
Abstract
The concept of progressive pulmonary fibrosis (PPF) has been introduced to predict the diverse prognosis of interstitial lung disease (ILD). However, the incidence and effect of PPF on outcomes in patients with connective tissue disease-associated interstitial lung disease (CTD-ILD) need to be elucidated. This study reviewed 197 patients with CTD-ILD. Symptomatic worsening, pulmonary function decline, and radiological deterioration were investigated to assess the fulfillment of PPF diagnostic criteria. Clinical outcomes, including mortality, were compared based on the presence or absence of PPF. The median follow-up duration was 17.4 months. The mean age of the patients was 64.0 years, and 60.9% were female. Among the underlying CTDs, rheumatoid arthritis (42.1%), inflammatory myositis (19.8%), and systemic sclerosis (13.2%) were the most common. Of the 197 patients, 37 (18.8%) met the diagnostic criteria for PPF during the follow-up period. Even after adjusting for other significant risk factors, PPF was independently associated with mortality [hazard ratio (HR) 3.856; 95% confidence interval (CI) 1.387–10.715; P = 0.010] and baseline albumin was marginally significantly associated with mortality (HR 0.549; CI 0.298–1.010; P = 0.054). The median survival was also significantly shorter in the PPF group than in the non-PPF group (72.3 ± 12.9 vs. 126.8 ± 15.5 months, P < 0.001). Baseline KL-6 ≥ 1000 (U/mL) was a significant risk factor for PPF (HR 2.885; CI 1.165–7.144; P = 0.022). In addition to increased mortality, the PPF group had significantly higher rates of respiratory-related hospitalizations, pneumonia, acute exacerbations, and weight loss than the non-PPF group. PPF is a significant prognostic indicator in patients with CTD-ILD. Thus, healthcare professionals should know that patients with CTD-ILD are at risk of PPF.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Background
Connective tissue disease (CTD) is a heterogeneous disease entity including various diseases such as rheumatoid arthritis, systemic sclerosis, inflammatory myositis, systemic lupus erythematosus, and Sjögren’s syndrome. These CTDs can cause multiple symptoms and signs and affect patients’ quality of life and survival [1, 2].
Particularly, interstitial lung disease (ILD) often coexists with CTD, and ILD has a significant effect on the morbidity and mortality of patients with CTD [3, 4]. ILD concurrent with CTD is called connective tissue disease-associated interstitial lung disease (CTD-ILD). The clinical course of CTD-ILD is very diverse, from asymptomatic to fulminant and life-threatening disease [5, 6]. Recently, several guidelines and statements have been suggested for the management of CTD-ILD [7,8,9,10]. However, until now, no gold standard has been established for CTD-ILD management.
The concept of progressive pulmonary fibrosis (PPF) has recently been introduced. PPF is defined as pulmonary fibrosis without idiopathic pulmonary fibrosis (IPF) that satisfied at least two of the symptoms, pulmonary function, or radiologic deterioration within the last 1 year [11]. Currently, PPF is being validated for its ability to predict the prognosis of patients with ILD in follow-up studies [12, 13].
According to previous studies, the prevalence of PPF in patients with CTD-ILD ranged from 23 to 38% [14,15,16]. However, to our knowledge, there is currently no large-scale, well-designed study available to investigate the clinical impacts of PPF in patients with CTD. Therefore, in this study, we investigated the clinical characteristics of patients with CTD who met the definition of PPF and the clinical effect of PPF on the course of CTD-ILD.
Methods
Study population
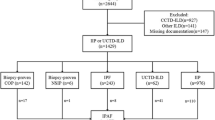
We retrospectively collected data of 197 patients with CTD-ILD from a tertiary referral hospital of 2700 beds in Seoul, South Korea. Patients who were diagnosed with CTD-ILD between April 1, 2007, and October 27, 2022, were included. The medical records of these patients were retrospectively analyzed in January 2023.
CTD was diagnosed by rheumatologists using specific criteria. Systemic sclerosis, rheumatoid arthritis, and systemic lupus erythematosus were diagnosed based on the diagnostic criteria established by the American College of Rheumatology [17,18,19]. Mixed connective tissue disease was diagnosed using the diagnostic criteria developed by Alarcon-Segovia [20]. Dermatomyositis and polymyositis were diagnosed according to the diagnostic and classification criteria established by Bohan and Peter [21]. Undifferentiated connective tissue disease was defined using classification criteria [22]. Sjögren’s syndrome was diagnosed based on the revised criteria proposed by the American-European Consensus Group [23].
Due to the absence of a single golden standard in the diagnosis of CTD-ILD, a multidisciplinary approach involving rheumatologists, pulmonologists, and radiologists was employed to diagnose CTD-ILD [24]. The diagnostic criteria for acute exacerbation of CTD-ILD were defined as follows, similar to those proposed by Collard et al. [25] for IPF patients: it is characterized by the occurrence or worsening of respiratory distress within the last 1 month in patients diagnosed with CTD-ILD. Additionally, chest computed tomography (CT) scans must reveal new bilateral ground-glass opacities and/or consolidation, and the deterioration cannot be fully explained by conditions such as cardiac failure or fluid overload.
