
Abstract
Genetic non-invasive sampling (gNIS) may provide valuable information for population monitoring, as it allows inferences of population density and key behavioural traits such as dispersal, kinship and reproduction. Despite its enormous potential, gNIS has rarely been applied to small mammals, for which live-trapping is still the most commonly used sampling method. Here we evaluated the applicability and cost-effectiveness of gNIS compared with live-trapping, to monitor a metapopulation of an Iberian endemic and elusive rodent: the Cabrera vole (Microtus cabrerae). We compared the genetic diversity, kinship and dispersal movements inferred using both methods. For that, we optimised microsatellite markers for individual identification of M. cabrerae, using both tissue (n = 31) and faecal samples (n = 323) collected from a metapopulation in south-western Iberia. An initial set of 20 loci was optimised for tissue samples, from which 11 were selected to amplify in faecal samples. Overall, gNIS revealed a higher number of identified individuals (65) than live-trapping (31), and the estimated genetic diversity was similar using data from tissues and gNIS. Kinship analysis showed a higher number of inferred relationships and dispersal events when including gNIS, and indicated absence of sex-biased dispersal. The total cost (fieldwork and genetic analysis) of each genotype obtained through live-trapping was three times greater than for gNIS. Our data strongly supports the high potential and cost-effectiveness of gNIS for monitoring populations of elusive and/or threatened small mammals. We also illustrate how this genetic tool can be logistically feasible in conservation.
Similar content being viewed by others

Introduction
Genetic data have increasingly been used in recent years for population monitoring and conservation (Schwartz et al. 2007a). Genetic information has helped to infer key ecological information in mammals, such as population density (Sollmann et al. 2013), kinship (Schmidt et al. 2016), dispersal behaviour (Mateo-Sánchez et al. 2015) and survival rates (De Barba et al. 2010; Woodruff et al. 2016). In general, genetic data are gathered using samples collected from live-trapping (e.g. tissue, blood), where individuals are physically captured and tagged for capture-mark-recapture analyses. For many years, capture-mark-recapture has provided accurate estimates of population density (Grenier et al. 2009), but genetic data can reveal cryptic patterns that are impossible to access through capture data alone, such as inbreeding rates and genetic diversity (Melosik et al. 2017; Taylor et al. 2017), or even the population structure and gene flow (Zimmerman et al. 2015; Walsh et al. 2016). The proper evaluation of these parameters is essential not only for assessing species ecology, but also for understanding how species respond to environmental change, anthropogenic habitat loss and for developing suitable conservation strategies (Swift and Hannon 2010; Lindenmayer and Fischer 2013).
While genetic analyses tied with live-trapping may improve population monitoring, the overall accuracy of the results depends on the number of samples, which can be low, particularly when focusing on traditional capture methods of rare and/or elusive mammals (Thompson 2013). Moreover, live-trapping is a potential source of stress and/or trauma for captured animals, which is a major concern when dealing with sensitive and threatened species (Powell and Proulx 2003; Pauli et al. 2010). An alternative to live-trapping for estimating population parameters is genetic non-invasive sampling (gNIS), which relies on the use of DNA retrieved from presence signs in the field (e.g. faeces, hairs, urine), without manipulating or even seeing the animals (see Waits and Paetkau 2005 and Beja-Pereira et al. 2009 for reviews).
gNIS has become a common method for population monitoring, especially in large mammals (e.g. carnivores and ungulates), and is often used to estimate population size (Woodruff et al. 2016; Gulsby et al. 2016), determine connectivity and gene flow (Walker et al. 2007; Stansbury et al. 2016) or even assess reproductive behaviour (Stanton et al. 2015; Schmidt et al. 2016). A notable advantage of the use of gNIS in mammals is the considerable increase in the number of available samples that can be processed when live-captures are low (Sollmann et al. 2013; Gillet et al. 2015; Mestre et al. 2015) or logistically difficult (Henry and Russello 2011; Silva et al. 2015). Furthermore, the use of gNIS in low density species may provide the same accuracy as that obtained from traditional live-trapping (Marucco et al. 2011; Mumma et al. 2015) and may even replace it altogether (Trinca et al. 2013).
Despite the great potential of gNIS, there is a general perception that this methodology requires complex logistics and data analysis (Lampa et al. 2013), and that it is more expensive than traditional live-trapping due to putative high laboratory costs. Nevertheless, few studies have explicitly compared the costs and practical effectiveness between these two approaches (Kilpatrick et al. 2013; Cheng et al. 2017). This comparison would provide important information for population monitoring, particularly in the case of species for which live-trapping is still the most common sampling method, such as small mammals (Watkins et al. 2010). While some studies have applied gNIS in small mammals, namely by using faecal samples and/or owl pellets to identify species (Agata et al. 2011; Galan et al. 2012; Barbosa et al. 2013), assess taxonomy (Kuch et al. 2002) and determine phylogeographic patterns (Jaarola and Searle 2004; Miller et al. 2006), it has only been applied once for population monitoring (Gillet et al. 2016).
In the present study, we evaluate the applicability and cost-effectiveness of gNIS for monitoring small mammals. We use as a model species a rodent with low capture rates, the Cabrera vole (Microtus cabrerae), which is an endangered species endemic to the Iberian Peninsula (Fig. 1a). This species inhabits fast disappearing damp grasslands (Pita et al. 2014), which have become highly fragmented during the past decades due to agriculture intensification and livestock grazing (Garrido-García and Soriguer-Escofet 2012; Laplana and Sevilla 2013). As a consequence, the species often shows a metapopulation-like spatial structure and extinction-(re)colonisation dynamics of available habitat patches (Pita et al. 2007). To understand the factors affecting their metapopulation dynamics, it is important to have information on population densities, fecundity, survival and dispersal rates, and the interaction among these, but this has largely been hampered by the difficulty of applying conventional capture-mark-recapture studies due to the low capture rates. Hence, in this work we assess the effectiveness of gNIS compared with live-trapping for monitoring a metapopulation of M. cabrerae in the south-western Iberian Peninsula, namely by gathering information on their genetic diversity, reproductive behaviour and dispersal. To achieve these goals, we use microsatellite data obtained from non-invasive (faeces) and invasive (tissue) samples, and compare inferences from both methods in terms of the number of identified individuals, genetic diversity parameters, kinship and dispersal events. In a companion paper, the same dataset was used to evaluate the use of gNIS for estimating population densities of the Cabrera vole (Sabino-Marques et al. 2018). We discuss the cost-effectiveness of live-trapping versus gNIS sampling methods, and the potential advantages and caveats of gNIS to monitor small mammal populations.

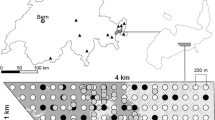
Maps of the study area, showing the potential dispersal barriers (roads and rivers) to Microtus cabrerae dispersal, and the habitat patches that are suitable for the presence of M. cabrerae. Patches where presence signs were found are marked as ‘occupied’, and were further sampled through live-trapping and genetic non-invasive sampling (gNIS). a Global distribution of M. cabrerae (grey shading). b Location of the 78 ha circular area (black circle) within Portugal (outlined), inside the distribution of M. cabrerae (grey shading). c Locations of the collected and extracted tissue samples (△, n = 31). d Locations of the collected and extracted faecal samples (■, n = 323)
Material and methods
Sampling and DNA extraction
Sampling was performed in a 78-ha circular study area near Vila Nova de Milfontes, south-western Portugal (centroid: 37.754720, −8.751256; Fig. 1b). This study area (Fig. 1c) is located within an agropastoral landscape, and included 37 suitable habitat patches (small damp areas with tall grass), initially surveyed for presence signs of M. cabrerae (latrines, grass clippings and grass tunnels; Pita et al. 2006). Samples from live-trapping were collected between November and December 2011 in 23 occupied patches, using Sherman traps in each patch for 9–10 nights (details in Sabino-Marques et al. 2018). A total of 371 traps were placed at likely capture sites and were monitored every 12 h. All individuals captured for the first time were marked with an individual passive integrated transponder (PIT) tag, sexed, weighed and a small ear biopsy was collected (tissue sample). Individuals weighing less than 28 g were considered juveniles (Fernández-Salvador et al. 2001). All individuals were released at the trapping site immediately after manipulation. Recaptured individuals identified through the PIT tag were released without further manipulation. A total of 31 tissue samples were collected in nine patches (Fig. 1c), and were stored in 96% ethanol. These samples were kept at −20 °C until extraction, using the EasySpin® Genomic DNA Minipreps Tissue Kit (Citomed, Lisbon, Portugal) following the manufacturer’s instructions.
