possibly to obtain information about new food sources, which might lead to close contact between roost members and unfamiliar individuals (Ramakers et al. 2016). Together this might increase the genetic diversity of TBHBV, especially in areas with limited high-quality roosts.
The analysis of non-synonymous mutations among the TBHBV genome revealed a clustering of amino acid substitutions within the c-terminus of the preS1 coding region and within the predicted antigenic loop of the surface protein. Equivalent sites are exposed to immunological pressure in the human HBV (Sloan et al. 2008; Glebe and Bremer 2013), therefore hinting at similarities within the immunological response in tent-making bats and humans infected with hepadnaviruses. In addition, the preS1 region represents a favored aim for non-synonymous mutations since the overlapping polymerase ORF does not code for any functional elements in this region, but rather serves as a spacer between the terminal protein and the reverse transcriptase (Glebe and Bremer 2013).
The combination of PCR and immunofluorescence analyses enabled us to distinguish between different stages of infections, if we assume a similar etiopathology of TBHBV as observed in HBV infections of humans. The occurrence of individuals with only an antibody response without the actual presence of viral DNA (n = 12) suggests that U. bilobatum may have overcome an infection with TBHBV (Seeger and Mason 2000). We identified also acute infections, in which the TBHBV DNA was present, while no antibody reaction could be detected (n = 16), indicating recently obtained infections (Seeger and Mason 2000; Liang 2009). Most of these individuals were caught within 3 months after the main mating period in February–March and September–October (Fleming et al. 1972). Bats tested positive for both TBHBV DNA- and TBHBV-specific antibodies (n = 4) may represent acute infections preceding seroclearance or chronic infections (Seeger and Mason 2000; Liang 2009). To actually confirm chronic infections, consecutive sampling of TBHBV DNA positive individuals over at least two time points is necessary, but difficult to achieve in the field.
The prevalence of current TBHBV infections detected in the BCI Canal Zone area (0.8%, n = 262) was lower than the prevalence previously reported for the same study area (9.3%, n = 54; Drexler et al. 2013). The original description of TBHBV (Drexler et al. 2013) was based on a much smaller sample size (54 vs. 262 individuals), and the high prevalence might have been influenced by the fact that four of the five positive bats were caught in the same night at the same fruiting fig tree (Cottontail, pers. comm.) and might represent roost mates. O’Mara et al. (2014)showed that U. bilobatum roost mates may pass on the information of a promising food source and might therefore share foraging sites and show group foraging behavior. The social organization of U. bilobatum might favor the transmission of TBHBV within a roosting group. Groups may consist of polygynous harems of 5–50 (Kunz 1982; Chaverri et al. 2010) individuals, with one male per 5–10 females, but the animals are also found to roost in multi-male or multi-female groups (Chaverri et al. 2010). Assuming a similar possibility for sexual transmission of TBHBV as in HBV, there are two general ways of transmission within a harem group: (1) the male is a chronic carrier, transmitting TBHBV onto the females, or (2) an infected female joins the harem, and passes the virus to the male, which thereafter infects the remaining females. Both hypotheses lead to a female-biased infection prevalence typical for polygynous mating systems (Ashby and Gupta 2015) as it was observed in this study, with females showing a higher prevalence (9.7%) than males (3.8%). Furthermore, chronically infected pregnant females with high viral load will most likely transmit the virus to their newborns during birth, which is a hallmark for all orthohepadnaviruses, including human HBV. Perinatal transmission will induce chronic infections in 90% of offspring (Gish et al. 2015). In conclusion, sex and reproductive state were significant factors affecting the probability of an infection with TBHBV.
