
Abstract
There are known differences in the properties of hair cells along the tonotopic axis of the avian auditory epithelium, the basilar papilla (BP). To determine the genetic basis of these differences, we compared gene expression between the high- (HF), middle-, and low-frequency (LF) thirds of 0-day-old chick auditory epithelia. RNA amplified from each sample was hybridized to whole-genome chicken arrays and GeneSpring software was used to identify differentially expressed genes. Two thousand six hundred sixty-three genes were found to be differentially expressed between the HF and LF segments, using a fold-change cutoff of 2 and a p value of 0.05. Many ion channel genes were differentially expressed between the HF and LF regions of the BP, an expression pattern that was previously established for some but not all of these genes. Quantitative PCR was used to verify tonotopic expression of 15 genes, including KCNMA1 (Slo) and its alternatively spliced STREX exon. Gene set enrichment analyses (GSEA) were performed on the microarray data and revealed many microRNA gene sets significantly enriched in the HF relative to the LF end, suggesting a tonotopic activity gradient. GSEA also suggested differential activity of the kinases protein kinase C and protein kinase A at the HF and LF ends, an interesting corollary to the observation that there is tonotopic expression of the STREX exon that confers on Slo sensitivity to the activity of kinases. Taken together, these results suggest mechanisms of induction and maintenance of tonotopicity and enhance our understanding of the complex nature of proximal–distal gene expression gradients in the chicken BP.
Similar content being viewed by others


Introduction
The basilar papilla (BP), the chicken auditory epithelium, is tonotopically organized much like its mammalian counterpart the organ of Corti. Hair cells residing at specific locations along the apical-basal axis are maximally responsive to sounds at a particular frequency. In non-mammalian vertebrates such as turtles (Crawford and Fettiplace 1981) and birds (Fuchs et al. 1988), this tuning seems to be largely mediated by intrinsic electrical resonance properties of hair cells. Electrical resonance in these cells is mediated by interplay between a depolarizing voltage-gated calcium current through calcium channels and a hyperpolarizing calcium-sensitive potassium current through BK channels.
Both hair cell physiological activity and gene expression are known to vary along the tonotopic axis of auditory epithelia. There are also differences in the properties of the transduction apparatus along the tonotopic axis. The mechanotransduction channels at the HF end of the turtle BP pass more current per channel than those in the LF end, allowing for faster adaptation (Ricci et al. 2003). As another example, there are changes in the electrical tuning apparatus along the length of the BP. Both models and data confirm the expression of inward and delayed rectifying potassium channels in hair cells in the low-frequency portion of the sensory epithelium, while BK channels and L-type calcium channels are present in higher density in hair cells in the high-frequency region (Art et al. 1995; Wu et al. 1995; Samaranayake et al. 2004). Differences in the density and properties of these channels are believed to give rise to the higher resonant frequency of cells in the HF end of the BP (Navaratnam et al. 1997; Rosenblatt et al. 1997; Samaranayake et al. 2004). Specifically, the higher resonant frequency of HF hair cells seems to be partly due to an increase in calcium influx and an increase in potassium efflux through an increased number of potassium and calcium channels (Art et al. 1995; Wu et al. 1995; Tucker et al. 1996).
In light of what is known about tonotopic gradients of gene expression and hair cell physiology, we endeavored to use a genome-wide comparison of gene expression between the high-frequency (HF), middle-frequency (MF), and low-frequency (LF) segments of the post-hatch chicken BP to identify tonotopically expressed genes. Genes found to have this expression pattern may be functionally significant and may therefore provide new insight into BP structure and function. Specifically, tonotopically expressed genes may represent differences in development, susceptibility to injury, or regenerative capacity along the tonotopic axis. We expected that such a data set would identify gene expression regulators such as transcription factors and microRNAs (miRNA) that are important for the development and/or maintenance of these transcriptional gradients. Affymetrix whole-genome chicken microarrays were therefore used to compare transcription profiles between the HF (basal), MF, and LF (apical) thirds of the post-hatch chicken BP. Many of the genes found to be differentially expressed between the two extreme ends of the epithelium were ion channels, some known to be tonotopically expressed and others with a previously uncharacterized pattern of expression. Given limitations in sensitivity and specificity of microarrays, gene set enrichment analyses (GSEA) were also performed. These analyses complement the microarray results as they are more specific and are less susceptible to systematic bias from false results. The GSEA results suggested tonotopic gradients of activity of a number of miRNAs and kinases, as well as expression of genes associated with the hematopoietic stem cell phenotype.
Methods
Basilar papilla tissue preparation
Animals were treated in accordance with policies established by the Yale Institutional Animal Care and Use Committee (protocol number 2007–10439). Cochlear ducts were carefully dissected out of 0-day-old chicks. The tegmentum vasculosum and tectorial membrane were immediately dissected off to expose the auditory epithelium, which was delicately freed from the basement membrane resting on cartilaginous plates using a tuberculin needle. The entire epithelium was then sectioned into even HF, MF, and LF thirds (Fig. 1). Three segments from three different BPs went into each sample to produce a total of three HF samples, three MF samples, and three LF samples. Samples were frozen at −80°C until RNA isolation could be performed.