Non-invasive samples (faeces) were collected from 21 out of 23 occupied habitat patches, the two missing due to logistic constraints (details in Sabino-Marques et al. 2018). A total of 487 faecal samples were collected in four consecutive days for each patch, to allow for capture-recapture of the genotyped individuals (total of 5 sampling days). The time spent during collection was proportional to the patch area, ranging between 20 and 120 min. The faecal samples were collected as isolated faeces or from small latrines (< 20 faeces) to avoid cross-contamination from conspecifics, given that voles often have communal latrines (Woodroffe et al. 1990). Up to 20 faeces per sample were collected using a sterilised sampling kit (collection tube with 96% ethanol, tweezers and latex gloves), and kept at −20 °C until extraction. Only faeces recognised in the field to be ‘fresh’ were collected, to maximise DNA extraction and amplification success. Of the 487 faecal samples collected, 323 (66.3%) were selected for DNA extraction based on number of faeces per sample (those with more faeces being preferred) and to represent the most comprehensive spatial coverage of each patch (Fig. 1d). At least 10 faecal samples were extracted per patch, except in the case of patches with less than 10 samples, from which all samples were extracted. DNA was extracted using the QIAamp® DNA Micro kit (Qiagen), using three to six faeces per sample. We followed the ‘forensic case work samples’ protocol, without vortexing during the digestion step, to minimise both the destruction of the faeces and the release of plant inhibitors (Wehausen et al. 2004).
Genotyping and sexing
In the 31 tissue samples from the captured individuals, we amplified a total of 20 microsatellite loci, from which 9 were cross-amplified loci from other species of the genus Microtus, and 11 were species-specific loci obtained through next-generation pyrosequencing with the 454 FLX Titanium sequencer (Roche Applied Science, Meylan, France; see Online Resources 1 and 2 for details on the microsatellite optimisation and amplification). We repeated the amplification for all loci with missing data and small allele peaks. The 20 loci were also re-amplified in at least 15% of the tissue samples to confirm the genotyping results and to estimate error rates.
From the set of 20 microsatellite loci used for the tissue samples, 11 were selected for amplification in the faecal samples, on the basis of small PCR fragment size (up to 250 base pairs), high amplification success rates for the tissue samples and also high estimates of genetic diversity (Table 1, Online Resources 1 and 2). For these 11 loci, we estimated the values of probability of identity among unrelated (PI = 3.3 × 10−12) and related individuals (PIsibs = 4.2 × 10−5), and the probability of exclusion (PE = 0.999), using the genotypes from the 31 tissue samples and the software GenAlEx 6.5 (Peakall and Smouse 2012). These values are within the recommended range for individual identification by gNIS (Mills et al. 2000; Waits et al. 2001; McKelvey and Schwartz 2004). The 11 loci were combined in amplification mixes with a smaller number of markers (two to three loci), as recommended for gNIS (Beja-Pereira et al. 2009; see Online Resources 1 and 2 for details).
For genotyping the faecal samples, we followed a stepwise approach, where we used the microsatellite primer mix with three loci (multiplex ‘Mix 1’) to screen the quality of the samples, before amplifying the remaining eight loci (multiplex ‘Mix 2’ and ‘Mix 3’, Fig. 2, Online Resources 1, 2 and 3). If the samples failed to amplify one of the three loci, or two loci displayed genotyping errors, the samples were excluded from further analyses. Amplification for the 11 loci was replicated a minimum of four times, and a maximum of eight times in instances when the initial four replicates displayed genotyping errors (Fig. 2, Online Resource 3). The PCR protocol for all microsatellites consisted of a touch-down annealing step ranging from 55 to 52 °C for 60 s and a final extension step at 60 °C for 30 min.

Workflow for genotyping non-invasive samples. Allele scoring and analysis were conducted independently by two users, to minimise bias (for detailed guidelines see Online Resource 3). Successful extractions and amplifications are shown as continuous lines (-), repetitions of the PCR amplification are shown as dashed lines (- -) and failed amplifications are shown as dotted lines (•••)
To confirm species identification and the sample quality, the complete cytochrome b (cyt-b) gene was amplified and sequenced for the 31 tissue samples using the primers L14727-SP and H15915-SP (Jaarola and Searle 2002), and for the faecal samples successfully amplified with ‘Mix 1’ a smaller fragment of cyt-b developed for all Iberian rodents was used (see Barbosa et al. 2013 for details). The small fragment of cyt-b was also amplified for at least 20% of all the discarded faecal samples (which failed to amplify with Mix 1, Fig. 2), to ensure that estimates of overall genotyping success rate for gNIS were not biased by attributing to M. cabrerae faeces belonging to non-target species (Fig. 2 and Online Resource 3).
The faecal samples that passed quality control were sexed by amplifying small fragments of two sex-linked introns, DBX5-S and DBY7-S (Online Resources 1 and 2). To account for possible genotyping errors, the PCR reaction for sexing was replicated three times and scored according to the defined criteria (Fig. 2 and Online Resource 3). The 31 tissue samples were also amplified for the two sex-linked introns in one replicate, to confirm genetically the sexing of the individuals. The PCR protocol consisted of a touch-down annealing step ranging from 59 to 53 °C for 45 s and a final extension of 60 °C for 5 min.
All PCR products were run on an ABI3130 capillary sequencer (Applied Biosystems), and while the complete sequence of cyt-b was sequenced for both strands in the tissue samples, for the faecal samples the small fragment was sequenced only for the reverse strand. The software GeneMapper version 4.0 (Applied Biosystems) was used for scoring the microsatellites and small-sized introns. The cyt-b gene sequences were analysed with the software Geneious 8 (Kearse et al. 2012).
A consensus genotype for the faecal samples was built based on a set of guidelines defined for quality control of samples and for scoring alleles (Online Resource 3). We compared the genotypes using the software Gimlet (Valière, 2002). We performed additional re-amplification of genotypes differing by up to two loci (mismatch) or having missing data in up to three loci, to verify the occurrence of genotyping errors (dropout or false alleles) and to correct or complete the genotype (Online Resource 3). If the new PCR reactions failed or produced the same result, the data for the relevant loci was classified as missing.
The genotyped samples from the tissue and faecal samples were assigned to individuals, taking into account that individuals could differ by up to two loci. This value was defined based on the number of mismatches that would minimise assignment errors, by using two approaches. First, we used the values of PI and PIsibs to estimate the probability that two random genotypes match at an increasing number of loci (as described in Lounsberry et al. 2015), which indicated that the probability of siblings sharing nine and eight loci was <1% and < 2.5%, respectively. Then, we analysed the frequencies of pairwise comparisons between all consensus genotypes (that could be from the same or from different individuals) matching at 0 to 11 loci. The results of this analysis indicated that separate samples from the same individual should share nine or more loci (Mckelvey and Schwartz 2004; see also Sabino-Marques et al. 2018 for details). Matches with incomplete genotypes with up to two missing loci were also accepted.
Data analyses
Data analyses were performed separately for the 31 tissue samples (TS dataset; 20 loci) and for the consensus genotypes from the 323 faecal samples (FC dataset; 11 loci). Tissue and faecal samples were also included in a combined dataset (TS + FC) and analysed for the 11 microsatellite loci. Species identification of the TS and FC samples was made by comparing their cyt-b sequences to the Barbosa et al. (2013) reference dataset. Cyt-b haplotype diversity for the TS and FC datasets was estimated in Arlequin 3.5 (Excoffier and Lischer 2010).
Amplification success rates per locus were calculated using the TS dataset and considering all PCR replicates performed during genotyping, for both multiplex and individual amplifications. The genotyping success rate for FC was calculated as the number of successfully amplified consensus genotypes over the sum of all faecal samples analysed (n = 323). Genotyping error rates (dropout and false allele) were estimated for all loci and for both TS and FC with the software Pedant 1.0 (Johnson and Haydon 2007). Since this software is limited to two replicates, and up to eight could be obtained for a single sample in the FC dataset, we followed the author’s instructions and randomly chose four replicates through a RANDOM function in Microsoft Excel, compared these replicates sequentially (e.g. rep1 vs rep2, rep2 vs rep3, etc.) and then averaged the results. For the TS dataset, there were at most only two replicates, simplifying the analysis.