In addition to these intrinsic factors, we identified forest cover as a significant external factor (Fig. 3). The loss of habitat and therefore also of natural roosting options changes the roosting behavior of U. bilobatum (Sagot et al. 2013). Their high flexibility, adaptability, and ability to roost in ornamental palms in close vicinity to humans (Rodríguez-Herrera et al. 2007; Sagot et al. 2013) and occasionally even under the eaves of houses (O’Mara et al. 2017) allow them to persist in areas with relatively low densities of natural roosting options. Roosting under house eaves could offer energetic benefits by eliminating the necessity of building a new tent every few months (Chaverri et al. 2010). Loss of natural habitat and the corresponding native roost plants may therefore lead to higher densities of bats in areas with coconut palms, which are most often cultivated in and around villages (Chaverri et al. 2007; Sagot and Stevens 2012; Sagot et al. 2013). Higher densities may also lead to larger social groups, in which females might be more receptive to stress than males as they show higher cortisol levels when restrained (O’Mara et al. 2017), possibly weakening the immune system (Rønsholt et al. 1998; Van der Poel et al. 2000), which coincides well with our observed female-biased infection prevalence. Lewis (1992) showed that groups in anthropogenic influenced areas are more stable due to this scarceness of possible roosts compared to natural habitats, favoring virus transmission within the group. Roosting in gardens and houses bring bats in close contact to humans, who, if noticing the bats, often attempt to expulse them by destroying their tents (Sagot and Stevens 2012; Streicker et al. 2012). However, this might result in raising infection levels among bats, as evicted bats subsequently try to enter other colonies, causing distress and raising fight frequency. Through enforced roost site changes, already infected bats may directly introduce TBHBV into healthy colonies (Kerth et al. 2002; Streicker et al. 2012) and may cause more rapid spreading in anthropogenic areas than in natural habitats.
As TBHBV is closely related to human hepatitis B, the close contact to humans of a possible reservoir species might sound alarming, but current studies reveal no imminent risk of any cross-species infections between bats and humans in their natural habitat. Further, most bats pose no risk to humans at all and often go unnoticed, but contribute critically important ecosystem services such as pollination, seed dispersal, and pest control (Kunz et al. 2011).
Nevertheless, this study emphasizes the necessity of a better understanding of host–virus systems in the wild. Many aspects, host sociality for example, remain unexplored, but could prove highly important in the propensity for disease transmission (Hayman et al. 2013; Luis et al. 2013). Although the number of sampled bats in our study was extensive, the low infection prevalence and therefore the low number of positive bats entering our models may limit the precision of our predictions. Nevertheless, our results are significant and we are confident that our interpretation is well supported. We demonstrate that the loss of natural habitat can promote the spreading of TBHBV and can increase the probability of U. bilobatum becoming infected with TBHBV. Anthropogenically altered habitats provide ideal circumstances for the spread of infection, for example, offering conditions promoting numerous, large roosting groups in close vicinity to each other, roosting conditions that typically avoided in pristine habitats lacking human disturbance. As demonstrated by Estrada-Peña et al. (2014), broad patterns of disease transmission can only be explained by the combination of environmental, biotic, molecular, and human socioeconomic phenomena. A robust understanding of the mechanisms promoting pathogen–host relationships, critical for accurate predictions of disease ecology, requires multidisciplinary investigation of both host ecology and pathogen ecology.
References
Acevedo-Whitehouse K, Duffus ALJ (2009) Effects of environmental change on wildlife health Philosophical Transactions of the Royal Society of London B: Biological Sciences 364:3429-3438
Aide TM, Clark ML, Grau HR, López-Carr D, Levy MA, Redo D, Bonilla-Moheno M, Riner G, Andrade-Núñez MJ, Muñiz M (2013) Deforestation and reforestation of Latin America and the Caribbean. Biotropica 45:262-271
Ashby B, Gupta S (2015) Sexually transmitted infections in polygamous mating systems Philosophical Transactions of the Royal Society B: Biological Sciences 368:20120048
Baker RJ, Clark CL (1987) Uroderma bilobatum Mammalian Species 279:1-4
Barton K (2016) MuMIn: multi-model inference. R package version 1.15.6. https://cran.r-project.org/package=MuMIn. Accessed 10 Oct 2016
Bates D, Sarkar D, Bates MD, Matrix LT (2007) lme4: linear mixed-effects models using S4 classes. R Package version 0.99875-9. https://cran.r-project.org/package=lme4. Accessed 10 Oct 2016
Brearley G, Rhodes J, Bradley A, Baxter G, Seabrook L, Lunney D, Liu Y, McAlpine C (2013) Wildlife disease prevalence in human-modified landscapes Biological Reviews 88:427-442
Brook CE, Dobson AP (2015) Bats as ‘special’ reservoirs for emerging zoonotic pathogens Trends in microbiology 23:172-180
Burnham KP, Anderson DR (2003) Model selection and multimodel inference: a practical information-theoretic approach. Berlin: Springer.
