
Abstract
Esophageal squamous cell carcinoma (ESCC) remains a serious health concern due to its high prevalence and mortality rates. Identifying prognostic biomarkers is essential to improving patient outcomes and treatment strategies. DOCK9, a gene implicated in various cellular functions, may play a significant role in ESCC progression and prognosis. We analyzed RNA microarray datasets and single-cell RNA sequencing data to identify survival-associated genes in ESCC. Using protein expression analysis, we examined DOCK9 in ESCC tissues and assessed its functional impact on human umbilical vein endothelial cells to understand its role in angiogenesis. Additionally, we developed a 21-gene prognostic risk model, focusing on the relevance of DOCK9. Our findings revealed that DOCK9 expression is significantly reduced in ESCC tissues and correlates with poor survival outcomes. Functionally, DOCK9 was found to regulate angiogenesis and modulate the tumor-associated fibroblast environment in ESCC. Furthermore, the DOCK9/CD31 ratio emerged as a potential marker to predict immune therapy response in ESCC. DOCK9 serves as a prognostic biomarker in ESCC, influencing both angiogenesis and immune response, and could guide future therapeutic strategies, particularly in immunotherapy. This study highlights DOCK9's relevance in ESCC prognosis, supporting its potential role in tailored therapies aimed at angiogenesis and immune modulation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Esophageal cancer (EC) is a highly prevalent malignancy worldwide, ranking as the seventh most common cancer and resulting in over 500,000 deaths annually [1]. EC is classified into two primary subtypes: esophageal squamous cell carcinoma (ESCC) and esophageal adenocarcinoma (EAC) [2]. ESCC accounts for approximately 90% of EC cases in China, while EAC is more common in Western countries [3,4,5]. ESCC is notorious for its high predisposition toward vascular invasion and lymph node metastasis [6], this makes the development of novel therapeutic interventions and early risk assessment crucial.
Targeted therapies have shown promising results in the management of ESCC. Treatments like cetuximab and nimotuzumab, which target the epidermal growth factor receptor (EGFR) [7, 8]; bevacizumab and apatinib, which target vascular endothelial growth factor (VEGF) [9,10,11] have all demonstrated significant efficacy in the treatment of ESCC. Additionally, immune checkpoint inhibitors have ushered in a new era in oncology, establishing themselves as a crucial fifth pillar of cancer treatment [12]. The advancement and application of immune checkpoint inhibitors (ICIs) in gastric cancer have sparked growing enthusiasm for combination strategies [13]. Recently, combinations of immuno-oncology therapies have emerged as a groundbreaking and highly effective strategy for treating cancer, leading to unprecedented response rates and survival benefits across various tumor types [14]. Combination therapy using different platinum-based drugs and immune checkpoint inhibitors (ICIs) exhibits varying levels of efficacy. The use of oxaliplatin, in particular, has shown a more potent synergistic antitumor effect when combined with ICIs compared to cisplatin, likely due to its ability to induce immunogenic cell death (ICD) in tumor cells [15]. There has been rapid progress in the field of tumor immunotherapy, with immune checkpoint inhibitors enhancing the anti-cancer immune response by suppressing co-inhibitory pathways that tumors exploit to evade immune surveillance [16]. Significantly transforming the therapeutic approach for ESCC [17]. Although immune checkpoint inhibitors such as Nivolumab [18], Camrelizumab [19], Pembrolizumab [20] which target PD-1/PD-L1, have shown promising results in ESCC therapy, a substantial number of patients are still unable to benefit from these immunotherapies. As such, the development of more effective immune checkpoint inhibitors is of utmost importance.
The Dedicator of Cytokinesis (DOCK) protein family, known for its role as activators of Rho GTPases—notably Rac and Cdc42 [21, 22], is classified into four distinct categories: DOCK-A, DOCK-B, DOCK-C, and DOCK-D [23]. These proteins modulate several cellular mechanisms, including forming the actin cytoskeleton, cellular adhesion, and migration [24, 25]. The Dedicator of Cytokinesis 9 (DOCK9), a member of the DOCK-D family, acts as a guanine nucleotide exchange factor (GEF) for Cdc42 and Rac1 and induces filopodia [26]. The literature on DOCK9 is limited, particularly in its relationship with cancer. Alkhateeb et al. suggested DOCK9 as a potential biomarker for predicting prostate cancer progression, especially from stage II onwards [27]. Concurrently, Zhu et al. demonstrated a correlation between DOCK9 and an increased risk of pancreatic adenocarcinoma [28]. However, the specific physiological function of the DOCK9 protein, particularly in ESCC, is not fully understood. As such, additional research is needed to uncover the mechanisms underlying the role of DOCK9 in cancer biology.
In this study, we employ bioinformatics analysis to evaluate the risk genes of survival rates in patients with ESCC. Furthermore, we investigate the potential therapeutic value of DOCK9 for ESCC patients by exploring its angiogenic and immune regulatory functions.
Methods and materials
Patients tissue collection
Six pairs of ESCC (Tumor) and adjacent non-tumor tissue were obtained from surgical resections of patients without preoperative treatment at People’s Hospital Affiliated to Jiangsu University (Zhenjiang, China). Surgically removed tissues were frozen in liquid nitrogen and stored at − 80 °C until analysis. Two pathologists independently identified all the resected samples. The patient's information sheet is shown in Supplementary Table 1.
Data source and differential gene expression (DEG) analysis
This study utilized RNA microarray data (GSE67269, GSE20347, GSE53625) and single-cell RNA-seq data (GSE160269, GSE188900) of ESCC patients, RNA-seq data GSE83299 of human dermal lymphatic endothelial cells (HDLECs), RNA microarray data GSE77597 of human umbilical vein endothelial cells (HUVECs) which were all acquired from the Gene Expression Omnibus (GEO) database (www.ncbi.nlm.nih.gov/geo). TCGA (The Cancer Genome Atlas) ESCC data were downloaded from the TCGA (https://tcga-data.nci.nih.gov/tcga/). The abundance of DOCK9 in multi-scRNA-seq of various tumors comes from the IMMUcan scDB database [29] (https://immucanscdb.vital-it.ch). The selection of each dataset was based on its unique insights into the pathology of ESCC and endothelial cell function, aimed at unveiling the potential role of DOCK9 in tumor development. All datasets were used by their respective databases' usage agreements and ethical review requirements. Data sources and basic information are provided in Supplementary Table 2.
Differential analysis of these datasets was carried out using various computational tools: the limma package for RNA microarray data, the edgeR package for RNA-seq data, and the FindMarkers function in the Seurat package for single-cell RNA-seq data. The criteria for differential gene expression were established as a false discovery rate (FDR) or p value of less than 0.05 and an absolute log2 fold change (|log2FC|) greater than 1 for RNA microarray and RNA-seq data. For single-cell RNA-seq data, the cutoff was set at an absolute log2FC greater than 0.4, with a p value less than 0.05. The top 50% was used as the high-expression group for high and low-group comparisons.
Least absolute shrinkage and selection operator (LASSO) regression analysis
A total of 43 genes selected from GSE53625, GSE67269, and GSE20347 of ESCC patients were subjected to LASSO regression analysis using the glmnet package. The goal was to identify prognostic biomarkers.