The number of alleles (Na), expected heterozygosity (HE) and observed heterozygosity (HO) for each locus were calculated using the software GenAlEx, and overall inbreeding (FIS) was estimated in the program INEST 2.0 (Chybicki and Burczyk 2009) using 2 × 105 iterations, with 50 iterations of thinning and a burn-in of 2 × 104 iterations. These estimates were obtained separately for the TS, FC and TS + FC datasets. Deviations from Hardy-Weinberg equilibrium (HWE) and linkage disequilibrium (LD) were calculated in genepop 4.3 (Rousset 2008) using a dememorisation of 1 × 104, with a batch length of 5 × 104 and batch number of 2000 for all datasets. The probability (p) values from the multiple HWE and LD tests were corrected using a false discovery rate (Benjamini and Hochberg 1995).
Kinship relations were assessed for the TS, FC and TS + FC datasets using the genetic parentage analysis software COLONY 2.0.5.9 (Jones and Wang 2010). In the analysis, we assumed a monogamous mating system, because previous ecological studies provided support for monogamous behaviour in the Cabrera vole (Fernández-Salvador et al. 2001; Pita et al. 2010; Gomes et al. 2013). Also, preliminary analyses in COLONY considering a polygamous system failed to provide consistent results among different runs (results not shown). Another relevant parameter in this analysis is the proportion of sampled parents in the dataset. Although this could not be estimated directly from our data, preliminary analyses showed that different proportions produced the same results (results not shown). Therefore, we used a default value of 0.5 for both parents in the kinship analyses, as suggested by the authors (Jones and Wang 2010).
To aid in kinship analysis, we compared the cyt-b haplotypes among individuals and used that information for excluding maternity and sibship in the input for COLONY. We also excluded as potential parents any juveniles identified during live-trapping. The ‘very long run’ option was used in COLONY, and six independent runs were performed by changing the seed numbers, with 20 replicates for each run. Only kinship relations consistent in four out of six runs with a likelihood of >95% were considered to be valid. For each dataset, we used the kinship results to detect possible inter-patch dispersal, by evaluating if a pair of related individuals (dyad) was found in different habitat patches.
We tested for sex-biased dispersal in our population using the software COANCESTRY 1.0.1.5 (Wang 2011) for all datasets. We estimated and compared the mean relatedness coefficient among males and females, using the ‘Queller and Goodnight’ estimator (Queller and Goodnight 1989). Moreover, we calculated the mean assignment index correction (AIc) values for each sex and tested for differences between sexes with a non-parametric Mann–Whitney U test in GenAlEx (Mossman and Waser 1999).
Sampling costs and logistics
The costs of live-trapping and gNIS were estimated for field and laboratory work as the sum of human resources (field and laboratory technicians), fieldwork expenses (lodging, fuel, sample collection material, etc.) and laboratory work costs (extraction kits, PCR, genotyping runs, etc.). Base values for human resources were obtained from Fundação para a Ciência e a Tecnologia (FCT) stipend values. We included the material that was especially ordered for this study (collection tubes, gloves, extraction kits, etc.). Equipment that is not specific to this study, such as vehicles and Sherman traps for field work, or thermocycler machines for laboratory work, were not considered for estimating costs. The costs and time associated with field preparation (assessment of suitable patches, monitoring for presence signs before trapping, etc.) and laboratory workflow optimisation (extraction kit trials, primer optimisation, etc.) were also not considered in the final estimates. The total costs were used to calculate the costs per genotype produced, per identified individual and per capture event (including recaptures).
Results
Genotyping success
We obtained complete microsatellite genotypes for all tissue samples (TS dataset), with amplification success among PCR replicates varying between 21.5 to 100% for multiplex reactions, and 50 to 100% for individual reactions (Online Resource 2). No evidence of genotyping errors was found in the TS samples. The cyt-b sequencing of the 31 samples in the TS dataset confirmed the species identification and revealed three different cyt-b haplotypes with four polymorphic sites and an estimated haplotype genetic diversity of 0.667 ± 0.030 (GenBank accession number: KY380621, KY380624, KY380629; see Barbosa et al. 2017). The amplification of the sex-linked introns revealed that the sex of 5 reproductively inactive individuals was misidentified in the field. Two individuals among those captured had the same genotype and cyt-b haplotype, even after re-extraction and re-amplification for all markers. Given the high probability that these individuals were identical twins, only one of them was used in genetic diversity and kinship analysis.
From the 323 selected faecal samples (FC dataset), we obtained consensus microsatellite genotypes for 115 of the samples (35.6%), all confirmed by cyt-b sequencing to be from M. cabrerae. From the cyt-b sequencing of 54 of the 208 discarded samples, we obtained 53 sequences of M. cabrerae, and one of the southwestern water vole (Arvicola sapidus). Only one cyt-b haplotype was found in all the M. cabrerae faecal samples analysed (n = 168), as the small cyt-b fragment amplified in these samples is located in a conservative region of the complete gene for M. cabrerae, and the three haplotypes detected in the TS dataset could not be differentiated in that region. From the total of 208 samples discarded during microsatellite genotyping (Fig. 2), six had three or more alleles for at least two loci, showing evidence of different individuals using the same latrine (conspecific contamination). The error rates were low for the 11 loci among the 115 genotyped samples, with dropout rates ranging from 0 to 1.8% and no evidence of false alleles (Table 1).
Genetic diversity
The genetic diversity and locus size ranges showed little variation among the three datasets (TS, FC and TS + FC, see Table 1). The Na per locus ranged from 2 to 12, HE varied between 0.20–0.93 and HO varied between 0.28–0.93. The overall FIS value was low in the three datasets (TS: 0.036; FC: 0.016; TS + FC: 0.005).
Only one species-specific locus (Mc07) in one dataset (the TS) showed significant (p < 0.05) departure from HWE due to heterozygote deficiency. We observed that all males showed only one allele for this locus, and a BLAST search (via http://blast.ncbi.nlm.nih.gov) against the assembled genome of the prairie vole (Microtus ochrogaster) retrieved the highest similarity to a genomic scaffold (NW_004949134) associated with the X chromosome. Given the likelihood that the locus Mc07 is on the X chromosome, we removed this locus from further analyses, but used the Mc07 genotype information to exclude potential paternities and maternities before running COLONY to improve performance of the software. LD was found in single comparisons in both the TS (Mc30-Mc34, p value = 0.000) and the FC (Mar03-Mar113, p value = 0.000) datasets. LD analyses were repeated using only unrelated individuals (according to the output from COLONY, see below), and these instances of LD disappeared, suggesting that the initial results were caused by related individuals sharing a higher number of alleles.
Kinship and dispersal
There were 81 capture events from 31 individuals during live-trapping in nine patches, with 50 recaptures observed in six patches. From the 115 genotypes obtained from the faecal samples (‘capture events’), we identified 65 individuals (‘captures’) in 14 patches, with 50 ‘recaptures’ observed in 11 of these patches (Table 2). In both the TS and FC datasets, none of the individuals dispersed outside the habitat patch (inter-patch dispersal). When pooling the 65 individuals (FC dataset) with the 31 tissue samples (TS dataset), we identified a total of 87 individuals (TS + FC dataset, Fig. 3a) in 15 habitat patches, with 109 ‘recaptures’ observed in 13 patches (Fig. 3b). Of these, 22 were ‘recaptures’ in the FC dataset of nine live-trapped individuals (TS dataset). The comparison of the consensus genotypes obtained for these nine individuals in the TS and FC datasets showed that only two of 22 genotypes in the FC dataset were different (one genotype had missing data at one locus and the other displayed dropout at two loci). From the overall ‘recaptures’ we identified one inter-patch dispersal event by a male to a nearby patch separated by 249 m (Fig. 4a).

Map of the study area, showing the location of individuals at: a first capture (white circles); b recapture (grey circles); combining data from both live-trapping and genetic non-invasive sampling (gNIS)

Location of females (white circles) and males (dark grey circles) identified from the 31 tissue and 115 faecal samples of Microtus cabrerae. Inter-patch dispersal events inferred by: a capture-recapture (TS + FC ═══) and kinship relations (TS + FC □ □ □) using all types of samples; b kinship relations for tissue (TS □□□) and faecal samples (FC □ □ □)
The parentage analysis in COLONY for the TS dataset revealed four related pairs of individuals (dyads), adding to the already described identical twins (total of five dyads, involving seven individuals out of the 31 individuals genotyped, 22.58%). These seven individuals clustered in three family groups (Online Resource 4), all consisting of siblings. Two siblings were found in two different patches at a distance of 177 m, corresponding to at least one possible inter-patch dispersal event (Table 2, Fig. 4b, Online Resource 4).