Calisher CH, Childs JE, Field HE, Holmes KV, Schountz T (2006) Bats: Important reservoir hosts of emerging viruses. Clinical Microbiology Reviews 19:531-545
Campbell P, Akbar Z, Adnan AM, Kunz TH (2006) Resource distribution and social structure in harem-forming Old World fruit bats: variations on a polygynous theme Animal Behaviour 72:687-698
Chaverri G, Gamba-Rios M, Kunz TH (2007) Range overlap and association patterns in the tent-making bat Artibeus watsoni Animal Behaviour 73:157-164
Chaverri G, Kunz TH, Regina M (2010) Ecological determinants of social systems: perspectives on the functional role of roosting ecology in the social behavior of tent-roosting bats. In: Regina M (ed) Advances in the Study of Behavior, vol 42. Academic Press, London, pp 275-318.
Chen C-J, Yang H-I, Iloeje UH, The R-HBVSG (2009) Hepatitis B virus DNA levels and outcomes in chronic hepatitis B Hepatology 49:S72-S84
Cottontail V, Wellinghausen N, Kalko E (2009) Habitat fragmentation and haemoparasites in the common fruit bat, Artibeus jamaicensis (Phyllostomidae) in a tropical lowland forest in Panamá. Parasitology 136:1133
Cunto GC, Bernard E (2012) Neotropical bats as indicators of environmental disturbance: what is the emerging message? Acta Chiropterologica 14:143-151
Drexler JF, Corman VM, Müller MA, Maganga GD, Vallo P, Binger T, Gloza-Rausch F, Rasche A, Yordanov S, Seebens A (2012) Bats host major mammalian paramyxoviruses Nature Communications 3:796
Drexler JF, Geipel A, König A, Corman VM, van Riel D, Leijten LM, Bremer CM, Rasche A, Cottontail VM, Maganga GD, Schlegel M, Müller MA, Adam A, Klose SM, Borges Carneiro AJ, Stöcker A, Franke CR, Gloza-Rausch F, Geyer J, Annan A, Adu-Sarkodie Y, Oppong S, Binger T, Vallo P, Tschapka M, Ulrich RG, Gerlich WH, Leroy E, Kuiken T, Glebe D, Drosten C (2013) Bats carry pathogenic hepadnaviruses antigenically related to hepatitis B virus and capable of infecting human hepatocytes Proceedings of the National Academy of Sciences 110:16151-16156
Drummond AJ, Rambaut A (2007) BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evolutionary Biology 7:214.
Ellis EC (2011) Anthropogenic transformation of the terrestrial biosphere Philosophical Transactions of the Royal Society of London A: Mathematical, Physical and Engineering Sciences 369:1010-1035
Ellis EC, Klein Goldewijk K, Siebert S, Lightman D, Ramankutty N (2010) Anthropogenic transformation of the biomes, 1700 to 2000 Global ecology and biogeography 19:589-606
ESRI (2011) ArcGIS Desktop: Release 10. CA: Environmental Systems Research Institute.