This study was conducted following the Declaration of Helsinki. The Institutional Review Board of Asan Medical Center endorsed this study protocol (IRB no. 2022-1564) The Institutional Review Board waived the need for informed consent because of anonymous clinical data and retrospective study design.
Clinical data
Clinical and survival data were collected from the medical records and the National Health Insurance of Korea database. We analyzed data from regular follow-up clinic visits or hospitalizations. Spirometry, forced vital capacity (FVC), diffusing capacity of the lung for carbon monoxide (DLco), and total lung capacity (TLC) measurements were reviewed following the guidelines provided by the American Thoracic Society/European Respiratory Society [26,27,28]. The outcomes were reported as a percentage of the expected normal values. The 6-min walk test (6MWT) was also performed following guidelines from the American Thoracic Society [29].
In addition, high-resolution computed tomography (HRCT) was performed in accordance with standard protocols at full inspiration without contrast enhancement. HRCT images were independently reviewed by two thoracic radiologists (YA and HNN) blinded to the clinical and pathologic information. HRCT images acquired at diagnosis and within a year following diagnosis were reviewed. On HRCT, initial radiologic classification (usual interstitial pneumonia (UIP) vs. non-UIP pattern) was determined and an increased extent of fibrosis was visually assessed with a side-by-side comparison [30]. Disagreement between readers was resolved via a consensus. A questionnaire was not used, but an electronic medical record within a year, to identify the worsening of symptoms and functional capacity.
PPF was diagnosed according to 2022 clinical practice guidelines [11], which defined PPF as meeting a minimum of two of the following three criteria: a decrease of at least 5% in the absolute value of the FVC or a decrease of at least 10% in DLco, worsening of symptoms, and radiological evidence indicating disease progression. To identify death and its dates, we used data from National Insurance Corporation from the diagnosis to January 1, 2023. In-hospital data were used for demographics, comorbidity, baseline laboratory test such as KL-6 and autoantibodies, administration of immunosuppressive agents and steroids, and their side effects.
Outcome analysis
Patients with CTD-ILD were divided into CTD-ILD groups with PPF and without PPF according to whether they met the diagnostic criteria for PPF, and the differences in mortality between the two groups were analyzed. Factors that affect mortality in patients with CTD-ILD and risk factors of PPF were also investigated. Subgroup analysis was performed on the most prevalent CTDs, rheumatoid arthritis, and inflammatory myositis, to investigate the effect of PPF on mortality differs according to CTD types.
Statistical analysis
All continuous variables are presented as means ± standard deviations, and categorical variables are presented as percentages. Student’s t-test or Mann–Whitney U-test was used to compare continuous variables, and the chi-square or Fisher’s exact test was employed to compare categorical variables. Cox proportional hazard analyses were performed to calculate the univariate and multivariate hazard ratios (HRs) of mortality according to PPF and disease entity. Cox proportional hazard analyses were also performed to determine risk factors and calculate the univariate and multivariate HRs of PPF according to potential risk factors. The Kaplan–Meier method with a log-rank test was used to estimate the cumulative mortality rates according to disease entity and presence of PPF and compare them. Variables with a P-value < 0.1 in the univariate analysis were only included in the multivariate analysis. All statistical tests were two-sided, and a P-value of < 0.05 was considered significant. All statistical analyses were performed using IBM SPSS version 24.0 (IBM Corp., Armonk, NY, USA).
Results
Patients
The median follow-up duration was 17.4 [interquartile range (IQR): 7.4–29.4] months. Of the 197 patients with CTD-ILD included in this study, 37 (18.8%) fulfilled the criteria for PPF during the follow-up period. When the 197 patients were categorized based on the three diagnostic criteria for PPF, 70 (35.5%) experienced symptomatic worsening, 41 (20.8%) exhibited radiological worsening, and 33 (16.8%) showed physiological worsening (decline in pulmonary function) (Fig. 1).

Venn diagram of the satisfaction status of each diagnostic criterion of PPF among 197 patients with connective tissue disease-associated interstitial lung disease
The baseline clinical characteristics of the two groups, divided based on the satisfaction of PPF diagnostic criteria, are presented in Table 1. In the PPF group, the baseline Krebs von den Lungen-6 (KL-6) level was significantly higher (median 1055.7, IQR 597.3–2213.5 vs. median 572.0, IQR 354.2–926.6, P < 0.001), whereas the albumin level was lower (median 3.0, IQR 2.4–3.7 vs. median 3.6, IQR 3.2–3.8, P < 0.001, respectively). Regarding the underlying CTD, the frequency of rheumatoid arthritis was significantly lower, whereas the frequency of inflammatory myositis was higher in the PPF group (27.0% vs. 45.6%, P = 0.039; 40.5% vs. 15.0%, P < 0.001, respectively). Pulmonary function parameters were also significantly lower in the PPF group (FVC, 60.8 ± 14.9 vs. 73.9 ± 17.4, P < 0.001; DLco, 46.1 ± 17.3 vs. 55.6 ± 17.4, P = 0.010; TLC, 67.1 ± 12.6 vs. 78.1 ± 15.8, P = 0.002). Moreover, the 6-min walking distance (6MWD) was significantly shorter in the PPF group (median 385.0, IQR 280.5–483.0 vs. median 455.0, IQR 360.0–507.5, P = 0.042).