Independent risk factors for overall survival and nomogram construction
Univariate and multivariate Cox regression analyses were conducted to identify independent risk factors for ESCC prognosis. Hazard ratios (HR) and 95% confidence intervals (CI) were calculated using the survival package in R. The results of the multivariate analysis were utilized to develop a nomogram for predicting 1, 3, and 5-year overall survival (OS) using the rms package in R. The predictive accuracy of the nomogram was assessed using the C-index and calibration plot.
Enrichment analysis
Gene Set Enrichment Analysis (GSEA) (version 4.3.2) was used to analyze patients in the GSE53625 dataset. Default weighted enrichment statistics with 1,000 random combinations were utilized. The significance criteria were set as p < 0.05, normalized enrichment score (NES) > 1.3, and FDR < 0.25.
Enrichment analysis of genes using the Metascape (http://metascape.org) [30] corrected p < 0.05 were considered significantly enriched.
Single-cell RNA-seq data analysis
The Seurat package (https://satijalab.org/seurat/, version 4.0.4) in R was utilized for the analysis of the single-cell RNA-seq data from the GSE160269 [31] and GSE188900 [32] datasets. Cell subtype annotations were made based on the original study's description and using highly expressed marker genes or well-known functional genes. For cell-filtering, parameters included nFeature_RNA > 1000, nFeature_RNA < 5000, and percent mt < 10%. The functions FindClusters and FindNeighbors in Seurat, with a resolution of 0.8, were used for cluster identification, using the markers from the original data classification. The harmony package was employed for the integration of scRNA-seq data. We conducted a weighted kernel density estimation of gene expression using the plot_density function in the Nebulosa package (v1.6.0) [33] with default parameters. We calculated GSVA scores by the R package GSVA and gene sets from "c2.cp.kegg.v7.4.-symbols," which were downloaded from the MSigDB database. We conducted a cell–cell communication analysis utilizing the CellChat (version 1.5) R package (https://github.com/sqjin/CellChat) with default parameters. The probability value signifies the quantitative strength of a particular ligand–receptor interaction. It is computed based on the law of mass action, which depends on the average expression of the ligand in one cell group and the receptor in another. For this analysis, endothelial cells with normalized DOCK9 RNA data > 1 were categorized as the high DOCK9 group, while those with normalized DOCK9 RNA data ≤ 1 were assigned to the low DOCK9 group.
Immunity analysis
The CIBERSORT algorithm was employed to examine the immune cell composition in samples with high and low levels of DOCK9 in TCGA-ESCC and GSE53625 datasets. A comparison of immune checkpoint gene expression levels was conducted between samples with high levels of DOCK9/CD31 and low levels of DOCK9/CD31 in both TCGA-ESCC and GSE53625 datasets. The Immunity Score was calculated using the ESTIMATE algorithm in TCGA-ESCC and GSE53625 datasets. A significance level of p < 0.05 was considered statistically significant. It is important to note that the high-level and low-level groups were defined using a 50% cutoff.
Western blotting
Patients' tissue proteins (ESCC and adjacent non-cancerous tissue samples) and cellular proteins (HUVEC) were extracted using RIPA Lysis Buffer (Beyotime, China), and the protein concentration was determined using the BCA protein assay kit (Beyotime, China). Subsequently, the protein samples were separated on 8%-12% SDS-PAGE gels and transferred to PVDF membranes (Millipore, USA). After blocking with 5% non-fat milk for 2–3 h at room temperature, the membranes were incubated overnight at 4 °C with specific primary antibodies: DOCK9 (proteintech, 18,987–1-AP, 1:1000), β-ACTIN (proteintech, 81115-1-RR,1:5000), and VEGFA (abcam, ab46154, 1:000). Following the primary antibody incubation, the membranes were incubated with secondary antibodies for 1 h. Finally, the protein blots were visualized using the ECL kit (Millipore, Germany).
HUVECs culture and transfection
HUVECs were obtained from the China Center for Type Culture Collection (CCTCC) and cultured in high-glucose DMEM (HiClone, USA) at 37 °C in a cell incubator with 5% CO2. Recombinant human Ang-2 (200 ng/ml [34], R&D Systems, MN, USA) was added to cells for 24 h to activate HUVECs, and all subsequent cell function experiments were performed after ANG-2 stimulation. When the cell density reached 50–70%, the cells were transfected with 20 nM si-DOCK9 (GEMA, Shanghai, China) using PolyPlus transfection reagent (Illkirch, France) and 10% fetal bovine serum (FBS, Gibco) in the complete medium. The medium was replaced 24 h after transfection.
Tube formation assay
The tube formation assay was performed to assess cellular angiogenic function. Matrigel (Corning, USA) and 10% FBS complete medium were mixed at a 1:1 ratio and added to a 24-well plate. Tube formation was evaluated using an inverted microscope (Olympus, IX73). Image quantification was performed using Image J software.
Wound healing assay
After the HUVECs were treated and reached confluence, a scratch was made in the cell monolayer using a 200 μl pipette tip. The cells were then washed with PBS, and images of the scratch area were captured with an inverted microscope (Olympus, IX73) using a 10 × objective. The scratched areas were photographed and marked for each well. The same scratched areas were re-photographed after a 24-h incubation in a 2% FBS medium. The wound closure was measured using Image J software.
CCK-8 assay
HUVEC cells were plated in a 96-well plate at a density of 1 × 104 cells/well. After 48 h of treatment with either Si-NC or si-DOCK9, a CCK-8 solution (Beijing Solarbio Science & Technology Co., Ltd.) was added to each well, and the cells were further incubated for two hours. The absorbance at 450 nm was measured using a microplate reader.
EdU staining assay
HUVECs were treated with either si-NC or si-DOCK9 for 48 h and then exposed to EdU solution (Thermo Fisher Scientific, Inc.). The cells were fixed with 4% paraformaldehyde and permeabilized with 0.5% Triton X-100. The cells were incubated with a reaction mixture for 20 min, followed by DAPI staining for 30 min in the dark. The images were observed under a fluorescence microscope (Olympus Corporation, Tokyo, Japan).
Tissue immunohistochemistry (IHC)
All patients gave informed consent for their tissue samples to be used for diagnosis and scientific research related to clinical treatment. Six ESCC specimens and corresponding adjacent tissues were collected. IHC was performed on consecutive paraffin sections using an Envision kit (Dako). Antigen retrieval was achieved with citrate pH 6.0 buffer, and endogenous peroxidase activity was blocked by pre-incubation in a 3% H2O2 solution in 1 × PBS with 10% goat serum. Slides were incubated overnight at four °C with the primary antibody diluted in the blocking solution. The primary antibody was anti-DOCK9 (proteintech, 18987-1-AP), and secondary peroxidase-conjugated antibodies were employed. Sections were counterstained with hematoxylin and observed under a Nikon Eclipse 80i microscope. Image J software was used for data analysis, including quantifying positive and highly positive areas.
Immunofluorescence
Before antibody incubation, clinical paraffin sections (4-μm-thick) were deparaffinized and rehydrated. Antigen retrieval for CD31 and DOCK9 was performed using microwave heating. At room temperature, the tissue sections were blocked with 10% goat serum for one hour. Then, the primary antibodies (CD31, abcam, ab182981; DOCK9, 1:200, proteintech, 18987-1-AP) were incubated with the sections at 4 °C. Image quantification and analysis for each sample were performed using Image J software.