Estrada-Peña A, Ostfeld RS, Peterson AT, Poulin R, de la Fuente J (2014) Effects of environmental change on zoonotic disease risk: an ecological primer Trends in Parasitology 30:205-214
Fahrig L (2003) Effects of Habitat Fragmentation on Biodiversity. Annual review of ecology, evolution, and systematics 34:487-515
Fleming TH, Hooper ET, Wilson DE (1972) Three central american bat communities: structure, reproductive cycles, and movement patterns. Ecology 53:555-569
Gish RG, Given BD, Lai C-L, Locarnini SA, Lau JYN, Lewis DL, Schluep T (2015) Chronic hepatitis B: Virology, natural history, current management and a glimpse at future opportunities. Antiviral Research 121:47-58
Glebe D, Bremer CM (2013) The molecular virology of hepatitis B virus. In: Seminars in liver disease, vol 2. New York: Thieme Medical Publishers, pp 103-112
Handley COW, Gardner AL (1991) Demography and natural history of the common fruit bat, Artibeus jamaicensis, on Barro Colorado Island, Panamá. Smithsonian Contributions to Zoology
Hansen MC, Potapov PV, Moore R, Hancher M, Turubanova SA, Tyukavina A, Thau D, Stehman SV, Goetz SJ, Loveland TR, Kommareddy A, Egorov A, Chini L, Justice CO, Townshend JRG (2013) High-resolution global maps of 21st-century forest cover change. Science 342:850–853 (Data available on-line from: http://earthenginepartners.appspot.com/science-2013-global-forest)
Hayman DTS, Bowen RA, Cryan PM, McCracken GF, O’Shea TJ, Peel AJ, Gilbert A, Webb CT, Wood JLN (2013) Ecology of Zoonotic Infectious Diseases in Bats: Current Knowledge and Future Directions Zoonoses and public health 60:2-21
He B, Fan Q, Yang F, Hu T, Qiu W, Feng Y, Li Z, Li Y, Zhang F, Guo H, Zou X, Tu C (2013) Hepatitis Virus in Long-Fingered Bats, Myanmar Emerging infectious diseases 19:638-640
He B, Zhang F, Xia L, Hu T, Chen G, Qiu W, Fan Q, Feng Y, Guo H, Tu C (2015) Identification of a novel orthohepadnavirus in pomona roundleaf bats in China Archives of Virology 160:335-337
Jones G, Jacobs DS, Kunz TH, Willig MR, Racey PA (2009) Carpe noctem: the importance of bats as bioindicators Endangered Species Research 8:93-115
Jones KE, Patel NG, Levy MA, Storeygard A, Balk D, Gittleman JL, Daszak P (2008) Global trends in emerging infectious diseases Nature 451:990-993
Keesing F, Holt RD, Ostfeld RS (2006) Effects of species diversity on disease risk Ecology Letters 9:485-498
Kerth G, Safi K, König B (2002) Mean colony relatedness is a poor predictor of colony structure and female philopatry in the communally breeding Bechstein’s bat (Myotis bechsteinii). Behavioral Ecology and Sociobiology 52:203-210
Kumar S, Stecher G, Tamura K (2016) MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Molecular Biology and Evolution 33:1870-1874
Kunz TH (1982) Roosting ecology of bats. In: Kunz TH (ed) Ecology of bats. Plenum Press, New York, pp 1 - 51.
Kunz TH, Braun de Torrez E, Bauer D, Lobova T, Fleming TH (2011) Ecosystem services provided by bats Annals of the New York Academy of Sciences 1223:1-38
Laurance WF, Camargo JLC, Luizão RCC, Laurance SG, Pimm SL, Bruna EM, Stouffer PC, Bruce Williamson G, Benítez-Malvido J, Vasconcelos HL, Van Houtan KS, Zartman CE, Boyle SA, Didham RK, Andrade A, Lovejoy TE (2011) The fate of Amazonian forest fragments: a 32-year investigation. Biological Conservation 144:56-67
Lewis SE (1992) Behavior of Peter’s tent-making bat, Uroderma bilobatum, at maternity roosts in Costa Rica Journal of Mammalogy 73:541-546
Liang TJ (2009) Hepatitis B: The virus and disease Hepatology 49:13-21
Littlejohn M, Locarnini S, Yuen L (2016) Origins and evolution of hepatitis B virus and hepatitis D virus. Cold Spring Harbor Perspectives in Medicine 6:a021360