When comparing baseline autoantibodies between the PPF group and the non-PPF group, the PPF group showed significantly lower frequencies of high ANA (Anti-nuclear antibody) titer (≥ 320:1) and anti-CCP (cyclic citrullinated peptide) antibody positivity (21.6% vs. 38.8%, P = 0.050 and 36.4% vs. 56.8%, P = 0.038, respectively, Additional file1: Supplementary Table 1).
Factors affecting mortality
During follow-up, 38 (19.3%) patients died. Table 2 presents the results of the Cox proportional hazard analysis for the risk factors of mortality. In the univariate analysis, several factors significantly correlated with mortality, including age [HR 1.039; 95% confidence interval (CI) 1.008–1.071; P = 0.014], male sex (HR 2.194; CI 1.150–4.187; P = 0.017), baseline KL-6 ≥ 1000 (U/mL) (HR 2.818; CI 1.205–6.592; P = 0.017), baseline albumin (HR 0.306; CI 0.221–0.424; P < 0.001), baseline radiologic UIP pattern (HR 2.528; CI 1.330–4.803; P = 0.005), PPF (HR 4.820; CI 2.539–9.152; P < 0.001), DLco (HR 0.959; CI 0.936–0.981; P < 0.001), 6MWD (HR 0.996; CI 0.992–0.999; P = 0.010), and 6MWT and lowest SpO2 (HR 0.855; CI 0.791–0.924; P < 0.001).
In the multivariate analysis, only PPF was independently associated with mortality (HR 3.856; CI 1.387–10.715; P = 0.010), after adjusting other risk factors, such as age (HR 1.044; P = 0.208), male sex (HR 1.737; P = 0.326), baseline KL-6 ≥ 1000U/mL (HR 2.453; P = 0.164), baseline albumin (HR 0.549; P = 0.054), radiologic UIP pattern (HR 2.075; P = 0.110), and baseline DLco (HR 0.979; P = 0.210). Figure 2 displays the Kaplan–Meier curves divided into two groups based on whether patients with CTD-ILD met the definition of PPF. The median survival was significantly shorter in the PPF group than in the non-PPF group (72.3 ± 12.9 vs. 126.8 ± 15.5 months, P < 0.001).

Kaplan–Meier plot of the cumulative mortality rate based on the diagnosis of progressive pulmonary fibrosis in patients with connective tissue disease-associated interstitial lung disease
Survival analysis in subgroups
Survival analyses were conducted for the two most frequent CTDs, namely, rheumatoid arthritis and inflammatory myositis. Among the 83 patients with rheumatoid arthritis, a significant difference was found in the survival curves between the PPF and non-PPF groups (P = 0.001, Additional file 2: Figure S1-A). Similarly, among the 39 patients with inflammatory myositis, significant differences were observed in the survival curves between the two groups (P = 0.003, Additional file 2: Figure S1-B).
Risk factors of PPF
In a Cox proportional hazard analysis conducted on 197 patients with CTD-ILD, the univariate analysis revealed several significant risk factors for PPF. These included baseline KL-6 ≥ 1000 (U/mL) (HR 4.371; CI 2.037–9.376; P < 0.001), baseline albumin levels (HR 0.422; CI 0.292–0.610; P < 0.001), baseline pulmonary function measurements of FVC (HR 0.963; CI 0.945–0.982; P < 0.001) DLco, (HR 0.963; CI 0.941–0.986; P = 0.002), and 6MWD (HR 0.997; CI 0.994–1.000; P = 0.042). In the multivariate analysis, the baseline KL-6 ≥ 1000 (U/mL) was statistically significant as a risk factor for PPF (HR 2.885; CI 1.165–7.144; P = 0.022), and the baseline lower FVC was marginally associated with PPF (HR 0.963; CI 0.927–1.001; P = 0.056) (Table 3).
Treatment and clinical outcomes
As shown in Table 4, among the 197 patients with CTD-ILD, 163 (82.7%) were on steroid therapy. A significantly higher proportion of patients with PPF was on steroid therapy than patients without PPF (35 of 37, 94.6% vs. 128 of 160, 80.0%, P = 0.034). Furthermore, the maximal dose of steroids was significantly higher in the PPF group (median 57.5 mg, IQR 30.0–75.0) than in the non-PPF group (median 30.0 mg, IQR 7.5–43.8, P < 0.001), and the PPF group had significantly shorter duration of steroid treatment (median 9.0 months, IQR 4.0–14.0) than the non-PPF group (median 14.0 months, IQR 6.0–36.8, P = 0.008).