Statistical analysis
Statistical analyses were conducted using GraphPad Prism version 8.0 software. The results are expressed as the mean ± standard deviation (SD). For comparisons between the two groups, an unpaired t test was utilized. In instances where two normally distributed datasets exhibited unequal variance, Welch's unequal variances t test was employed. Survival analysis was undertaken using the Kaplan–Meier method, and comparisons were made via the log-rank test. Either Spearman or Pearson correlation analysis was conducted to assess the correlation.
Results
Analysis process of this study
In this study, depicted in Fig. 1, we sought novel targets for ESCC therapy. We identified 619 co-differentially expressed genes (DEGs) from GSE67269 and GSE20347 RNA microarrays. After univariate COX survival and LASSO regression analyses of the GSE53625 dataset (179 ESCC patients), we selected 21 genes to construct a prognostic risk model. The model highlighted DOCK9, a gene of significant weight, leading us to explore its role in ESCC at the single-cell level. Using TCGA data, we conducted an immune analysis and assessed the ratio of DOCK9 to CD31 through Kaplan–Meier survival analysis, GSEA, CIBERSORT, ESTIMATE, and immune checkpoint analysis. We confirmed DOCK9 expression and distribution in ESCC patients and investigated its role in HUVECs.

Analysis workflow of this study
Screening genes and constructing a prognostic risk model for ESCC
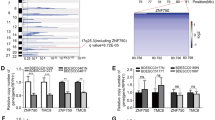
Our preliminary analysis focused on two microarray datasets, GSE67269 and GSE20347, aiming to identify DEGs in ESCC patients by comparing cancerous tissues with adjacent non-cancerous tissues. This exploration revealed 683 DEGs in the GSE67269 dataset and 1345 DEGs in the GSE20347 dataset. We employed a Venn diagram to highlight an overlap of 619 co-differentially expressed genes between these datasets (Fig. 2A).

Screening Genes and Constructing a Prognostic Risk Model for ESCC. A Overlapping DEGs between GSE67269 and GSE20347 datasets via a Venn diagram. B Summary table of 619 DEGs univariate Cox regression analysis results. C Lambda filter representation from LASSO Regression analysis. D Identification of 21 survival-related genes from LASSO analysis. E Heatmap of expression patterns for 21 risk score-associated genes. F Kaplan–Meier survival curve comparison between high and low-risk groups. G The ROC curve demonstrates the predictive power of the risk scores
Following this, we conducted a univariate Cox regression analysis on the GSE53625 dataset, leading to the identification of 43 prognostically significant genes (p < 0.1) (Fig. 2B). We further employed a LASSO regression model, facilitating the development of a prognostic gene signature comprised of 21 out of the previously identified 43 genes (Fig. 2C, D). These 21 screened genes consistently demonstrated differential expression between cancer and adjacent non-cancerous tissues across all three ESCC microarray datasets, and they also exhibited statistically significant variations in expression in the GSE53625 dataset (Supplementary Figure 1A).
Subsequently, we evaluated ESCC prognosis using the risk score derived from the 21-gene signature within the GSE53625 dataset. Patients were divided into high- and low-risk groups based on median risk score values. The survival status and risk score distribution revealed a higher mortality rate among high-risk ESCC patients during the follow-up period relative to the low-risk group (Fig. 2E). Kaplan–Meier analysis affirmed that patients in the high-risk group (n = 66) experienced significantly poorer survival outcomes compared to those in the low-risk group (n = 113) (p < 0.0001) (Fig. 2F). Additionally, the prognostic model demonstrated robust predictive capability for 12-month (AUC = 0.77), 36-month (AUC = 0.78), and 60-month (AUC = 0.82) survival rates in ESCC patients, as illustrated by the Receiver Operating Characteristic (ROC) curve (Fig. 2G).
Risk modeling is an independent prognostic factor that effectively predicts ESCC risk
To evaluate the independent prognostic value of the established risk score, we performed univariate and multivariate Cox regression analyses on the GSE53625 dataset, considering various clinicopathological features such as age, sex, TNM stage, grade, tumor location, tobacco use, and alcohol use. In the univariate Cox regression analysis, age, tumor TNM stage III, poor tumor grade, and the risk score were identified as high-risk factors associated with poor prognosis in ESCC (Fig. 3A). We included all variables with p values less than 0.2 in the multivariate Cox regression analysis. Notably, this analysis confirmed that age, TNM stage III, poor tumor grade, upper tumor location, and risk score were independent high-risk factors for poor prognosis in ESCC patients. In particular, the risk score showed a significant independent prognostic value (p < 0.0001, HR 4.48, 95% CI 3.06–6.54) (Fig. 3B).

Risk modeling is an independent prognostic factor that effectively predicts ESCC risk. A, B Univariate and multivariate Cox regression analysis of the GSE53625 dataset. C Nomogram predicting overall survival probabilities at 12, 36, and 60 months. D Calibration curve of the survival nomogram model. E ROC analysis for 12-, 36-, and 60-month survival. F, G GSEA representations of Hallmark and REACTOME gene sets for high and low-risk ESCC patients
We developed a predictive nomogram to visualize patient survival probabilities based on the multivariate Cox regression analysis results. The nomogram's calibration curve confirmed its accuracy in predicting overall patient survival (Fig. 3C, D). We also performed a ROC analysis to assess the diagnostic value of the risk score, which revealed that the risk score had strong predictive accuracy with AUC values of 0.79, 0.84, and 0.87 for 12-, 36-, and 60-month ROC, respectively (Fig. 3E).
In addition, we used the HALLMARK and REACTOME databases to investigate the activated biological pathways in the low-risk and high-risk score groups. Gene Set Enrichment Analysis (GSEA) results showed the top two pathways from the HALLMARK database suggested a strong association between the high-risk score and Epithelial–mesenchymal transition (EMT) and Angiogenesis (Fig. 3F). Similarly, the top two pathways from the REACTOME database indicated a close connection between the high-risk score and Collagen formation and Extracellular matrix organization in the GSE53625 dataset (Fig. 3G).
DOCK9 is highly expressed in endothelial cells of ESCC
Next, we performed a multivariate Cox regression analysis of the 21 prognostic genes from the GSE53625 database. This analysis revealed that only DOCK9 and SLC39A6 had significant associations with overall survival (OS) in the ESCC samples. Notably, DOCK9 displayed a hazard ratio exceeding one and a p value < 0.05, suggesting that higher expression levels of DOCK9 may indicate a poorer prognosis for ESCC patients (Fig. 4A). Considering that DOCK9 occupied the top position in the LASSO regression analysis coefficient score (Fig. 4A).

DOCK9 is highly expressed in endothelial cells of ESCC. A Multivariate Cox regression results of 21 risk modeling genes. B 2D UMAP plot showing cell type classification in the single-cell RNA-seq data GSE160269. C, D Visualization of DOCK9 expression density and relative expression in cell clusters of GSE160269 dataset. E 2D UMAP plot of cell types in the GSE188900 dataset. F, G DOCK9 expression across cell clusters within the GSE188900 dataset
To further delve into the potential role of DOCK9 in ESCC, we analyzed two single-cell RNA-seq datasets from ESCC patients: GSE160269 and GSE188900. In GSE160269, a total of 187,682 filtered cells derived from four standard human esophageal specimens and 60 ESCC specimens were classified into eight distinct cell populations based on cell markers (Fig. 4B). Remarkably, DOCK9 demonstrated high expression specifically within endothelial cells and lymphatic endothelial cells (Fig. 4C, D). In the GSE188900 dataset, comprising 26,125 filtered cells from one standard human esophageal specimen and 6 ESCC specimens, they were categorized into eleven cell populations based on cell markers (Fig. 4E). Similarly, DOCK9 exhibited high expression specifically within endothelial cells and lymphatic endothelial cells (Fig. 4F, G). Intriguingly, using the IMMUcan scDB database (https://immucanscdb.vital-it.ch), we found that DOCK9 consistently displayed high expression in endothelial cells in other tumor tissues as well (Supplementary Figure 2A).