For the FC dataset, we identified 27 dyads in COLONY, involving 29 out of the 65 individuals genotyped (44.62%). We found 10 family groups among the 27 dyads, with two instances of paternity (father-offspring), four instances of maternity (mother-offspring) and 21 pairs of siblings (Online Resource 4). There were a minimum of four possible inter-patch dispersal events (91-245 m) detected in three family groups (Table 2, Fig. 4b, Online Resource 4).
For the TS + FC data, the genetic parentage analysis in COLONY recovered a total of 42 dyads (including the identical twins; five of these dyads were found in the TS dataset and 19 found in the FC dataset), involving 48 individuals out of the 87 genotyped (55.17%). We identified 18 family groups, in which five dyads involved paternity, three involved maternity and the remaining 34 were siblings (Online Resource 4). We found a total of six possible inter-patch dispersal events (121–377 m), including that already detected from the TS dataset and another from the FC dataset (Fig. 4a, Online Resource 4).
In the TS dataset, we found a significant difference (mean difference = −0.038, 95% confidence interval, CI = −0.036 to 0.036) in the mean relatedness (MR) among males (MR = −0.010, SE = 0.02) and females (MR = −0.048, SE = 0.03) in COANCESTRY, but no evidence of sex-biased dispersal in GenAlEx (Mann-Whitney U test: Z = 1.721, p = 0.085, two-tailed). For the FC and TS + FC datasets, no significant differences were found in the mean relatedness (FC: mean difference = −0.002, 95% confidence interval, CI = −0.018 to 0.018; TS + FC: mean difference = −0.005, 95% confidence interval, CI = −0.013 to 0.013), among males (FC: MR = −0.019, SE = 0.03; TS + FC: MR = −0.012, SE = 0.03) and females (FC: MR = −0.021, SE = 0.03; TS + FC: MR = −0.017, SE = 0.03) in COANCESTRY, nor in the mean AIc values (FC: Z = −0.364, p = 0.716, two-tailed; TS + FC: Z = 0.278, p = 0.393, two-tailed) obtained with GenAlEx.
Sampling costs and logistics
For live-trapping, a total of 21 days were spent in the field for capture-recapture sessions, representing a cost of 9224.00€ (Table 3). In the laboratory, a total of 37 h were spent analysing the 31 tissue samples (bench work and analysis), with a final cost of 1194.00€. These costs include all replicates and re-extractions done to confirm the genotypes and error rates. For gNIS, a total of five days were spent in the field for faecal sample collection, representing a cost of 2130.00€ (Table 3). A total of 118 h were spent in the laboratory for genotyping the faecal samples, with a final cost of 9722.00€. Considering the results obtained by each method, for live-trapping the estimated cost per individual (and respective genotype) was 336.06€. However, the costs dropped to 128.62€, when we consider the cost per capture event, since animals were tagged with a PIT tag which allowed recapture information to be obtained without genotyping. For gNIS, each genotype corresponds to a capture event, so the estimated cost per genotype and ‘capture’ event was 103.06€, while the cost per individual increased to 182.34€.
Discussion
Workflow and genotyping success
The easy identification of M. cabrerae faeces in our study area allowed for a targeted collection of faecal samples, thereby yielding a species identification accuracy of 99.4%. The only misidentification was a sample belonging to A. sapidus, which is a larger vole that occurs in the same habitat types (Pita et al. 2016), and from which the faeces of juveniles may be mistaken for those of adult M. cabrerae (Garrido-García and Soriguer 2014). This result ensures that failure in microsatellite genotyping was mostly due to low DNA yield rather than non-target sampling. There was also a low rate of conspecific contamination (1.9% of 323 samples), which likely resulted from our strategy to avoid collecting faeces from large and potentially communal latrines.
The genotyping success rate in gNIS (35.6%) was similar to nuclear amplification rates from another study with rodents (43%, Barbosa et al. 2013), but smaller when compared with other herbivorous mammals (e.g. Lepus americanus: 54–69%, Schwartz et al. 2007b; North African ungulates: 80%, Silva et al. 2015). This presumably reflects faecal pellet size and the impact that has on quantities and accessibility of the epithelial cells that provide DNA, with the smaller pellets of rodents resulting in a smaller quantity of DNA that can be extracted per sample. Besides faecal pellet size, the low genotyping success rate might also be due to the rapid DNA degradation owing to the dampness of the grassland habitat in which samples were collected. This is supported by the low genotyping success rates observed in other mammals inhabiting damp habitats (e.g. otters in streamside habitats: Lutra lutra: 41–46%, Prigioni et al. 2006, Lampa et al. 2013; Lontra canadensis: 24%, Mowry et al. 2011), or in mammals for which faeces were exposed to rainy conditions (Odocoileus virginianus: 28%, Goode et al. 2014). The presence of PCR inhibitors—which are common in herbivore faecal samples (Huber et al. 2002, Wehausen et al. 2004)—may also have contributed to reduced genotyping success, even in fresh samples with high DNA yield. In our study, the inhibitors were minimised by extracting only intact faeces and by avoiding vortexing during the digestion step, thus minimising the release of plant content and its inhibitors during the extraction protocol. However, novel extraction protocols may help decrease the PCR inhibitors while improving the DNA amplification rates, by removing inhibitors during the extraction process (Costa et al. 2017), or even reduce them during the collection process (Ramón-Laca et al. 2015).
In contrast to nuclear DNA, the mitochondrial gene cyt-b was successfully amplified in all reactions (n = 169). The higher success in mitochondrial DNA amplification was expected and is similar to that found in other studies, due to the much higher copy numbers of mitochondrial DNA molecules (Beja-Pereira et al. 2009).
When analysing non-invasive samples, it is important to screen for DNA quantity and quality, to avoid unnecessary laboratory costs on samples that will likely show high genotyping error rates and low amplification success with nuclear genes (Beja-Pereira et al. 2009; Lampa et al. 2013). Mitochondrial genes are often used in quality control owing to their greater amplification success rates because of high copy numbers in the cells. However, the higher success in mtDNA amplification may not reflect the quantity and quality of the nDNA present in the faecal samples. Therefore, we chose a small subset of 3 microsatellites (‘Mix 1’) for quality control, following initial optimisation tests where we observed that samples which failed to amplify ‘Mix 1’ also failed to amplify the remaining eight microsatellite loci and sex-linked introns (Online Resource 1). By creating a workflow where the faecal samples were initially screened for these three loci, we were able to restrict the remaining steps to the best quality samples, thereby reducing costs associated with genotyping (Fig. 2 and Online Resource 3). Future modifications to our workflow could include incorporation of real-time PCR (qPCR), with greater precision than simple PCR for quality control (although more expense as well). Another improvement with qPCR could be species specific primers located in the sex chromosomes, allowing species identification and sexing in a single reaction (Moran et al. 2008; O'Neill et al., 2013).
Overall, therefore, by taking a variety of methodological approaches aimed to maximise genotyping success in gNIS, we were able to establish a stepwise workflow starting from field sample collection to laboratory procedures and data analyses. Furthermore, we ensured low genotyping errors with a careful selection and optimisation of the loci for gNIS and the use of technical replicates. High genotyping error rates can result in incorrect genotypes, with subsequent errors in individual and population assignments and downstream analyses (Lampa et al. 2013). Therefore, we highlight the importance of establishing standardised protocols, including technical replication, to reduce bias (including possible human errors) during laboratory work and genotype scoring.
Genetic diversity, kinship and dispersal
Our results showed that either the set of 20 or 11 microsatellites (in tissue and faecal samples, respectively) are highly informative for estimating the genetic diversity and kinship in M. cabrerae. The estimate of average genetic diversity was similar when considering both sets of microsatellites, demonstrating that the smaller set (11) used for faecal samples was sufficient to provide accurate diversity values (Table 1). The metapopulation studied showed moderate to high levels of genetic diversity, with low inbreeding rates.