Among all patients with CTD-ILD, 47.2% (93 of 197) received immunosuppressant therapy. Immunosuppressive agents were significantly more commonly administered to the PPF group than to the non-PPF group (73.0%, 27 of 37 vs. 41.3%, 66 of 160, P < 0.001). Mycophenolate mofetil was the most commonly used immunosuppressant, followed by azathioprine, cyclosporine, cyclophosphamide, and tocilizumab. The use of mycophenolate mofetil was significantly higher in the PPF group than in the non-PPF group (59.5% vs. 18.1%, P < 0.001).
Regarding clinical outcomes, of the total 197 patients, 38 (19.3%) died. During the follow-up period, significantly more deaths were observed in the PPF group than in the non-PPF group (18 of 37, 48.6% vs. 20 of 160, 12.5%, P < 0.001). Comparing other outcomes, respiratory-related hospitalizations were significantly higher in the PPF group than in the non-PPF group (81.1% vs. 20.0%, P < 0.001). Pneumonia and acute exacerbation were also significantly more prevalent in the PPF group than in the non-PPF group (78.4% vs. 16.3%, P < 0.001 and 64.9% vs. 14.4%, P < 0.001, respectively). Significant weight loss defined as a decrease of ≥ 5% in body weight was also significantly higher in the PPF group than in the non-PPF group (36.4% vs. 15.2%, P = 0.033, Table 4).
Discussion
This study investigated the frequency and clinical outcomes of PPF in patients with CTD-ILD. Approximately one-fifth of patients (18.8% of study populations) met the PPF criteria, and PPF was independently associated with mortality (HR 3.856; CI 1.387–10.715; P = 0.010). In addition, despite more treatment, the PPF group showed worse survival than the non-PPF group.
According to previous studies, the prevalence of PPF in patients with CTD-ILD ranged from 23 to 38% [14,15,16]. In the present study, 37 (18.8%) patients met the diagnostic criteria of PPF, which is relatively lower than those in previous studies. These results might be attributed to the difference in the study design. Lee et al. [14] analyzed 107 patients with CTD and identified PPF in 38.3% of these patients. In addition, Chiu et al. [15] reported that 53 (23%) of their 230 patients with CTD-ILD had a PPF with median follow-up of 6 years. The lower prevalence of PPF in the present study than in previous studies may be attributed to several factors. First, the shorter follow-up period in the present study than in previous studies might have contributed to the lower observed prevalence (median follow-up duration: 17.4, IQR; 7.4–29.4 months) when compared with the median follow-up duration of 49.9 ± 36.5 months reported by Lee et al. and of 6 (IQR 6–9) years by Chiu et al. [14, 15]. In addition, the application of stricter criteria for radiologic deterioration, assessed by two radiologists, could have resulted in a more stringent evaluation. Despite potential variations in the criteria used to define PPF, a relatively large-scale study involving 1,749 patients demonstrated that 16.6% (42 of 253 individuals) of patients with CTD exhibited a progressive fibrosing ILD phenotype [31]. To determine the exact proportion of PPF in patients with CTD-ILD, further follow-up studies with a larger sample size are necessary.
In the present study, PPF was independently associated with mortality even after adjusting for other factors, such as baseline albumin level and baseline radiologic pattern, and our finding supports previous reports [12, 13, 16, 32]. Previously, several staging systems using baseline characteristics were suggested for predicting prognosis in patients with CTD-ILD, particularly in systemic sclerosis and rheumatoid arthritis [33,34,35]. To the best of our knowledge, a few studies have clinically compared the PPF and non-PPF groups only among patients with CTD. Khor et al. [13] analyzed 753 patients with non-IPF fibrotic ILD (including 372 patients with CTD) and reported that PPF was independently associated with increased mortality and lung transplantation (HR 2.08). In addition, Pugashetti et al. [12] enrolled 1341 patients (including 516 patients with CTD) and showed that ≥ 10% relative FVC decline was related to reduced survival in the patients with non-IPF ILD, and additional PPF criteria were associated with reduced transplant-free survival. The present study indicates that important prognostic factors in CTD-ILD include not only initial characteristics such as baseline lung function, CT pattern, and ILD extent but also the clinical course including changes in lung function, symptoms, and radiological findings.
Interestingly, this study showed that despite the higher usage of treatments such as steroids and immunosuppressive agents in the PPF group than in the non-PPF group, they had a worse prognosis. Although several drugs have beneficial effects on CTD-ILD, no gold standard has been established for the management of patients with CTD-ILD [36,37,38]. Specifically, in patients with a progressive course, such as those in the PPF group, more studies are needed to evaluate the effectiveness of treatments, including antifibrotic agents [39].