DOCK9's potential role in tumor angiogenesis and immune responses
In order to better understand the role of DOCK9 in endothelial cells, we integrated endothelial cells from ESCC obtained from the GSE160269 and GSE188900 datasets and divided them into 11 endothelial cell subclusters (Fig. 5A). We further differentiated the combined single-cell data into tumor endothelial cells (TECs) and normal endothelial cell (NECs) clusters using UMAP algorithms. The presence of PDGFB and ANGPT2 marked tumor endothelial cells. In contrast, normal endothelial cells were marked by ICAM1, VCAM1, HLA-DQA1, and ACKR1 (Fig. 5B). We visualized the relationships among these subclusters by calculating the average gene expression in each subcluster, determining the GSVA scores, generating Pearson correlation heat maps, and creating dot plots to display the top five highly variable genes for each subcluster (Supplementary Figure 2B).

DOCK9's Potential Role in Tumor Angiogenesis and Immune Responses: A UMAP plots provide an overview of the combined endothelial cells from the GSE160269 and GSE188990 datasets, effectively visualizing the multi-dimensional data in two dimensions. B UMAP plots exhibit the identification of TECs and NECs as cell subclusters based on specific marker genes, providing a clear visual distinction between these two groups. C The violin plot demonstrates the relative RNA expression of marker genes in TECs and NECs, presenting a clear visualization of distribution and density. D A violin plot showcases the relative RNA level of DOCK9 in TECs and NECs, highlighting a clear difference in expression between the two groups. E UMAP plots illustrate the expression density of DOCK9 in the combined endothelial cells, providing a detailed visual summary of DOCK9 expression across the cell landscape. F A heatmap and enrichment analysis histogram presents the expression distribution and functional enrichment of the top 10 high-expression and low-expression genes in TEC1, offering a visual understanding of gene expression and functional enrichment patterns. G, H Bubble plots demonstrate selected ligand–receptor interactions between high DOCK9 expression endothelial cells and low DOCK9 expression endothelial cells. Ligand-expressing cells (endothelial cells) are marked in red, receptor-expressing cells in blue, and ligand receptors specific to the high-DOCK9 group are labeled, clearly visualizing the intercellular communication patterns. I A violin plot shows the expression of DOCK9 across different TNM stages, visually portraying the differences in expression across these stages. J A violin plot illustrates the expression of DOCK9 across different N stages, effectively visualizing the differences in expression, particularly the high expression in stage N3. K, L Heatmaps display the proportions of 22 immune cell types in both TCGA-ESCC and GSE53625 samples as estimated by the CIBERSORT algorithm, providing a vivid visualization of the immune cell composition in the two sample groups
Further analysis showed a significantly higher DOCK9 expression ratio in TECs compared to NECs (Fig. 5D), with the highest expression seen in TEC1 (Fig. 5E). To explore the function of DOCK9 in TEC1, we split the endothelial cells into two groups. TEC1 cells were labeled as pct 1 and all other endothelial cells as pct 2. We then generated a heatmap and an enrichment analysis histogram to demonstrate the distribution of gene expression and the top 10 DEGs between the two groups. The genes with significantly higher expression in TEC1 cells were primarily related to angiogenesis, extracellular matrix organization, and NABA CORE MATRISOME (Fig. 5F).
We performed cell–cell communication analysis on the GSE160269 dataset to explore cellular interactions and the correlation between DOCK9 expression and intercellular signaling in ESCC. The results showed that endothelial cells with high DOCK9 expression communicated with other endothelial cells through VEGFC-VEGFR3 and VEGFC-VEGFR2R3 pathways. PDGFB was only observed in high-DOCK9 expression endothelial cell communication with fibroblasts and pericytes. In addition, CXCL12 showed typical communication patterns with endothelial cells in lymphatic endothelial cells (LECs) and T cells (Fig. 5G, H, and Supplementary Figure 2C). Notably, there were fewer differences between the high and low DOCK9 groups in terms of cellular communication signals received by endothelial cells, with the OSM-(LIFR + IL6ST) pathway having higher expression in the low DOCK9 group and the VEGFC-VEGFR3 and VEGFC-VEGFR2R3 pathways remaining prominent in the high DOCK9 group (Supplementary Figure 2D).
Our analysis implicates DOCK9 in promoting endothelial cell angiogenesis, the modulation of tumor-associated fibroblast dynamics, and the progression of T cell development. This led us to hypothesize a potential link between DOCK9 expression and ESCC stage progression. Upon investigating the GSE160269 dataset, which comprises 60 ESCC tumor samples and four adjacent normal specimens, we scrutinized the correlation between DOCK9 expression and ESCC TNM or N stages. Although our findings did not reveal significant differences in DOCK9 expression across various TNM stages (Fig. 5I), we observed notably higher DOCK9 expression in the N3 stage than N0, N1, and N2 stages. This suggests a plausible association between elevated DOCK9 expression and lymph node metastasis in ESCC (Fig. 5J).
To further investigate the relationship between DOCK9 and the ESCC immune microenvironment, we compared the composition of 22 tumor-infiltrating immune cells between the high-DOCK9 and low-DOCK9 groups in ESCC using the CIBERSORT algorithm. Both the TCGA and GSE53625 datasets showed that the high-DOCK9 group had a significantly lower proportion of CD8+ T cells compared to the low-DOCK9 group (Fig. 5K, L, and Supplementary Figure 3A). Analysis of immune checkpoints associated with DOCK9 expression showed a correlation between high DOCK9 expression and low PDCD1 (PD-1) expression only in TCGA-ESCC data (Supplementary Figure 3C).
DOCK9/CD31 as potential prognostic indicator and immune therapy correlate
Given the distinct expression of DOCK9 in endothelial cells and its variable expression across different tissues, an analysis focusing solely on DOCK9 at the transcriptome level may not adequately capture its role. Consequently, we incorporated several well-known endothelial cell markers into our analysis, namely CDH5, CD34, CD31, and VWF, derived from the combined scRNA-seq data (Supplementary Figure 3C). We found that CD31, a known vascular endothelial cell marker, did not demonstrate significant differences in expression between TECs and NECs (Fig. 6A). To assess the expression of DOCK9 in TECs, we calculated the ratio of DOCK9 to CD31 expression. Intriguingly, patients with a higher DOCK9/CD31 ratio demonstrated a worse prognosis compared to those with a lower DOCK9/CD31 ratio (Fig. 6C). This relationship was not apparent when CD31 expression was evaluated independently (Fig. 6B).

DOCK9/CD31 as Potential Prognostic Indicator and Immune Therapy Correlate: A Violin and scatter plots shows the relative expression levels of endothelial markers in merged single-cell sequencing data. B, C Kaplan–Meier survival analysis demonstrates the relation between CD31 expression, DOCK9/CD31 ratio, and overall survival. D GSEA of pathways enriched in high and low DOCK9/CD31 groups. E Scatter plot of ESTIMATE scores correlation with relative DOCK9/CD31 expression. F, G Boxplots shows the immune checkpoint expression in high and low DOCK9/CD31 ratio groups
Further analysis using GSEA on the GSE53625 dataset revealed that a low DOCK9/CD31 ratio was associated with adaptive immune response involving α and β T cell differentiation. In contrast, high DOCK9/CD31 ratio was correlated with cell cycle processes (Fig. 6D).