Overall, the use of gNIS doubled the number of individuals detected (n = 65) in comparison with live-trapping (n = 31). The higher number of individuals identified from the TS to the FC and TS + FC datasets also resulted in an increase in kinship relations detected (Table 2, Online Resource 4). Interestingly, we observed inconsistencies among the kinship relations detected with the FC and TS + FC datasets. The greater sample size in TS + FC might have increased the accuracy of the COLONY analysis, resulting in an overall consistency of the runs for this dataset (only 4 out of 6 runs were consistent in FC). Also incorporated into the analyses of the TS and TS + FC datasets was information drawn from live-trapping, such as the individual’s age class, and additional genetic data obtained from tissue samples (cyt-b haplotypes and genotypes from the X-linked locus). These parameters allowed the exclusion of unlikely dyads from the analysis in TS and TS + FC datasets in comparison to the FC dataset. Generalising from our findings and those of other studies (see Harrison et al. 2013), it is clear that the accuracy of kinship assignments can be improved by increased sampling effort, but this comes at increased cost per unique individual, because greater sampling leads to increased recapture rates. It is also clear that incorporation of a highly variable mitochondrial gene and sex-linked microsatellites is of value in kinship assignments (but again increasing the overall cost). Even better for precision of kinship assignments would be to incorporate high throughput sequencing to allow the capture of whole mitogenomes from faecal samples (van der Valk et al. 2017).
Despite the differences in kinship analyses results between datasets, dispersal distances obtained in the three datasets had overlapping results (TS: 177 m; FC: 91–245 m; TS + FC: 121–378 m). This allowed unseen dispersal events in the landscape to be detected with a maximum dispersal distance of 378 m. This maximum distance is however smaller than those previously detected through telemetry (448 m, Pita et al. 2010) or inferred from stochastic patch occupancy modelling (median: 1147–4837 m, Mestre et al. 2017), likely due to the small size of our study area.
We found no significant differences in sex-biased dispersal in all datasets, and the comparison of mean relatedness among sexes showed no evidence of a philopatric sex. The exception was the TS dataset, in which females were significantly less related than males. However, this was probably an artefact of small sample sizes, as other analysis using the same dataset did not provide evidence for sex-biased dispersal. Overall, lack of philopatry and sex-biased dispersal may be taken as an indication of social monogamy in mammals (Perrin et al. 2000; Lawson Handley and Perrin 2007; Le Galliard et al. 2012; Brom et al. 2016), which is in line with previous observations on the mating system of the Cabrera vole. In fact, social monogamy was previously suggested based on the lack of sexual dimorphism in the species (Ventura et al. 1998), as well as through studies on ecology and home ranges (Fernández-Salvador et al. 2001; Pita et al. 2010; Gomes et al. 2013). However, further research is required to confirm the genetic monogamy of M. cabrerae.
Effectiveness and cost
Using gNIS we were able to detect more individuals with less field effort than conventional live-trapping, which makes the method more efficient timewise for monitoring programs. In fact, using gNIS we detected on average 13.0 different individuals/day (23.0 ‘capture’ events/day, including ‘recaptures’), while live-trapping yielded only 1.5 different individuals/day (3.9 capture events/day) (Table 3). As a consequence, the final costs per obtained genotype, including total fieldwork and genetic analyses, were three times higher when using tissue samples obtained from live-trapping (336.06€) than when using gNIS (103.06€). However, the costs per ‘capture’ event were only slightly higher for live-trapping (128.62€) than for gNIS (103.06€). The higher costs of traditional tissue sampling were associated with the low capture rates observed during live-trapping. In contrast, faecal samples were easier to collect, took less time in the field and provided larger sample sizes, which resulted in more cost-effective field logistics. Thus, even though the laboratory analyses were cheaper and less labour intensive for tissue samples than for gNIS, because the good DNA quality of the tissue samples enables higher genotyping rates with less replicates per locus, gNIS is more cost-effective when considering the price per genotype, because a higher number of individuals were identified. The higher sample sizes obtained with gNIS thus produce better estimates of attributes based on genetic data of major applicability to conservation and monitoring programs, including sexing, kinship, dispersal and social behaviour.
The threshold at which the advantages of gNIS surpass those of live-trapping must be carefully assessed on a case-by-case basis. Based on the research questions and the required information, researchers should consider the time needed in the field for collecting samples and the number and type of samples to be analysed in the laboratory. These factors will impact the costs associated with each sampling strategy, but also the amount of information obtained. For instance, the cost-effectiveness of our gNIS study was high because the faecal samples of M. cabrerae were easy to identify and collect in the field. Also, the small latrines of M. cabrerae seem to be used almost always by a single individual, which makes genotyping easier than if there was conspecific or interspecific contamination. Population density may also affect the cost-effectiveness of gNIS. This is illustrated by two studies with lagomorphs evaluating the costs associated with each sampling method, with results similar to ours obtained for the endangered New England cottontail (Sylvilagus transitionalis) (Kilpatrick et al. 2013), while the cost-effectiveness of gNIS did not surpass that of live-trapping in high density populations of the snowshoe hare (Lepus americanus) (Cheng et al. 2017). Therefore, for abundant small mammal species with high capture rates, we suggest that live-trapping may still be the most cost-effective monitoring technique, as gNIS would not bring any further advantage for field and laboratory logistics, sample size and data analyses. However, this may change in the near future due to the ever declining costs and increasing performance of molecular analysis.
While dedicated laboratory facilities help during non-invasive amplification (e.g. separate room, UV lamps - see Beja-Pereira et al. 2009 and Lampa et al. 2013 for a detailed review), new high-throughput sequencing techniques could also improve the amount of data obtainable through gNIS, and avoid the issues encountered with microsatellites (Guichoux et al. 2011, Shafer et al. 2015). However, microsatellite markers are still widely used in gNIS, as the high-throughput methods require large quantities and good quality of DNA, hampering the implementation of widely used techniques such as genotyping-by-sequencing (Russello et al. 2015). Nevertheless, high-throughput sequencing has been applied for microsatellite genotyping of faecal samples in brown bears, and provided a higher amplification success with lower genotyping error rates than traditional Sanger sequencing (De Barba et al. 2017). High-throughput methods applied to microsatellites can help reduce the cost and time spent on the laboratory, as well as overcome the technical difficulties present in allele scoring, providing a second-life for microsatellites and improving the cost-effectiveness of gNIS for monitoring.
Conclusion
gNIS has been increasingly used in recent years, with many of its drawbacks being overcome by careful sample collection procedures and the optimisation of laboratory work, namely extraction protocols, use of species-specific genetic markers, and design of a proper workflow for increasing genotyping success. While rarely applied to small mammals, our study and the companion paper by Sabino-Marques et al. (2018) have shown that it is possible to use gNIS to monitor these species when presence signs are easily observed, with a clear overall cost-effectiveness compared with traditional live-trapping approaches. In particular, we found that using gNIS provides more information at reduced field effort and lower cost, thus allowing more precise estimates of population parameters such as genetic diversity, inbreeding, dispersal rates, and kinship structure, among others. Furthermore, this information can be obtained without the need to capture, handle and manipulate the individuals (tagging them or taking biopsies), which can be a great source of stress or even mortality (Powell and Proulx 2003; Pauli et al. 2010). Therefore, we propose that gNIS may generally provide a practical and ethically preferable alternative to live-trapping for elusive or rare species with low capture rates, reducing logistical difficulties and expenses. gNIS is thus a promising tool for monitoring small mammals, and we strongly suggest the consideration of this approach for threatened and rare species.