It is challenging to directly compare the group that received steroid and/or immunosuppressive therapy with the group that did not, as more than 80% of the overall patients underwent such treatment. Additionally, analyzing the cause of death is also difficult, as in more than half of the deceased patients (20 of 38), the exact cause of death remains unknown. Nonetheless, as shown in Table 4, individuals with PPF encountered a greater frequency of acute exacerbations, hospitalizations due to respiratory issues, and notably, a heightened susceptibility to pneumonia. In light of these findings, it is reasonable to infer that the unfavorable clinical outcome in PPF patients could have been impacted not solely by the severity of the underlying ILD but also by the presence of infections.
Although the criteria for defining the progressive fibrosing phenotype may vary across studies, previous studies have proposed several clinical variables in predicting this phenotype [40, 41]. Hambly et al. [40] analyzed 2746 patients with fibrotic ILD and identified age, male sex, gastroesophageal reflux disease, and impaired lung function as indicators of progression. Among studies on CTD, a retrospective study found male sex and South Asian ethnicity independently predicted a decline in FVC, whereas male sex, smoking history, and systemic sclerosis as the specific CTD type were associated with a decline in DLco [41]. In the present study, the baseline KL-6 ≥ 1000 (U/mL) was identified as a statistically significant risk factor for PPF (HR 2.885; CI 1.165–7.144; P = 0.022), and the baseline FVC was borderline significantly associated with PPF (HR 0.963; CI 0.927–1.001; P = 0.056). In a previous study of anti-synthetase syndrome-associated ILD with 72 participants, the baseline KL-6 was reported as a significant risk factor for PPF [42]. Although research on the prevalence and risk factors of ILD occurrence in patients with CTD is relatively scarce [41], risk factors for disease progression in CTD-ILD are not yet well elucidated; thus, further research is needed.
Another intriguing finding is that there was a significant relationship between baseline albumin levels and mortality as well as PPF in the univariate analysis (HR 0.306; CI 0.221–0.424; P < 0.001, HR 0.422; CI 0.292–0.610; P < 0.001, respectively), even though it was not significant in the multivariate analysis (HR 0.549; CI 0.298–1.010; P = 0.054, HR 0.608; CI 0.329–1.124; P = 0.113, respectively). Previous studies report that low albumin levels are associated with increased mortality in various respiratory diseases, possibly reflecting poor nutritional status [24, 43]. Additionally, while the exact mechanism is not well understood, there has been recent research indicating that low albumin levels predict PPF in Sjögren's syndrome-related interstitial pneumonia patients, probably reflecting ongoing inflammation [44]. Although baseline albumin levels were not an independent predictor of PPF in this study's multivariate analysis, further research on this topic is needed in the future.
Generally, a UIP pattern on imaging has been reported as a hallmark of pulmonary fibrosis associated with poor prognosis [45, 46]. In our current study, while the baseline radiologic UIP pattern showed a significant association with mortality in univariate analysis (HR 2.528; CI 1.330–4.803; P = 0.005), it only had a borderline significant relationship with mortality in multivariate analysis (HR 2.705; CI 0.799–9.158; P = 0.110, Table 2). However, the radiologic UIP pattern did not have a significant association with the development of PPF (HR 1.336; CI 0.659–2.709; P = 0.422, Table 3). Therefore, we believe that while baseline image patterns might be important in patients with CTD, factors related to clinical course, like PPF, appear to be more clinically meaningful in these patients.
The limitations of this study include its retrospective, single-center design. In addition, a selection bias may have occurred because the study excluded individuals with short follow-up. Furthermore, standardized methods such as questionnaires were not used to assess the worsening of symptoms. In addition, this retrospective study was unable to adhere to a well-designed protocol regarding the intervals of pulmonary function tests and CT. In South Korea, the antifibrotic agent including nintedanib is not covered by healthcare insurance. Therefore, owing to its high cost, the majority of patients with PPF had not received nintedanib; thus, the effectiveness of antifibrotic agents could not be analyzed. However, despite the retrospective design of this study, the review of CT scans by two radiologists for the diagnosis of PPF is considered an advantage.
Conclusions
In conclusion, our findings indicate that PPF could be a significant prognostic indicator in individuals with CTD-ILD. As a result, healthcare professionals should be aware that patients with CTD-ILD are at risk of developing PPF.
Abbreviations
- ANA:
-
Anti-nuclear antibody
- CCP:
-
Cyclic citrullinated peptide
- CI:
-
95% Confidence interval
- CT:
-
Computed tomography
- CTD:
-
Connective tissue disease
- CTD-ILD:
-
Connective tissue disease-associated interstitial lung disease
- DLco:
-
Diffusing capacity of the lung for carbon monoxide
- FVC:
-
Forced vital capacity
- HR:
-
Hazard ratio
- HRCT:
-
High-resolution computed tomography
- ILD:
-
Interstitial lung disease
- IPF:
-
Idiopathic pulmonary fibrosis
- IQR:
-
Interquartile range
- UIP:
-
Usual interstitial pneumonia
- KL-6:
-
Krebs von den Lungen-6
- PPF:
-
Progressive pulmonary fibrosis
- SpO2:
-
Peripheral oxygen saturation
- TLC:
-
Total lung capacity
- 6MWD:
-
6-Minute walk test distance
- 6MWT:
-
6-Minute walk test
References
Garen T, Lerang K, Hoffmann-Vold A-M, Andersson H, Midtvedt Ø, Brunborg C, Kilian K, Gudbrandsson B, Gunnarsson R, Norby G, et al. Mortality and causes of death across the systemic connective tissue diseases and the primary systemic vasculitides. Rheumatology. 2018;58:313–20.