Using the ESTIMATE algorithm, we calculated immune scores for esophageal squamous cell carcinoma (ESCC) patients within the TCGA database. These immune scores exhibited a substantial inverse correlation with the DOCK9/CD31 ratio (r = − 0.539, p < 0.0001). This finding was mirrored in the GSE53625 dataset (r = − 0.579, p < 0.0001) (Fig. 6E).
Moreover, our evaluation of immune checkpoint RNA expression in TCGA-ESCC patients and the GSE53625 dataset, across high and low DOCK9/CD31 groups, revealed that the expressions of CTLA4, PDCD1LG2, and TIGIT were significantly lower in the high DOCK9/CD31 group (Fig. 6F, G).
Decreased DOCK9 expression in ESCC patients and co-localization with CD31
To validate the preceding data analysis, we obtained cancerous and adjacent paraneoplastic tissues from six patients diagnosed with ESCC (Fig. 7A). Western blot analysis showed a significant decrease in DOCK9 expression within cancerous tissues compared to the adjacent tissues (Fig. 7B). Immunohistochemical verification in the six ESCC patients further corroborated the higher expression of DOCK9 in adjacent tissues than in the cancerous tissues (p = 0.0361) (Fig. 7C). Interestingly, we observed a significant negative correlation between CD31 and DOCK9 expressions in the paraneoplastic tissues of ESCC (Fig. 7D), while no such correlation was apparent in cancerous tissues as per the GSE53625 dataset (Fig. 7E). Following our prior results, tissue immunofluorescence revealed a lack of significant co-localization between DOCK9 and CD31 in paraneoplastic tissues. However, substantial co-localization was detected in the cancerous tissues, where DOCK9 expression was significantly reduced (Fig. 7F). These findings underscore the potentially crucial role of DOCK9 in the function of tumor-associated endothelial cells in ESCC patients.

Decreased DOCK9 Expression in ESCC Patients and Co-localization with CD31: A A schematic diagram outlines the collection of human ESCC and adjacent tissues. B A western blot analysis demonstrates the expression of DOCK9 in human ESCC and adjacent tissues. C Representative immunohistochemical images at 200X and 400X magnification show the expression of DOCK9 in both adjacent and ESCC tissues; the bar chart shows the statistical results of immunohistochemical staining in six patients. D, E Scatter plots present the Spearman correlation between CD31 and ANGPT2 in adjacent and ESCC tissues from the GSE53625 dataset. F Combined and separate immunofluorescence staining of adjacent and ESCC tissues, with DOCK9 indicated in green, CD31 in red, and merged areas in orange. Areas of high magnification in the merged images of adjacent tissue (i) and ESCC tissue (ii, iii) are provided for more detailed examination (n = 6)
Silencing DOCK9 decreased the activation of ANG-2 induced HUVECs
Our findings revealed a significant concentration of ANGPT2 in the TEC1 subcluster derived from the merged endothelial cell data (Supplementary Figure 4A). This observation aligns with the high expression of DOCK9 within this subcluster. Kaplan–Meier survival analysis further demonstrated significantly poorer survival in the group with high ANGPT2 expression among the 179 ESCC patients in the GSE53625 dataset (p = 0.02, HR 1.57).
Moreover, when the ANGPT2 receptor Tie2 (TEK) was knocked down in human dermal lymphatic endothelial cells (HDLECs, GSE83299 RNA-seq data), ANGPT2, PDGFB, and VEGFC expressions were markedly elevated. Interestingly, only DOCK9, among the DOCK family genes 1–11, showed a statistically significant increase in expression (Supplementary Figure 4D). Based on these observations, we hypothesize that the elevated DOCK9 expression in tumor endothelial cells is linked to ANGPT2/Tie2 axis activity. Next, we utilized RNA microarray data from HUVECs (GSE77597) to ANGPT/Tie2. The data analysis demonstrated that DOCK9 expression was downregulated by ANG-1 and the Tie2 activator, while ANG-2 blocking antibody groups seem to have no significant change compared with the control (Supplementary Figure 4E).
To emulate the activation state of TEC1 in tumor endothelial cells, we administered ANG-2 to HUVECs. Notably, DOCK9 expression significantly increased following the addition of ANG-2 to the HUVECs (Fig. 8B). To probe the role of DOCK9 in ANG2-stimulated HUVECs, we used small interfering RNA (siRNA) to knock down the expression of DOCK9. The efficiency of siRNA was verified by Western blot (Supplementary Figure 4F). Moreover, the knockdown of DOCK9 expression in the ANG-2-treated HUVEC group resulted in a considerable decline in both the angiogenic capability of the cells and the protein expression level of VEGFA. The wound healing assay results indicated that silencing DOCK9 impaired the migration ability of endothelial cells after exogenous ANG-2 stimulation (Fig. 8E, F). Additionally, both the CCK-8 (Fig. 8G) and EDU assays (Fig. 8H) revealed that the proliferation capacity of endothelial cells was significantly attenuated following DOCK9 knockdown.

Silencing DOCK9 decreased the activation of ANG-2 induced HUVECs: A A schematic diagram illustrates the setup of in vitro experiments. B Western blot analysis shows the expression of DOCK9 in HUVECs treated with exogenous ANG-2. C Western blot analysis demonstrates the expression levels of DOCK9 and VEGFA in HUVECs after si-DOCK9 treatment. D Tube formation assay quantifies the tube-forming capacity of HUVECs treated with si-DOCK9. Accompanying bar graphs display the differences and p values between the experimental groups for capillary length and branch points. Scar bar = 100 μm (n = 3). E The wound healing (scratch) assay assesses the migratory ability of HUVECs + ANG-2 groups treated with either si-DOCK9 or si-NC at 24 h and 48 h post-treatment. Scar bar = 200 μm (n = 3). F t test analysis determines the statistical significance of the wound healing assay results. G The scatter plot illustrates the results of the CCK8 assay in HUVECs. H EdU assay depicts the proliferative capacity of HUVECs after si-DOCK9 treatment. Scar bar = 200 μm (n = 3)
Discussion
Despite advancements in ESCC treatment, patient survival rates over five years remain disappointingly low [35]. This study harnessed RNA microarray data from the GEO database to pinpoint potential biomarkers and formulate risk models for forecasting the survival of ESCC patients across one-, three-, and five-year survival rates. The clinical value of DOCK9 as a biomarker should be highlighted here, such as the predictive power of personalized therapy, immunotherapy, and potential new drug targets. At the same time, combining the dual roles of angiogenesis and immune regulation may lead to the development of comprehensive treatment strategies.