Locarnini S, Littlejohn M, Aziz MN, Yuen L (2013) Possible origins and evolution of the hepatitis B virus (HBV). In: Seminars in Cancer Biology, vol 6. Amsterdam: Elsevier, pp 561-575
Lüdecke D (2017) ggeffects: Create Tidy Data Frames of Marginal Effects for ‘ggplot’ from Model Outputs vol R package version 0.2.1. URL https://CRAN.R-project.org/package=ggeffects. Accessed 10 Oct 2016
Luis AD, Hayman DTS, O’Shea TJ, Cryan PM, Gilbert AT, Pulliam JRC, Mills JN, Timonin ME, Willis CKR, Cunningham AA, Fooks AR, Rupprecht CE, Wood JLN, Webb CT (2013) A comparison of bats and rodents as reservoirs of zoonotic viruses: are bats special?. Proceedings of the Royal Society of London B: Biological Sciences 280:20122753
Mantilla-Meluk H (ed) (2014) Defining Species and Species Boundaries in Uroderma (Chiroptera: Phyllostomidae) with a Description of a New Species, vol 325. Occasional Papers of the Museum of Texas Tech University. Museum of Texas Tech University
Menne S, Cote PJ (2007) The woodchuck as an animal model for pathogenesis and therapy of chronic hepatitis B virus infection World Journal of Gastroenterology 13:104
Meyer CF, Kalko EKV, Kerth G (2009) Small-scale fragmentation effects on local genetic diversity in two phyllostomid bats with different dispersal abilities in Panama Biotropica 41:95-102
O’Mara MT, Dechmann DKN, Page RA (2014) Frugivorous bats evaluate the quality of social information when choosing novel foods Behavioral Ecology 25:1233-1239
O’Mara MT, Wikelski M, Voigt CC, Ter Maat A, Pollock HS, Burness G, Desantis LM, Dechmann DK (2017) Cyclic bouts of extreme bradycardia counteract the high metabolism of frugivorous bats eLife 6:e26686
Oksanen J, Blanchet F, Friendly M, Kindt R, Legendre P, McGlinn D, Minchin PR, O’Hara R, Simpson G, Solymos P, Stevens M, Szoecs E, Wagner H (2016) vegan: Community ecology package. R package version 2.4-1. URL https://CRAN.R-project.org/package=vegan. Accessed 10 Oct 2016
Paradis E, Claude J, Strimmer K (2004) APE: analyses of phylogenetics and evolution in R language. Bioinformatics 20:289-290
Parker J, Rambaut A, Pybus OG (2008) Correlating viral phenotypes with phylogeny: Accounting for phylogenetic uncertainty Infection, Genetics and Evolution 8:239-246
R Development Core Team (2016) R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria.
Ramakers JJ, Dechmann DK, Page RA, O’Mara MT (2016) Frugivorous bats prefer information from novel social partners Animal Behaviour 116:83-87
Rasche A, Souza BFdCD, Drexler JF (2016) Bat hepadnaviruses and the origins of primate hepatitis B viruses Current Opinion in Virology 16:86-94
Rodríguez-Herrera B, Medellín R, Timm R (eds) (2007) Murciélagos neotropicales que acampan en hojas. Neotropical tent-roosting bats. Ecosistemas de Costa Rica. INBio.
Rønsholt L, Sörensen KJ, Bruschke CJM, Wellenberg GJ, van Oirschot JT, Johnstone P, Whitby JE, Bourhy H (1998) Clinically silent rabies infection in (zoo) bats. Veterinary Record 142:519-520
Sagot M, Rodríguez-Herrera B, Stevens RD (2013) Macro and Microhabitat Associations of the Peter’s Tent-Roosting Bat (Uroderma bilobatum): Human-Induced Selection and Colonization? Biotropica 45:511-519
Sagot M, Stevens RD (2012) The Evolution of Group Stability and Roost Lifespan: Perspectives from Tent-Roosting Bats Biotropica 44:90-97
Seeger C, Mason WS (2000) Hepatitis B virus biology Microbiology and Molecular Biology Reviews 64:51-68
Seim I, Fang X, Xiong Z, Lobanov AV, Huang Z, Ma S, Feng Y, Turanov AA, Zhu Y, Lenz TL, Gerashchenko MV, Fan D, Hee Yim S, Yao X, Jordan D, Xiong Y, Ma Y, Lyapunov AN, Chen G, Kulakova OI, Sun Y, Lee S-G, Bronson RT, Moskalev AA, Sunyaev SR, Zhang G, Krogh A, Wang J, Gladyshev VN (2013) Genome analysis reveals insights into physiology and longevity of the Brandt’s bat Myotis brandtii. Nature Communications 4:2212