Li L, Zuo X, Luo H, Li Y, You Y, Duan L, Zhang W, Zhao H, Li T, Ning W, et al. Mortality trend of inpatients with connective tissue diseases: 2005–2014. Zhong Nan Da Xue Xue Bao Yi Xue Ban. 2017;42:927–33.
Oldham JM, Adegunsoye A, Valenzi E, Lee C, Witt L, Chen L, Husain AN, Montner S, Chung JH, Cottin V, et al. Characterisation of patients with interstitial pneumonia with autoimmune features. Eur Respir J. 2016;47:1767–75.
Yunt ZX, Chung JH, Hobbs S, Fernandez-Perez ER, Olson AL, Huie TJ, Keith RC, Janssen WJ, Goldstein BL, Lynch DA, et al. High resolution computed tomography pattern of usual interstitial pneumonia in rheumatoid arthritis-associated interstitial lung disease: Relationship to survival. Respir Med. 2017;126:100–4.
Park JH, Kim DS, Park IN, Jang SJ, Kitaichi M, Nicholson AG, Colby TV. Prognosis of fibrotic interstitial pneumonia: idiopathic versus collagen vascular disease-related subtypes. Am J Respir Crit Care Med. 2007;175:705–11.
Fujisawa T, Suda T, Nakamura Y, Enomoto N, Ide K, Toyoshima M, Uchiyama H, Tamura R, Ida M, Yagi T, et al. Differences in clinical features and prognosis of interstitial lung diseases between polymyositis and dermatomyositis. J Rheumatol. 2005;32:58–64.
Kondoh Y, Makino S, Ogura T, Suda T, Tomioka H, Amano H, Anraku M, Enomoto N, Fujii T, Fujisawa T, et al. 2020 guide for the diagnosis and treatment of interstitial lung disease associated with connective tissue disease. Respir Investig. 2021;59:709–40.
Lee AS, Scofield RH, Hammitt KM, Gupta N, Thomas DE, Moua T, Ussavarungsi K, St Clair EW, Meehan R, Dunleavy K, et al. Consensus guidelines for evaluation and management of pulmonary disease in Sjögren’s. Chest. 2021;159:683–98.
Jee AS, Sheehy R, Hopkins P, Corte TJ, Grainge C, Troy LK, Symons K, Spencer LM, Reynolds PN, Chapman S, et al. Diagnosis and management of connective tissue disease-associated interstitial lung disease in Australia and New Zealand: a position statement from the Thoracic Society of Australia and New Zealand. Respirology. 2021;26:23–51.
Rahaghi FF, Hsu VM, Kaner RJ, Mayes MD, Rosas IO, Saggar R, Steen VD, Strek ME, Bernstein EJ, Bhatt N, et al. Expert consensus on the management of systemic sclerosis-associated interstitial lung disease. Respir Res. 2023;24:6.
Raghu G, Remy-Jardin M, Richeldi L, Thomson CC, Inoue Y, Johkoh T, Kreuter M, Lynch DA, Maher TM, Martinez FJ, et al. Idiopathic pulmonary fibrosis (an update) and progressive pulmonary fibrosis in adults: an official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. 2022;205:e18–47.
Pugashetti JV, Adegunsoye A, Wu Z, Lee CT, Srikrishnan A, Ghodrati S, Vo V, Renzoni EA, Wells AU, Garcia CK, et al. Validation of proposed criteria for progressive pulmonary fibrosis. Am J Respir Crit Care Med. 2023;207:69–76.
Khor YH, Farooqi M, Hambly N, Kolb M, Ryerson CJ. Patient characteristics and survival for progressive pulmonary fibrosis using different definitions. Am J Respir Crit Care Med. 2023;207:102–5.
Lee J, Kim K, Jo YS. Comparison of the diagnostic criteria for progressive pulmonary fibrosis in connective tissue disease related interstitial lung disease. Respir Med. 2023;212: 107242.
Chiu YH, Koops MFM, Voortman M, van Es HW, Langezaal LCM, Welsing PMJ, Jamnitski A, Wind AE, van Laar JM, Grutters JC, Spierings J. Prognostication of progressive pulmonary fibrosis in connective tissue disease-associated interstitial lung diseases: a cohort study. Front Med (Lausanne). 2023;10:1106560.
Chiu YH, Spierings J, de Jong PA, Hoesein FM, Grutters JC, van Laar JM, Voortman M. Predictors for progressive fibrosis in patients with connective tissue disease associated interstitial lung diseases. Respir Med. 2021;187: 106579.