Our research highlights a novel aspect of DOCK9's role in ESCC. While DOCK9 did not reach statistical significance in the univariate setting (Fig. 2B), its selection was supported by the multivariate Cox regression analysis, which yielded a P value of 0.0041 and a Hazard Ratio of 1.93, making it the top prognostic factor in Fig. 4A. Additionally, LASSO coefficient analysis identified DOCK9 as having the highest LASSO coefficient score in the predictive risk model, further indicating its independent prognostic role in esophageal cancer. Therefore, this study mainly focuses on the role of DOCK9 in esophageal cancer. Unlike its elevated expression observed in various non-cancerous tissues [36], we found a pronounced decrease in DOCK9 expression within ESCC samples compared to adjacent non-tumorous tissues. While DOCK9 expression is globally downregulated in tumor tissues, our single-cell RNA sequencing analysis revealed that DOCK9 is specifically upregulated in tumor-associated endothelial cells (TECs) (Fig. 5D). This localized increase in TECs suggests that DOCK9 plays a critical role in tumor angiogenesis and progression, which may explain its association with worse prognosis. Furthermore, these findings are consistent with our immunofluorescence (IF) staining results (Fig. 7F), which demonstrated elevated DOCK9 expression in TECs within tumor tissues, reinforcing its potential role in tumor vascularization. Additionally, our functional assays in endothelial cells (Fig. 8) provide further experimental validation of DOCK9’s role in angiogenesis. These results suggest that DOCK9 is not only a prognostic marker but may also actively contribute to endothelial cell function and tumor-associated blood vessel formation in ESCC. Studies by Hirata et al. demonstrated that DOCK9 contributes to the invasiveness of glioblastoma cells through its effect on Rac1 and Cdc42 activities [37]. Additionally, Passon et al. observed that DOCK9 could serve as a prognostic variable in papillary thyroid carcinomas [38]. These findings suggest that DOCK9 may be associated with tumor invasion and prognosis. This is consistent with our risk model and survival analysis in ESCC, which identify DOCK9 as a marker associated with high-risk prognosis despite its decreased expression in the cancer.
Angiogenesis is pivotal in tumor development, notably in ESCC [39]. While anti-angiogenic therapies show promise, the challenge of tumor heterogeneity often leads to treatment failure, highlighting the need to identify patients who could benefit most from these interventions.
Tumor progression and metastasis are widely recognized as phenomena not solely reliant on a tumor cell-autonomous mechanism. They are also significantly influenced by the tumor microenvironment, which encompasses an array of abnormal stromal cells [40]. Here, we mined single-cell data from ESCC patients to investigate the potential mechanism of DOCK9. Notably, the single-cell RNA-seq analysis of two datasets involving 66 ESCC patients revealed that DOCK9 was enriched in endothelial cells, particularly TECs. Our study, through the combined approach of bioinformatics analysis and tissue immunofluorescence, has discovered an abundant expression of DOCK9 in TECs in ESCC for the first time. Cell–cell communication analysis indicates increased signaling from VEGFC to endothelial cells, as well as from PDGFB to fibroblasts and pericytes, in the context of high DOCK9 expression within endothelial cells. VEGFC and PDGFB are generally considered oncogenes associated with tumor angiogenesis and lymph node metastasis. Interestingly, when endothelial cells act as acceptors, LIFR as a receptor thought to have primarily tumor-suppressive effects [41, 42] is significantly increased in the low-DOCK9 group. These analyses are mainly related to the extracellular matrix, angiogenesis and also have similarities with GSEA analysis results from the risks model.
Endothelial cell dysfunction plays a crucial role in tumor growth and metastasis. In tumors, endothelial cells undergo phenotypic transformation into TECs due to chronic stimulation by growth factors and hypoxia, leading to abnormal tumor blood vessel morphology and increased permeability [43]. Angiopoietin is a kind of endothelial growth factor secreted by activated endothelial cells [44]. Angiopoietin-1 (ANGPT1) and Angiopoietin-2 (ANGPT2) can bind to the receptor tyrosine kinase TIE2 (TEK) present on endothelial cells. They act as natural activators or antagonists of Tie2, respectively, and are involved in modulating angiogenesis, vascular homeostasis, and remodeling [45, 46]. The ANGPT2/TIE2 axis is an attractive target for tumor anti-angiogenic therapy [47]. ANGPT2 is significantly overexpressed in the TECs of ESCC patients. Its expression has significant survival implications in a cohort of 179 ESCC patients from GSE53625 (p = 0.02, HR 1.57, Supplementary Figure 4B). Data analysis showed that, after knockdown of Tie2 in HDLECs, the expression of DOCK9, ANGPT2, PDGFB, and VEGFC all stably increased, but the expression of the rest of the DOCK family did not significantly change. In HUVECs, adding ANG-1 or Tie2 activator could down-regulate the expression of DOCK9. In cell experiments, after adding ANG-2 to HUVECs, the protein expression of DOCK9 significantly increased. These results suggest that the increased DOCK9 in TECs of ESCC is closely related to the ANGPT/TIE2 axis. In subsequent experiments under conditions of exogenous ANG-2 stimulation of HUVECs, we found that the knockdown of DOCK9 could significantly inhibit the tube formation, migration, and proliferation capabilities of HUVECs. These results suggest that ANGPT2/Tie2 may promote the formation of TECs in ESCC endothelial cells by enhancing the expression of DOCK9.
Previous studies have also reported an association between DOCK9 and poor prognosis in high-grade soft tissue sarcoma and identificate DOCK9 as an immune-associated gene suggests its involvement in immune-related processes in cancer [48]. Of note is that high DOCK9 endothelial cells have more signaling to T cells for CXCL12-CXCR4 ligand receptors in cell–cell communication analysis. It has been shown that CXCR4 can inhibit T cells from CXCL12-rich stroma and decreases T cell infiltration to tumor sites [49]. The CYBERSOCRT analysis and ESTIMATE score of TCGA and GSE53625 datasets provided further evidence linking DOCK9 expression to the ESCC immune microenvironment, particularly in reducing CD8+ T cells. GOBP enrichment analysis of 21 risk model genes also showed a high correlation with immune responses (Supplementary Figure 1B). This suggests that DOCK9 may potentially affect the progression of ESCC by influencing immune responses.
The analysis of immune checkpoints did not significantly underscore the role of DOCK9 in immunotherapy, except for a statistically significant (p = 0.04) correlation with PDCD1 expression in the TCGA database. This might suggest that DOCK9 alone is insufficient for determining patient prognosis or providing accurate immune-related analyses. It could be due to the general high expression of DOCK9 in NECs and TECs, with its more critical role in TECs not being directly represented due to individual patient differences.
To address this issue, we compared four common endothelial cell markers, including CD31, CD34, VWF, and CDH5, in TECs and NECs based on merged endothelium scRNA-seq. Interestingly, CD31 (PECAM1), a pan-endothelial marker, did not demonstrate significant differential expression between NECs and TECs. Consequently, for the first time, we propose using the DOCK9 to CD31 ratio as an effect score to consider the pro-oncogenic effect of DOCK9 at the tissue transcriptome level. This ratio offers a more comprehensive approach to assessing the impact of DOCK9 expression based on its function in TECs. In the ESCC dataset of GSE53625, we observed that the high DOCK9/CD31 group had a poor prognosis, and the KM analysis of ANGPT2/CD31 (p = 0.02, HR 1.59, Supplementary Figure 4C) is mainly consistent with ANGPT2 alone. This suggests the success of our model using CD31 as the denominator and indirectly reflects the potential impact of DOCK9 on TECs. Moreover, GSEA and ESTIMATE scores indicated a correlation between a high DOCK9/CD31 ratio and a low immune response. Upon introducing DOCK9/CD31, immune checkpoint analysis, which initially showed no significant differences with DOCK9 expression alone, revealed notable distinctions. Specifically, CTLA4, PDCD1LG2, and TIGIT significantly decreased in patients with high DOCK9/CD31 expression, further supporting DOCK9's potential immunomodulatory mechanism.