Sharifi M, Mozafari F, Taghinezhad N, Javanbakht H (2008) Variation in Ectoparasite Load Reflects Life History Traits in the Lesser Mouse-eared Bat Myotis blythii (Chiroptera: Vespertilionidae) in Western Iran. Journal of Parasitology 94:622-625
Sloan RD, Ijaz S, Moore PL, Harrison TJ, Teo C-G, Tedder RS (2008) Antiviral resistance mutations potentiate hepatitis B virus immune evasion through disruption of its surface antigen a determinant Antiviral therapy 13:439
Streicker DG, Recuenco S, Valderrama W, Gomez Benavides J, Vargas I, Pacheco V, Condori Condori RE, Montgomery J, Rupprecht CE, Rohani P, Altizer S (2012) Ecological and anthropogenic drivers of rabies exposure in vampire bats: implications for transmission and control Proceedings of the Royal Society of London B: Biological Sciences 279:3384
Suzán G, Armién A, Mills JN, Marcé E, Ceballos G, Ávila M, Salazar-Bravo J, Ruedas L, Armién B, Yates TL (2008) Epidemiological Considerations of Rodent Community Composition in Fragmented Landscapes in Panama Journal of Mammalogy 89:684-690
Symonds MRE, Moussalli A (2011) A brief guide to model selection, multimodel inference and model averaging in behavioural ecology using Akaike’s information criterion. Behavioral Ecology and Sociobiology 65:13-21
Timm RM, Lewis SE (1991) Tent construction and use by Uroderma bilobatum in coconut palms (Cocos nucifera) in Costa Rica. Bulletin of the American Museum of Natural History 206:251-260
Van der Poel WHM, Van der Heide R, Van Amerongen G, Van Keulen LJM, Wellenberg GJ, Bourhy H, Schaftenaar W, Groen J, Osterhaus ADME (2000) Characterisation of a recently isolated lyssavirus in frugivorous zoo bats Archives of Virology 145:1919-1931
Villalobos-Chaves D, Bonaccorso FJ, Rodríguez-Herrera B, Cordero-Schmidt E, Arias-Aguilar A, Todd CM (2016) The Influence of Sex and Reproductive Status on Foraging Behavior and Seed Dispersal by Uroderma convexum (Chiroptera: Phyllostomidae). In: Ortega J (ed) Sociality in Bats. Springer International Publishing, Cham, pp 281-301.
Wang B-J, Tian Y-J, Meng Z-J, Jiang M, Wei B-Q, Tao Y-Q, Fan W, Li A-Y, Bao J-J, Li X-Y, Zhang Z-M, Wang Z-D, Wang H, Roggendorf M, Lu M-J, Yang D-L (2011) Establishing a new animal model for hepadnaviral infection: susceptibility of Chinese Marmota-species to woodchuck hepatitis virus infection Journal of General Virology 92:681-691
World Health Organization (2016) Hepatitis B fact sheet 2016. URL: http://www.who.int/mediacentre/factsheets/fs204/en/. Accessed July 2016
World Health Organization (2017) Global Hepatitis Report 2017. Geneva
Zuur AF (2010) AED: Data files used in Mixed effects models and extensions in ecology with R
Zuur AF, Ieno EN, Elphick CS (2010) A protocol for data exploration to avoid common statistical problems Methods in Ecology and Evolution 1:3-14
Acknowledgements
We want to thank the Smithsonian Tropical Research Institute, especially Oris Acevedo and Belkys Jimenez, for providing the infrastructure and logistics for field work. We are grateful to all people of the BCI and Gamboa BatLabs for help and assistance in the field. We further want to thank Nina Schwensow for a crash course in statistical analysis of molecular data and two anonymous reviewers who helped to improve the manuscript. This study was funded by German Research Foundation (DFG) SPP 1596 Grants DR 810/1-1, GL 595/4-1, and TS 81/6-1 (to J.F.D., M.T., and D.G.), which had no influence in study design or interpretation of the results.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Hiller, T., Rasche, A., Brändel, S.D. et al. Host Biology and Anthropogenic Factors Affect Hepadnavirus Infection in a Neotropical Bat. EcoHealth 16, 82–94 (2019). https://doi.org/10.1007/s10393-018-1387-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10393-018-1387-5