Preliminary criteria for the classification of systemic sclerosis (scleroderma). Subcommittee for scleroderma criteria of the American Rheumatism Association Diagnostic and Therapeutic Criteria Committee. Arthritis Rheum 1980, 23:581–590
Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS, Healey LA, Kaplan SR, Liang MH, Luthra HS, et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 1988;31:315–24.
Tan EM, Cohen AS, Fries JF, Masi AT, McShane DJ, Rothfield NF, Schaller JG, Talal N, Winchester RJ. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1982;25:1271–7.
Alarcón-Segovia D, Cardiel MH. Comparison between 3 diagnostic criteria for mixed connective tissue disease. Study of 593 patients. J Rheumatol. 1989;16:328–34.
Bohan A, Peter JB. Polymyositis and dermatomyositis (first of two parts). N Engl J Med. 1975;292:344–7.
Mosca M, Neri R, Bombardieri S. Undifferentiated connective tissue diseases (UCTD): a review of the literature and a proposal for preliminary classification criteria. Clin Exp Rheumatol. 1999;17:615–20.
Vitali C, Bombardieri S, Jonsson R, Moutsopoulos HM, Alexander EL, Carsons SE, Daniels TE, Fox PC, Fox RI, Kassan SS, et al. Classification criteria for Sjögren’s syndrome: a revised version of the European criteria proposed by the American-European Consensus Group. Ann Rheum Dis. 2002;61:554–8.
Vij R, Strek ME. Diagnosis and treatment of connective tissue disease-associated interstitial lung disease. Chest. 2013;143:814–24.
Collard HR, Ryerson CJ, Corte TJ, Jenkins G, Kondoh Y, Lederer DJ, Lee JS, Maher TM, Wells AU, Antoniou KM, et al. Acute exacerbation of idiopathic pulmonary fibrosis. An international working group report. Am J Respir Crit Care Med. 2016;194:265–75.
Miller MR, Hankinson J, Brusasco V, Burgos F, Casaburi R, Coates A, Crapo R, Enright P, van der Grinten CP, Gustafsson P, et al. Standardisation of spirometry. Eur Respir J. 2005;26:319–38.
Macintyre N, Crapo RO, Viegi G, Johnson DC, van der Grinten CP, Brusasco V, Burgos F, Casaburi R, Coates A, Enright P, et al. Standardisation of the single-breath determination of carbon monoxide uptake in the lung. Eur Respir J. 2005;26:720–35.
Wanger J, Clausen JL, Coates A, Pedersen OF, Brusasco V, Burgos F, Casaburi R, Crapo R, Enright P, van der Grinten CP, et al. Standardisation of the measurement of lung volumes. Eur Respir J. 2005;26:511–22.
Holland AE, Spruit MA, Troosters T, Puhan MA, Pepin V, Saey D, McCormack MC, Carlin BW, Sciurba FC, Pitta F, et al. An official European Respiratory Society/American Thoracic Society technical standard: field walking tests in chronic respiratory disease. Eur Respir J. 2014;44:1428–46.
Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, Colby TV, Cordier JF, Flaherty KR, Lasky JA, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183:788–824.
Simpson T, Barratt SL, Beirne P, Chaudhuri N, Crawshaw A, Crowley LE, Fletcher S, Gibbons MA, Hallchurch P, Horgan L, et al. The burden of progressive fibrotic interstitial lung disease across the UK. Eur Respir J. 2021;58
Nasser M, Larrieu S, Boussel L, Si-Mohamed S, Diaz F, Marque S, Massol J, Revel D, Thivolet-Bejui F, Chalabreysse L, et al. Epidemiology and mortality of non-idiopathic pulmonary fibrosis (IPF) progressive fibrosing interstitial lung disease (PF-ILD) using the French national health insurance system (SNDS) database in France: the PROGRESS study. Eur Respir J. 2020;56:444.
Goh NS, Desai SR, Veeraraghavan S, Hansell DM, Copley SJ, Maher TM, Corte TJ, Sander CR, Ratoff J, Devaraj A, et al. Interstitial lung disease in systemic sclerosis: a simple staging system. Am J Respir Crit Care Med. 2008;177:1248–54.
Jacob J, Hirani N, van Moorsel CHM, Rajagopalan S, Murchison JT, van Es HW, Bartholmai BJ, van Beek FT, Struik MHL, Stewart GA, et al. Predicting outcomes in rheumatoid arthritis related interstitial lung disease. Eur Respir J. 2019;53
Kim HC, Lee JS, Lee EY, Ha YJ, Chae EJ, Han M, Cross G, Barnett J, Joseph J, Song JW. Risk prediction model in rheumatoid arthritis-associated interstitial lung disease. Respirology. 2020;25:1257–64.
Tashkin DP, Roth MD, Clements PJ, Furst DE, Khanna D, Kleerup EC, Goldin J, Arriola E, Volkmann ER, Kafaja S, et al. Mycophenolate mofetil versus oral cyclophosphamide in scleroderma-related interstitial lung disease (SLS II): a randomised controlled, double-blind, parallel group trial. Lancet Respir Med. 2016;4:708–19.