Our findings highlight DOCK9's significant role in ESCC, mainly through its involvement in angiogenesis and the immune microenvironment. Despite its decreased expression, the novel insight that DOCK9 acts as a high-risk prognostic marker in ESCC underscores its potential as a biomarker for patient stratification in clinical settings. This could facilitate the identification of patients who are most likely to benefit from targeted anti-angiogenic therapies, thus personalizing treatment approaches and potentially improving patient outcomes. Furthermore, the discovery that silencing DOCK9 can inhibit angiogenic and proliferative activities driven by ANG2 in HUVECs suggests that targeting the DOCK9/ANGPT2/TIE2 signaling axis may offer a new therapeutic strategy for ESCC treatment. Our analysis of the DOCK9/CD31 ratio further supports the importance of assessing tumor endothelial cell and DOCK9 levels as part of the prognostic evaluation in ESCC, opening new avenues for research into targeted therapies that could significantly impact patient survival and quality of life.
Despite this study revealing the potential role of DOCK9 in esophageal squamous cell carcinoma related to tumor angiogenesis and immune regulation through extensive bioinformatics analysis and some experiments, as well as its potential as a prognostic biomarker and therapeutic target, we must acknowledge its limitations. Firstly, the functional effects of DOCK9 and the therapeutic potential of targeting DOCK9 in ESCC still need further validation through animal model studies and clinical trials, which is crucial for translating our bioinformatics and in vitro findings into potential therapeutic applications. Secondly, although the publicly available RNA microarray and single-cell RNA-seq data used in this study are valuable, the inherent data quality and variability limitations mean our findings reflect these inherent constraints. Future studies utilizing larger samples, diverse population data, and standardized testing methods could provide more substantial evidence for these results. Lastly, the current analysis primarily established correlational relationships, highlighting the connection between DOCK9 and the progression and prognosis of ESCC patients, but the specific mechanisms of DOCK9 in tumor development, angiogenesis, and the immune environment remain unclear. This will not only validate the efficacy of DOCK9 as a prognostic marker and therapeutic target but may also unveil new pathways for intervention. Future studies are crucial to further unravel the therapeutic methods and diagnostic approaches for esophageal cancer. This will involve in-depth mechanistic studies, the development of hub gene inhibitors or activators, the application of liquid biopsy techniques, the use of artificial intelligence optimization models, and clinical trials exploring combination treatment strategies.
In conclusion, our study sheds light on the significant role of DOCK9 in ESCC and its potential as a prognostic biomarker and therapeutic target.
Conclusion
Our research identified DOCK9 as a critical predictive element for patient outcomes. We discovered a substantial downregulation of DOCK9 in ESCC and a marked expression in endothelial cells associated with ESCC, correlating closely with angiogenesis and the tumor immune microenvironment. Further, we observed that DOCK9 knockdown attenuates endothelial cell response to ANG-2 in HUVECs, indicating its potential as a target for innovative anti-angiogenic and immunotherapeutic strategies in ESCC.
Data availability
Data included in method part in article.
Abbreviations
- ANG:
-
Angiopoietin
- AUC:
-
Area under the curve
- DEGs:
-
Differentially expressed genes
- ESCC:
-
Esophageal squamous cell carcinoma
- FDR:
-
False discovery rate
- GEO:
-
Gene Expression Omnibus
- GSEA:
-
Gene set enrichment analysis
- GSVA:
-
Gene set variation analysis
- HDLECs:
-
Human dermal lymphatic endothelial cells
- HUVECs:
-
Human umbilical vein endothelial cells
- ICIs:
-
Immune checkpoint inhibitors
- LASSO:
-
Least absolute shrinkage and selection operator
- NECs:
-
Normal endothelial cells
- NES:
-
Normalized enrichment score
- ROC:
-
Receiver operating characteristic
- scRNA-seq:
-
Single-cell RNA sequencing
- TCGA:
-
The Cancer Genome Atlas
- TECs:
-
Tumor endothelial cells
References
Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424.
Pennathur A, Gibson MK, Jobe BA, Luketich JD. Oesophageal carcinoma. Lancet. 2013;381(9864):400–12.
Rustgi AK, El-Serag HB. Esophageal carcinoma. N Engl J Med. 2014;371(26):2499–509.
Domper Arnal MJ, Ferrández Arenas Á, Lanas Arbeloa Á. Esophageal cancer: Risk factors, screening and endoscopic treatment in Western and Eastern countries. World J Gastroenterol. 2015;21(26):7933–43.
Napier KJ, Scheerer M, Misra S. Esophageal cancer: a review of epidemiology, pathogenesis, staging workup and treatment modalities. World J Gastrointest Oncol. 2014;6(5):112–20.
Smyth EC, Lagergren J, Fitzgerald RC, et al. Oesophageal cancer. Nat Rev Dis Primers. 2017;3:17048.
Zhu H, Wang C, Wang J, et al. A subset of esophageal squamous cell carcinoma patient-derived xenografts respond to cetuximab, which is predicted by high EGFR expression and amplification. J Thorac Dis. 2018;10(9):5328–38.
Han X, Lu N, Pan Y, Xu J. Nimotuzumab combined with chemotherapy is a promising treatment for locally advanced and metastatic esophageal cancer. Med Sci Monit. 2017;23:412–8.
Ma S, Lu CC, Yang LY, et al. ANXA2 promotes esophageal cancer progression by activating MYC-HIF1A-VEGF axis. J Exp Clin Cancer Res. 2018;37(1):183.
Liu G, Wang Y, Wang C, et al. Clinical efficacy and safety of apatinib as maintenance treatment in patients with advanced esophageal squamous cell carcinoma. Expert Rev Clin Pharmacol. 2020;13(12):1423–30.
Li J, Wang L. Efficacy and safety of apatinib treatment for advanced esophageal squamous cell carcinoma. Onco Targets Ther. 2017;10:3965–9.
Guven DC, Erul E, Kaygusuz Y, et al. Immune checkpoint inhibitor-related hearing loss: a systematic review and analysis of individual patient data. Support Care Cancer. 2023;31(12):624.
Ricci AD, Rizzo A, Rojas Llimpe FL, et al. Novel HER2-directed treatments in advanced gastric carcinoma: another paradigm shift? Cancers (Basel). 2021;13(7):1664.
Rizzo A, Santoni M, Mollica V, et al. Peripheral neuropathy and headache in cancer patients treated with immunotherapy and immuno-oncology combinations: the MOUSEION-02 study. Expert Opin Drug Metab Toxicol. 2021;17(12):1455–66.
Ricci AD, Rizzo A, Brandi G. DNA damage response alterations in gastric cancer: knocking down a new wall. Future Oncol. 2021;17(8):865–8.
Sahin TK, Ayasun R, Rizzo A, Guven DC. Prognostic value of neutrophil-to-eosinophil ratio (NER) in cancer: a systematic review and meta-analysis. Cancers (Basel). 2024;16(21):3689.
Thuss-Patience P, Stein A. Immunotherapy in squamous cell cancer of the esophagus. Curr Oncol. 2022;29(4):2461–71.