References
Agata K, Alasaad S, Almeida-Val VMF, Álvarez-Dios JA, Barbisan F, Beadell JS et al (2011) Permanent genetic resources added to molecular ecology resources database 1 December 2010–31 January 2011. Mol Ecol Resour 11:586–589. https://doi.org/10.1111/j.1755-0998.2011.03004.x
Barbosa S, Paupério J, Searle JB, Alves PC (2013) Genetic identification of Iberian rodent species using both mitochondrial and nuclear loci: application to noninvasive sampling. Mol Ecol Resour 13:43–56. https://doi.org/10.1111/1755-0998.12024
Barbosa S, Paupério J, Herman JS, Ferreira CM, Pita R, Vale-Gonçalves HM, Cabral JA, Garrido-García JA, Soriguer RC, Beja P, Mira A, Alves PC, Searle JB (2017) Endemic species may have complex histories: within-refugium phylogeography of an endangered Iberian vole. Mol Ecol 26:951–967. https://doi.org/10.1111/mec.13994
Beja-Pereira A, Oliveira R, Alves PC, Schwartz MK, Luikart G (2009) Advancing ecological understandings through technological transformations in noninvasive genetics. Mol Ecol Resour 9:1279–1301. https://doi.org/10.1111/j.1755-0998.2009.02699.x
Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Roy Stat Soc B Met 57:289–300. https://doi.org/10.2307/2346101
Brom T, Massot M, Legendre S, Laloi D (2016) Kin competition drives the evolution of sex-biased dispersal under monandry and polyandry, not under monogamy. Anim Behav 113:157–166. https://doi.org/10.1016/j.anbehav.2016.01.003
Cheng E, Hodges KE, Sollmann R, Mills LS (2017) Genetic sampling for estimating density of common species. Ecol Evol 7:6210–6219. https://doi.org/10.1002/ece3.3137
Chybicki IJ, Burczyk J (2009) Simultaneous estimation of null alleles and inbreeding coefficients. J Hered 100:106–113. https://doi.org/10.1093/jhered/esn088
Costa V, Rosenbom S, Monteiro R, O’Rourke SM, Beja-Pereira A (2017) Improving DNA quality extracted from fecal samples - a method to improve DNA yield. Eur J Wildl Res 63:3. https://doi.org/10.1007/s10344-016-1058-1
De Barba M, Waits LP, Garton EO, Genovesi P, Randi E, Mustoni A, Groff C (2010) The power of genetic monitoring for studying demography, ecology and genetics of a reintroduced brown bear population. Mol Ecol 19:3938–3951. https://doi.org/10.1111/j.1365-294X.2010.04791.x
De Barba M, Miquel C, Lobréaux S, Quenette PY, Swenson JE, Taberlet P (2017) High-throughput microsatellite genotyping in ecology: improved accuracy, efficiency, standardization and success with low-quantity and degraded DNA. Mol Ecol Resour 17:492–507. https://doi.org/10.1111/1755-0998.12594
Excoffier L, Lischer HEL (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10:564–567. https://doi.org/10.1111/j.1755-0998.2010.02847.x
Fernández-Salvador R, García-Perea R, Ventura J (2001) Reproduction and postnatal growth of the Cabrera vole, Microtus cabrerae, in captivity. Can J Zool 79:2080–2085. https://doi.org/10.1139/cjz-79-11-2080
Galan M, Pagès M, Cosson J-F (2012) Next-generation sequencing for rodent barcoding: species identification from fresh, degraded and environmental samples. PLoS One 7:e48374. https://doi.org/10.1371/journal.pone.0048374
Garrido-García JA, Soriguer RC (2014) Topillo de Cabrera Iberomys cabrerae (Thomas, 1906). Guía virtual de los indicios de los mamíferos de la Península Ibérica, Islas Baleares y Canarias. http://www.secem.es/wp-content/uploads/2015/07/020.-Iberomys-cabrerae.pdf Accessed 9 May 2017
Garrido-García JA, Soriguer-Escofet RC (2012) Cabrera’s vole Microtus cabrerae Thomas, 1906 and the subgenus Iberomys during the Quaternary: evolutionary implications and conservation. Geobios 45:437–444. https://doi.org/10.1016/j.geobios.2011.10.014
Gauffre B, Galan M, Bretagnolle V, Cosson JF (2007) Polymorphic microsatellite loci and PCR multiplexing in the common vole, Microtus arvalis. Mol Ecol Notes 7:830–832. https://doi.org/10.1111/j.1471-8286.2007.01718.x
Gillet F, Cabria MT, Némoz M, Blanc F, Fournier-Chambrillon C, Sourp E, Vial-Novella C, Aulagnier S, Michaux JR (2015) PCR-RFLP identification of the endangered Pyrenean desman, Galemys pyrenaicus (Soricomorpha, Talpidae), based on faecal DNA. Mammalia 79:473–477. https://doi.org/10.1515/mammalia-2014-0093
Gillet F, Bruno LR, Blanc F, Bodo A, Fournier-Chambrilon C, Fournier P, Jacob F, Lacaze V, Némoz M, Aulagnier S, Michaux JR (2016) Genetic monitoring of the endangered Pyrenean desman (Galemys pyrenaicus) in the Aude River, France. Belg J Zool 146:44–52
Gomes LAP, Salgado PMP, Barata EN, Mira APP (2013) The effect of pair bonding in Cabrera vole’s scent marking. Acta Ethol 16:181–188. https://doi.org/10.1007/s10211-013-0151-7
Goode MJ, Beaver JT, Muller LI, Clark JD, Manen FT, Harper CA, Basinger PS (2014) Capture-recapture of white-tailed deer using DNA from fecal pellet groups. Wildlife Biol 20:270–278. https://doi.org/10.2981/wlb.00050
Grenier MB, Buskirk SW, Anderson-Sprecher R (2009) Population indices versus correlated density estimates of black-footed ferret abundance. J Wildl Manag 73:669–676. https://doi.org/10.2193/2008-269
Guichoux E, Lagache L, Wagner S, Chaumeil P, Léger P, Lepais O, Lepoittevin C, Malausa T, Revardel E, Salin F, Petit RJ (2011) Current trends in microsatellite genotyping. Mol Ecol Resour 11:591–611. https://doi.org/10.1111/j.1755-0998.2011.03014.x
Gulsby WD, Killmaster CH, Bowers JW, Laufenberg JS, Sacks BN, Statham MJ, Miller KV (2016) Efficacy and precision of fecal genotyping to estimate coyote abundance. Wildl Soc Bull 40:792–799. https://doi.org/10.1002/wsb.712
Harrison HB, Saenz-Agudelo P, Planes S, Jones GP, Berumen ML (2013) Relative accuracy of three common methods of parentage analysis in natural populations. Mol Ecol 22:1158–1170. https://doi.org/10.1111/mec.12138
Henry P, Russello MA (2011) Obtaining high-quality DNA from elusive small mammals using low-tech hair snares. Eur J Wildl Res 57:429–435. https://doi.org/10.1007/s10344-010-0449-y
Huber S, Bruns U, Arnold W (2002) Sex determination of red deer using polymerase chain reaction of DNA from feces. Wildl Soc Bull 30:208–212. https://doi.org/10.2307/3784655
Ishibashi Y, Yoshinaga Y, Saitoh T, Abe S, Iida H, Yoshida MC (1999) Polymorphic microsatellite DNA markers in the field vole Microtus montebelli. Mol Ecol 8:163–164
Jaarola M, Searle JB (2002) Phylogeography of field voles (Microtus agrestis) in Eurasia inferred from mitochondrial DNA sequences. Mol Ecol 11:2613–2621. https://doi.org/10.1046/j.1365-294X.2002.01639.x
Jaarola M, Searle JB (2004) A highly divergent mitochondrial DNA lineage of Microtus agrestis in southern Europe. Heredity 92:228–234. https://doi.org/10.1038/sj.hdy.6800400
Jaarola M, Ratkiewicz M, Ashford RT, Brunhoff C, Borkowska A (2007) Isolation and characterization of polymorphic microsatellite loci in the field vole, Microtus agrestis, and their cross-utility in the common vole, Microtus arvalis. Mol Ecol Notes 7:1029–1031. https://doi.org/10.1111/j.1471-8286.2007.01763.x
Johnson PCD, Haydon DT (2007) Maximum-likelihood estimation of allelic dropout and false allele error rates from microsatellite genotypes in the absence of reference data. Genetics 175:827–842. https://doi.org/10.1534/genetics.106.064618
Jones OR, Wang J (2010) COLONY: a program for parentage and sibship inference from multilocus genotype data. Mol Ecol Resour 10:551–555. https://doi.org/10.1111/j.1755-0998.2009.02787.x
Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, Thierer T, Ashton B, Meintjes P, Drummond A (2012) Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28:1647–1649. https://doi.org/10.1093/bioinformatics/bts199
Kilpatrick HJ, Goodie TJ, Kovach AI (2013) Comparison of live-trapping and noninvasive genetic sampling to assess patch occupancy by New England cottontail (Sylvilagus transitionalis) rabbits. Wildl Soc Bull 37:901–905. https://doi.org/10.1002/wsb.330