Khanna D, Lin CJF, Furst DE, Goldin J, Kim G, Kuwana M, Allanore Y, Matucci-Cerinic M, Distler O, Shima Y, et al. Tocilizumab in systemic sclerosis: a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Respir Med. 2020;8:963–74.
Maher TM, Tudor VA, Saunders P, Gibbons MA, Fletcher SV, Denton CP, Hoyles RK, Parfrey H, Renzoni EA, Kokosi M, et al. Rituximab versus intravenous cyclophosphamide in patients with connective tissue disease-associated interstitial lung disease in the UK (RECITAL): a double-blind, double-dummy, randomised, controlled, phase 2b trial. Lancet Respir Med. 2023;11:45–54.
Distler O, Highland KB, Gahlemann M, Azuma A, Fischer A, Mayes MD, Raghu G, Sauter W, Girard M, Alves M, et al. Nintedanib for systemic sclerosis-associated interstitial lung disease. N Engl J Med. 2019;380:2518–28.
Hambly N, Farooqi MM, Dvorkin-Gheva A, Donohoe K, Garlick K, Scallan C, Chong SG, MacIsaac S, Assayag D, Johannson KA, et al. Prevalence and characteristics of progressive fibrosing interstitial lung disease in a prospective registry. Eur Respir J. 2022;60
Joy GM, Arbiv OA, Wong CK, Lok SD, Adderley NA, Dobosz KM, Johannson KA, Ryerson CJ. Prevalence, imaging patterns and risk factors of interstitial lung disease in connective tissue disease: a systematic review and meta-analysis. Eur Respir Rev. 2023;32
Fu H, Zheng Z, Zhang Z, Yang Y, Cui J, Wang Z, Xue J, Chi S, Cao M, Chen J. Prediction of progressive pulmonary fibrosis in patients with anti-synthetase syndrome-associated interstitial lung disease. Clin Rheumatol. 2023;42:1917–29.
Ergun R, Arsava BE. Acute phase reactants as predictors for hospital mortality in acute respiratory failure due to COPD exacerbation. Eur Respir J. 2013;42:P2451.
Chen YH, Lee TJ, Hsieh HJ, Hsieh SC, Wang HC, Chang YC, Yu CJ, Chien JY. Clinical outcomes and risk factors of progressive pulmonary fibrosis in primary Sjögren’s syndrome-associated interstitial lung disease. BMC Pulm Med. 2023;23:268.
Flaherty KR, Thwaite EL, Kazerooni EA, Gross BH, Toews GB, Colby TV, Travis WD, Mumford JA, Murray S, Flint A, et al. Radiological versus histological diagnosis in UIP and NSIP: survival implications. Thorax. 2003;58:143–8.
Solomon JJ, Chung JH, Cosgrove GP, Demoruelle MK, Fernandez-Perez ER, Fischer A, Frankel SK, Hobbs SB, Huie TJ, Ketzer J, et al. Predictors of mortality in rheumatoid arthritis-associated interstitial lung disease. Eur Respir J. 2016;47:588–96.
Acknowledgements
None.
Funding
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF), which is funded by the Ministry of Education (2021R1A4A5032806). The funder had no role in study design, data collection, and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
HCK takes responsibility for the content of the manuscript, including the data and analysis. JKL, SML and HCK contributed to the study conception and design. JKL, YA, HNN, SML, BY, CKL, YGK, SH, SMA, and HCK contributed to the acquisition of data. JKL, YA, HNN, SML, and HCK contributed to the analysis and interpretation of data. JKL and HCK drafted the manuscript. JKL, YA, HNN, SML, BY, CKL, YGK, SH, SMA, and HCK had access to the final version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest related to this study.
Ethical approval
This study adhered to the principles outlined in the Declaration of Helsinki and was carried out with the endorsement of the Institutional Review Board of Asan Medical Center (IRB no. 2022-1564).
Informed consent
The need for informed consent was waived by the Institutional Review Board due to the utilization of anonymous clinical data and the retrospective nature of the study.
Consent for publication
Not applicable.
Availability of data and materials
The datasets utilized and/or examined in the present study can be obtained from the corresponding author upon a reasonable request.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Additional file 1: Supplementary TABLE 1.
Baseline autoantibodies of 197 patients with connective tissue disease-associated interstitial lung disease based on progressive pulmonary fibrosis

Additional file 2: Figure S1.
Kaplan–Meier plot of the cumulative mortality rate based on the diagnosis of progressive pulmonary fibrosis in patients with rheumatoid arthritis-related interstitial lung disease (ILD) (A) and patients with inflammatory myositis-related ILD (B).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

{kind=link}
Cite this article
Lee, J.K., Ahn, Y., Noh, H.N. et al. Clinical effect of progressive pulmonary fibrosis on patients with connective tissue disease-associated interstitial lung disease: a single center retrospective cohort study. Clin Exp Med 23, 4797–4807 (2023). https://doi.org/10.1007/s10238-023-01212-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10238-023-01212-z