Okada M, Kato K, Cho BC, et al. Three-year follow-up and response-survival relationship of nivolumab in previously treated patients with advanced esophageal squamous cell carcinoma (ATTRACTION-3). Clin Cancer Res. 2022;28(15):3277–86.
Wang X, Zhang B, Chen X, et al. Lactate dehydrogenase and baseline markers associated with clinical outcomes of advanced esophageal squamous cell carcinoma patients treated with camrelizumab (SHR-1210), a novel anti-PD-1 antibody. Thorac Cancer. 2019;10(6):1395–401.
Kojima T, Shah MA, Muro K, et al. Randomized phase III KEYNOTE-181 study of pembrolizumab versus chemotherapy in advanced esophageal cancer. J Clin Oncol. 2020;38(35):4138–48.
Fukui Y, Hashimoto O, Sanui T, et al. Haematopoietic cell-specific CDM family protein DOCK2 is essential for lymphocyte migration. Nature. 2001;412(6849):826–31.
Yang J, Zhang Z, Roe SM, et al. Activation of Rho GTPases by DOCK exchange factors is mediated by a nucleotide sensor. Science. 2009;325(5946):1398–402.
Côté JF, Vuori K. Identification of an evolutionarily conserved superfamily of DOCK180-related proteins with guanine nucleotide exchange activity. J Cell Sci. 2002;115(Pt 24):4901–13.
Benson CE, Southgate L. The DOCK protein family in vascular development and disease. Angiogenesis. 2021;24(3):417–33.
Gadea G, Blangy A. Dock-family exchange factors in cell migration and disease. Eur J Cell Biol. 2014;93(10–12):466–77.
Ruiz-Lafuente N, Minguela A, Parrado A. DOCK9 induces membrane ruffles and Rac1 activity in cancer HeLa epithelial cells. Biochem Biophys Rep. 2018;14:178–81.
Alkhateeb A, Rezaeian I, Singireddy S, et al. Transcriptomics signature from next-generation sequencing data reveals new transcriptomic biomarkers related to prostate cancer. Cancer Inform. 2019;18:1176935119835522.
Zhu J, Shu X, Guo X, et al. Associations between genetically predicted blood protein biomarkers and pancreatic cancer risk. Cancer Epidemiol Biomark Prev. 2020;29(7):1501–8.
Camps J, Noël F, Liechti R, et al. Meta-analysis of human cancer single-cell RNA-seq datasets using the IMMUcan database. Cancer Res. 2023;83(3):363–73.
Zhou Y, Zhou B, Pache L, et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun. 2019;10(1):1523.
Zhang X, Peng L, Luo Y, et al. Dissecting esophageal squamous-cell carcinoma ecosystem by single-cell transcriptomic analysis. Nat Commun. 2021;12(1):5291.
Pan X, Wang J, Guo L, et al. Identifying a confused cell identity for esophageal squamous cell carcinoma. Signal Transduct Target Ther. 2022;7(1):122.
Alquicira-Hernandez J, Powell JE. Nebulosa recovers single-cell gene expression signals by kernel density estimation. Bioinformatics. 2021;37(16):2485–7.
Azad T, Janse Van Rensburg HJ, Lightbody ED, et al. A LATS biosensor screen identifies VEGFR as a regulator of the Hippo pathway in angiogenesis. Nat Commun. 2018;9(1):1061.
Li L, Jiang D, Zhang Q, et al. Integrative proteogenomic characterization of early esophageal cancer. Nat Commun. 2023;14(1):1666.
Parrado A. Expression of DOCK9 and DOCK11 analyzed with commercial antibodies: focus on regulation of mutually exclusive first exon isoforms. Antibodies (Basel). 2020;9(3):27.
Hirata E, Yukinaga H, Kamioka Y, et al. In vivo fluorescence resonance energy transfer imaging reveals differential activation of Rho-family GTPases in glioblastoma cell invasion. J Cell Sci. 2012;125(Pt 4):858–68.
Passon N, Bregant E, Sponziello M, et al. Somatic amplifications and deletions in genome of papillary thyroid carcinomas. Endocrine. 2015;50(2):453–64.
Chen Y, Wang D, Peng H, et al. Epigenetically upregulated oncoprotein PLCE1 drives esophageal carcinoma angiogenesis and proliferation via activating the PI-PLCε-NF-κB signaling pathway and VEGF-C/ Bcl-2 expression. Mol Cancer. 2019;18(1):1.
Maishi N, Hida K. Tumor endothelial cells accelerate tumor metastasis. Cancer Sci. 2017;108(10):1921–6.
Liu SC, Hsu T, Chang YS, et al. Cytoplasmic LIF reprograms invasive mode to enhance NPC dissemination through modulating YAP1-FAK/PXN signaling. Nat Commun. 2018;9(1):5105.
Okamura Y, Nomoto S, Kanda M, et al. Leukemia inhibitory factor receptor (LIFR) is detected as a novel suppressor gene of hepatocellular carcinoma using double-combination array. Cancer Lett. 2010;289(2):170–7.
Dudley AC. Tumor endothelial cells. Cold Spring Harb Perspect Med. 2012;2(3): a006536.
Poto R, Cristinziano L, Modestino L, et al. Neutrophil extracellular traps, angiogenesis and cancer. Biomedicines. 2022;10(2):431.
Eklund L, Olsen BR. Tie receptors and their angiopoietin ligands are context-dependent regulators of vascular remodeling. Exp Cell Res. 2006;312(5):630–41.
Davis S, Aldrich TH, Jones PF, et al. Isolation of angiopoietin-1, a ligand for the TIE2 receptor, by secretion-trap expression cloning. Cell. 1996;87(7):1161–9.
Xie JY, Wei JX, Lv LH, et al. Angiopoietin-2 induces angiogenesis via exosomes in human hepatocellular carcinoma. Cell Commun Signal. 2020;18(1):46.
Bae JY, Choi KU, Kim A, et al. Evaluation of immune-biomarker expression in high-grade soft-tissue sarcoma: HLA-DQA1 expression as a prognostic marker. Exp Ther Med. 2020;20(5):107.
Bockorny B, Semenisty V, Macarulla T, et al. BL-8040, a CXCR4 antagonist, in combination with pembrolizumab and chemotherapy for pancreatic cancer: the COMBAT trial. Nat Med. 2020;26(6):878–85.
Acknowledgements
This research was funded by the Doctoral Research Start-up Project Fund of the People's Hospital Affiliated to Jiangsu University, China (KFB2023011) and additional support was provided by Gusu Health Talent Program (GSWS2023003).
Funding
The authors have not disclosed any funding.
Author information
Authors and Affiliations
Contributions
Yaqiang Pan, Yangyong Sun, and Ying Xiao contributed equally to this work, performing the original manuscript—drafting, primary data analysis and interpretation. Jifei Ding conducted experimental validation, functional experiment and assisted in data collection. Ge Hu designed the study and supervised the research process. Chang Chen and Zhiqiang Lin contributed to the study design, supervised data analysis, and provided critical revisions to the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Ethics statement
This study was reviewed and approved by the Jiangsu University of Medical Sciences Ethics Committee, with the approval number: K-20230079-W. All participants provided informed consent to participate in the study.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Pan, Y., Sun, Y., Xiao, Y. et al. DOCK9 as a predictive biomarker linked to angiogenesis and immune response in esophageal squamous cell carcinoma. Clin Exp Med 25, 126 (2025). https://doi.org/10.1007/s10238-025-01653-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10238-025-01653-8