Kuch M, Rohland N, Betancourt JL, Latorre C, Steppan S, Poinar HN (2002) Molecular analysis of a 11 700-year-old rodent midden from the Atacama Desert, Chile. Mol Ecol 11:913–924. https://doi.org/10.1046/j.1365-294X.2002.01492.x
Lampa S, Henle K, Klenke R, Hoehn M, Gruber B (2013) How to overcome genotyping errors in non-invasive genetic mark-recapture population size estimation—a review of available methods illustrated by a case study. J Wildl Manag 77:1490–1511. https://doi.org/10.1002/jwmg.604
Laplana C, Sevilla P (2013) Documenting the biogeographic history of Microtus cabrerae through its fossil record. Mammal Rev 43:309–322. https://doi.org/10.1111/mam.12003
Lawson Handley LJ, Perrin N (2007) Advances in our understanding of mammalian sex-biased dispersal. Mol Ecol 16:1559–1578. https://doi.org/10.1111/j.1365-294X.2006.03152.x
Le Galliard J-F, Rémy A, Ims RA, Lambin X (2012) Patterns and processes of dispersal behaviour in arvicoline rodents. Mol Ecol 21:505–523. https://doi.org/10.1111/j.1365-294X.2011.05410.x
Lindenmayer DB, Fischer J (2013) Habitat fragmentation and landscape change: an ecological and conservation synthesis. Island Press, Washington
Lounsberry ZT, Forrester TD, Olegario MT, Brazeal JL, Wittmer HU, Sacks BN (2015) Estimating sex-specific abundance in fawning areas of a high-density Columbian black-tailed deer population using fecal DNA. J Wildl Manag 79:39–49. https://doi.org/10.1002/jwmg.817
Marucco F, Boitani L, Pletscher DH, Schwartz MK (2011) Bridging the gaps between non-invasive genetic sampling and population parameter estimation. Eur J Wildl Res 57:1–13. https://doi.org/10.1007/s10344-010-0477-7
Mateo-Sánchez MC, Balkenhol N, Cushman S, Pérez T, Domínguez A, Saura S (2015) Estimating effective landscape distances and movement corridors: comparison of habitat and genetic data. Ecosphere 6:1–16. https://doi.org/10.1890/ES14-00387.1
McKelvey KS, Schwartz MK (2004) Genetic errors associated with population estimation using non-invasive molecular tagging: problems and new solutions. J Wildl Manag 68:439–448. https://doi.org/10.2193/0022-541X(2004)068[0439:GEAWPE]2.0CO;2
Melosik I, Ziomek J, Winnicka K, Reiners TE, Banaszek A, Mammen K, Mammen U, Marciszak A (2017) The genetic characterization of an isolated remnant population of an endangered rodent (Cricetus cricetus L.) using comparative data: implications for conservation. Conserv Genet 18:759–775. https://doi.org/10.1007/s10592-017-0925-y
Mestre F, Pita R, Paupério J, Martins FMS, Alves PC, Mira A, Beja P (2015) Combining distribution modelling and non-invasive genetics to improve range shift forecasting. Ecol Model 297:171–179. https://doi.org/10.1016/j.ecolmodel.2014.11.018
Mestre F, Risk BB, Mira A, Beja P, Pita R (2017) A metapopulation approach to predict species range shifts under different climate change and landscape connectivity scenarios. Ecol Model 359:406–414. https://doi.org/10.1016/j.ecolmodel.2017.06.013
Miller MP, Bellinger MR, Forsman ED, Haig SM (2006) Effects of historical climate change, habitat connectivity, and vicariance on genetic structure and diversity across the range of the red tree vole (Phenacomys longicaudus) in the Pacific Northwestern United States. Mol Ecol 15:145–159. https://doi.org/10.1111/j.1365-294X.2005.02765.x
Mills LS, Citta JJ, Lair KP, Schwartz MK, Tallmon DA (2000) Estimating animal abundance using noninvasive DNA sampling: promise and pitfalls. Ecol Appl 10:283–294. https://doi.org/10.1890/1051-0761(2000)010[0283:EAAUND]2.0.CO;2
Moran S, Turner PD, O’Reilly C (2008) Non-invasive genetic identification of small mammal species using real-time polymerase chain reaction. Mol Ecol Resour 8:1267–1269. https://doi.org/10.1111/j.1755-0998.2008.02324.x
Mossman CA, Waser PM (1999) Genetic detection of sex-biased dispersal. Mol Ecol 8:1063–1067. https://doi.org/10.1046/j.1365-294x.1999.00652.x
Mowry RA, Gompper ME, Beringer J, Eggert LS (2011) River otter population size estimation using noninvasive latrine surveys. J Wildl Manag 75:1625–1636. https://doi.org/10.1002/jwmg.193
Mumma MA, Zieminski C, Fuller TK, Mahoney SP, Waits LP (2015) Evaluating noninvasive genetic sampling techniques to estimate large carnivore abundance. Mol Ecol Resour 15:1133–1144. https://doi.org/10.1111/1755-0998.12390
O'Neill D, Turner PD, O'Meara DB, Chadwick EA, Coffey L, O'Reilly C (2013) Development of novel real-time TaqMan® PCR assays for the species and sex identification of otter (Lutra lutra) and their application to noninvasive genetic monitoring. Mol Ecol Resour 13:877–883. https://doi.org/10.1111/1755-0998.12141
Pauli JN, Whiteman JP, Riley MD, Middleton AD (2010) Defining noninvasive approaches for sampling of vertebrates. Conserv Biol 24:349–352. https://doi.org/10.1111/j.1523-1739.2009.01298.x
Peakall R, Smouse PE (2012) GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research—an update. Bioinformatics 28:2537–2539. https://doi.org/10.1093/bioinformatics/bts460
Perrin N, Mazalov V, Otto AESP (2000) Local competition, inbreeding, and the evolution of sex-biased dispersal. Am Nat 155:116–127. https://doi.org/10.1086/303296
Pita R, Mira A, Beja P (2006) Conserving the Cabrera vole, Microtus cabrerae, in intensively used Mediterranean landscapes. Agric Ecosyst Environ 115:1–5. https://doi.org/10.1016/j.agee.2005.12.002
Pita R, Beja P, Mira A (2007) Spatial population structure of the Cabrera vole in Mediterranean farmland: the relative role of patch and matrix effects. Biol Conserv 134:383–392. https://doi.org/10.1016/j.biocon.2006.08.026
Pita R, Mira A, Beja P (2010) Spatial segregation of two vole species (Arvicola sapidus and Microtus cabrerae) within habitat patches in a highly fragmented farmland landscape. Eur J Wildl Res 56:651–662. https://doi.org/10.1007/s10344-009-0360-6
Pita R, Mira A, Beja P (2014) Microtus cabrerae (Rodentia: Cricetidae). Mamm Species 46(912):48–70. https://doi.org/10.1644/912.1
Pita R, Lambin X, Mira A, Beja P (2016) Hierarchical spatial segregation of two Mediterranean vole species: the role of patch-network structure and matrix composition. Oecologia 182:253–263. https://doi.org/10.1007/s00442-016-3653-y
Powell RA, Proulx G (2003) Trapping and marking terrestrial mammals for research: integrating ethics, performance criteria, techniques, and common sense. ILAR J 44:259–276. https://doi.org/10.1093/ilar.44.4.259
Prigioni C, Remonti L, Balestrieri A, Sgrosso S, Priore G, Mucci N, Randi E (2006) Estimation of European otter (Lutra lutra) population size by fecal DNA typing in southern Italy. J Mammal 87:855–858. https://doi.org/10.1644/05-MAMM-A-294R1.1
Queller DC, Goodnight KF (1989) Estimating relatedness using genetic markers. Evolution 43:258–275. https://doi.org/10.2307/2409206
Ramón-Laca A, Soriano L, Gleeson D, Godoy JA (2015) A simple and effective method for obtaining mammal DNA from faeces. Wildlife Biol 21:195–203. https://doi.org/10.2981/wlb.00096
Rousset F (2008) genepop’007: a complete re-implementation of the genepop software for Windows and Linux. Mol Ecol Resour 8:103–106. https://doi.org/10.1111/j.1471-8286.2007.01931.x
Russello MA, Waterhouse MD, Etter PD, Johnson EA (2015) From promise to practice: pairing non-invasive sampling with genomics in conservation. PeerJ 3:e1106. https://doi.org/10.7717/peerj.1106
Sabino-Marques H, Fereira CM, Paupério J, Costa P, Barbosa S, Encarnação C, Alpizar-Jara R, Alves PC, Searle JB, Mira A, Beja P, Pita R (2018) Combining genetic non-invasive sampling with spatially explicit capture-recapture models for density estimation of a patchily distributed small mammal. Eur J Wildl Res. https://doi.org/10.1007/s10344-018-1206-x
Schmidt K, Davoli F, Kowalczyk R, Randi E (2016) Does kinship affect spatial organization in a small and isolated population of a solitary felid: the Eurasian lynx? Integr Zool 11:334–349. https://doi.org/10.1111/1749-4877.12182
Schwartz MK, Luikart G, Waples RS (2007a) Genetic monitoring as a promising tool for conservation and management. Trends Ecol Evol 22:25–33. https://doi.org/10.1016/j.tree.2006.08.009
Schwartz MK, Pilgrim KL, McKelvey KS, Rivera PT, Ruggiero LF (2007b) DNA markers for identifying individual snowshoe hares using field-collected pellets. Northwest Sci 81:316–322. https://doi.org/10.3955/0029-344X-81.4.316
Shafer ABA, Wolf JBW, Alves PC, Bergstörm L, Bruford MW, Brännström I et al (2015) Genomics and the challenging translation into conservation practice. Trends Ecol Evol 30:78–87. https://doi.org/10.1016/j.tree.2014.11.009
Sikes RS, Gannon WL, Animal Care and Use Committee of the American Society of Mammalogists (2011) Guidelines of the American Society of Mammalogists for the use of wild mammals in research. J Mammal 92:235–253. https://doi.org/10.1644/10-MAMM-F-355.1
Silva TL, Godinho R, Castro D, Abáigar T, Brito JC, Alves PC (2015) Genetic identification of endangered north African ungulates using noninvasive sampling. Mol Ecol Resour 15:652–661. https://doi.org/10.1111/1755-0998.12335
Sollmann R, Tôrres NM, Furtado MM, Almeida AT, Palomares F, Roques S, Silveira L (2013) Combining camera-trapping and noninvasive genetic data in a spatial capture–recapture framework improves density estimates for the jaguar. Biol Conserv 167:242–247. https://doi.org/10.1016/j.biocon.2013.08.003
Stansbury CR, Ausband DE, Zager P, Mack CM, Waits LP (2016) Identifying gray wolf packs and dispersers using noninvasive genetic samples. J Wildl Manag 80:1408–1419. https://doi.org/10.1002/jwmg.21136
Stanton DWG, Hart J, Kümpel NF, Vosper A, Nixon S, Bruford MW, Ewen JG, Wang J (2015) Enhancing knowledge of an endangered and elusive species, the okapi, using non-invasive genetic techniques. J Zool 295:233–242. https://doi.org/10.1111/jzo.12205
Swift TL, Hannon SJ (2010) Critical thresholds associated with habitat loss: a review of the concepts, evidence, and applications. Biol Rev 85:35–53. https://doi.org/10.1111/j.1469-185X.2009.00093.x
Taylor HR, Colbourne RM, Robertson HA, Nelson NJ, Allendorf FW, Ramstad KM (2017) Cryptic inbreeding depression in a growing population of a long-lived species. Mol Ecol 26:799–813. https://doi.org/10.1111/mec.13977
Thompson W (2013) Sampling rare or elusive species: concepts, designs, and techniques for estimating population parameters. Island Press, Washington
Trinca CS, Jaeger CF, Eizirik E (2013) Molecular ecology of the Neotropical otter (Lontra longicaudis): non-invasive sampling yields insights into local population dynamics. Biol J Linn Soc 109:932–948. https://doi.org/10.1111/bij.12077
Valière N (2002) Gimlet: a computer program for analysing genetic individual identification data. Mol Ecol Notes 2:377–379. https://doi.org/10.1046/j.1471-8286.2002.00228.x-i2
van der Valk T, Lona Durazo F, Dalén L, Guschanski K (2017) Whole mitochondrial genome capture from faecal samples and museum-preserved specimens. Mol Ecol Resour 17:e111–e121. https://doi.org/10.1111/1755-0998.12699
Ventura J, López-Fuster MJ, Cabrera-Millet M (1998) The Cabrera vole, Microtus cabrerae, in Spain: a biological and morphometric approach. Neth J Zool 48:83–100. https://doi.org/10.1163/156854298X00237
Waits LP, Paetkau D (2005) Noninvasive genetic sampling tools for wildlife biologists: a review of applications and recommendations for accurate data collection. J Wildl Manag 69:1419–1433. https://doi.org/10.2193/0022-541X(2005)69[1419:NGSTFW]2.0.CO;2
Waits LP, Luikart G, Taberlet P (2001) Estimating the probability of identity among genotypes in natural populations: cautions and guidelines. Mol Ecol 10:249–256. https://doi.org/10.1046/j.1365-294X.2001.01185.x
Walker RS, Novaro AJ, Perovic P, Palacios R, Donadio E, Lucherini M, Pia M, López MS (2007) Diets of three species of Andean carnivores in high-altitude deserts of Argentina. J Mammal 88:519–525. https://doi.org/10.1644/06-MAMM-A-172R.1
Walser B, Heckel G (2007) Microsatellite markers for the common vole (Microtus arvalis) and their cross-species utility. Conserv Genet 9:479–481. https://doi.org/10.1007/s10592-007-9355-6
Walsh SE, Woods WE, Hoffman SM (2016) Effects of range contraction and habitat fragmentation on genetic variation in the woodland deer mouse (Peromyscus maniculatus gracilis). Am Midl Nat 176:272–281. https://doi.org/10.1674/0003-0031-176.2.272
Wang J (2011) COANCESTRY: a program for simulating, estimating and analysing relatedness and inbreeding coefficients. Mol Ecol Resour 11:141–145. https://doi.org/10.1111/j.1755-0998.2010.02885.x
Watkins AF, McWhirter JL, King CM (2010) Variable detectability in long-term population surveys of small mammals. Eur J Wildl Res 56:261–274. https://doi.org/10.1007/s10344-009-0308-x
Wehausen JD, Ramey RR, Epps CW (2004) Experiments in DNA extraction and PCR amplification from bighorn sheep feces: the importance of DNA extraction method. J Hered 95:503–509. https://doi.org/10.1093/jhered/esh068
Woodroffe GL, Lawton JH, Davidson WL (1990) Patterns in the production of latrines by water voles (Arvicola terrestris) and their use as indices of abundance in population surveys. J Zool 220:439–445. https://doi.org/10.1111/j.1469-7998.1990.tb04317.x
Woodruff SP, Lukacs PM, Christianson D, Waits LP (2016) Estimating Sonoran pronghorn abundance and survival with fecal DNA and capture–recapture methods. Conserv Biol 30:1102–1111. https://doi.org/10.1111/cobi.12710
Zimmerman M, Oddy D, Stolen E, Breininger D, Pruett CL (2015) Microspatial sampling reveals cryptic influences on gene flow in a threatened mammal. Conserv Genet 16:1403–1414. https://doi.org/10.1007/s10592-015-0749-6
Acknowledgements
This study was funded by Fundo Europeu de Desenvolvimento Regional (FEDER) funds through the Programa Operacional Factores de Competitividade (COMPETE), and national funds through the Portuguese Foundation for Science and Technology (FCT), within the scope of the projects PERSIST (PTDC/BIA-BEC/105110/2008), NETPERSIST (PTDC/AAG-MAA/3227/2012), and MateFrag (PTDC/BIA-BIC/6582/2014). This study was supported by FCT individual grants (HSM - SFRH/BD/73765/2010; SB - SFRH/BD/77726/2011, RP - SFRH/BPD/73478/2010 and SFRH/BPD/109235/2015) and an EDP Biodiversity Chair (PB). JP was supported by a postdoctoral grant funded by the project ‘Genomics and Evolutionary Biology’ co-financed by North Portugal Regional Operational Programme 2007/2013 (ON.2 - O Novo Norte), under the National Strategic Reference Framework, through the ERDF and by the European Union’s Horizon 2020 research and innovation programme under project EnvMetaGen (grant agreement no 668981). We would like to thank Susana Lopes, Patrícia Ribeiro, Sofia Mourão, Diana Castro and Pedro Cardoso for all their help in laboratory work. We are also grateful to the anonymous reviewers for their valuable comments.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed. All procedures were performed according to Portuguese law, under permits no. 76, 77 and 80/2011/CAPT from the Portuguese biodiversity conservation agency (ICNF – Instituto de Conservação da Natureza e das Florestas) and conformed to the guidelines approved by the American Society of Mammalogists (Sikes et al. 2011).
Additional information
Data accessibility
Microsatellite sequences are deposited at GenBank with the accession numbers MH264520-MH264530; detailed information on samples and genotypes are deposited at OSF (doi: 10.17605/OSF.IO/38RUW)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Ferreira, C.M., Sabino-Marques, H., Barbosa, S. et al. Genetic non-invasive sampling (gNIS) as a cost-effective tool for monitoring elusive small mammals. Eur J Wildl Res 64, 46 (2018). https://doi.org/10.1007/s10344-018-1188-8
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10344-018-1188-8