Shown in panel A is a schematic diagram showing the manner in which the auditory epithelia were sectioned. Low-, middle-, and high-frequency segments were pooled as described in the text to allow for comparison of genome-wide expression across the three indicated regions. Shown in panel B is a proportional Venn diagram representing overlap of differentially expressed genes for the high- versus low-frequency (HF vs. LF; 2,663), high- versus middle-frequency (HF vs. MF; 303), and middle- versus low-frequency (MF vs. LF; 1,306) comparisons. Far more genes are differentially expressed between the HF and LF segments than either of the middle-frequency comparisons, suggesting that many of the HF vs. LF genes are expressed in a gradient along the tonotopic axis of the basilar papilla.
RNA isolation
Frozen samples were removed from the −80°C freezer and immediately placed in the lysis buffer from the RNAqueous® Kit (Ambion, Austin, TX). The tissue was not actively homogenized or disrupted given the small amount of tissue. Total RNA isolation was performed as per the manufacturer’s instructions. The quality of each sample was confirmed by gel and Bioanalyzer analysis. All samples had an A260/A280 ratio of at least 1.9, as well as 18S and 28S bands with no obvious evidence of degradation or genomic contamination and RNA integrity numbers (based on 28S/18S ratio and other parameters) of at least 9 on a ten-point scale (Schroeder et al. 2006). Total RNA concentration was determined by measuring absorbance at 260 nm on a spectrophotometer.
Microarray hybridizations
Microarray hybridizations were performed as previously reported (Frucht et al. 2010). Briefly, double-stranded cDNA and biotin-labeled cRNA were synthesized from 1 to 5 μg of total RNA using a two-cycle target labeling kit (Affymetrix 2004). Biotin-labeled cRNA was purified using the GeneChip Cleanup Module prior to fragmenting to a size of 35–200 bases. Hybridization of the samples with Affymetrix whole-genome chicken microarrays was then performed at the Yale University Keck Facility according to the manufacturer’s protocol (Affymetrix 2009). This array contains many internal controls, including negative hybridization controls that serve to minimize the detection of signals resulting from nonspecific binding to probes. All microarray data is MIAME-compliant and has been uploaded to the EMBL-EBI ArrayExpress database (http://www.ebi.ac.uk/arrayexpress/) along with extensive annotations on both experimental and analytical methods as well as all raw and normalized data (accession: E-MEXP-2806).
Differential gene expression analysis
Gene expression analyses were performed using GeneSpring GX 9.0 software (Agilent Technologies, Santa Clara, CA). After GC-RMA normalization without baseline transformation, the microarray data were then filtered by expression (20–100% in at least one of the three samples from each segment of the BP) to exclude genes expressed only at very low levels in all samples. In cases of probeset redundancy, median expression values were used. Differentially expressed genes were identified by using a fold-change cutoff of 2 and then performing an unpaired t test (p < 0.05) after Benjamini–Hochberg correction for multiple comparisons. R version 2.10.1 (www.r-project.org) was used to generate a heat map using the HeatPlus add-on in the Bioconductor package.
Quantitative PCR validation of microarray expression data
In order to validate the microarray data, quantitative PCR was performed on 15 detected genes. Six of these genes were upregulated in the HF relative to the LF region, and nine were upregulated in the LF relative to the HF region. The differentially expressed genes validated by quantitative PCR (qPCR) were highly expressed in at least one region and chosen to span a range in fold-change differences between the HF and LF regions, from 2.33 to 30.61.
The 15 genes that were validated are brain-derived neurotrophic factor, glutamate receptor 2, medium neurofilament, aquaporin 5, chordin-like 1, docking protein 7, ret proto-oncogene, SLIT, and NTRK-like family member 4, wnt inhibitory factor 1, integrin A1, potassium large conductance calcium-activated channel, subfamily M, alpha member 1 (KCNMA1, Slo; total and STREX exon), potassium voltage-gated channel, shaker-related subfamily, beta member 1 (KCNAB1), potassium voltage-gated channel, shaker-related subfamily, beta member 2 (KCNAB2), transient receptor potential cation channel, subfamily C, member 1 (TRPC1), and sodium channel, voltage-gated, type II, alpha subunit (SCN2A). The RNA samples were the same ones used for microarray analysis for all genes except KCNMA1 (total and STREX containing only) and TRPC1. Total Slo (KCNMA1) expression and expression of the Slo exon STREX were determined in the HF, MF, and LF segments of the BP to assess for expression gradients. At least three samples, each comprised of three BP segments, were used. Expression of the 18S ribosomal subunit was also assessed to allow for normalization of total RNA levels between samples using the 2–ΔΔC T method (Schmittgen and Livak 2008). Primer 3 (Rozen and Skaletsky 2000) was used to design intron spanning primer pairs that were validated by melting curve analysis.
Five micrograms of RNA was isolated from each sample as described above and used to create first-strand cDNA using oligo-dT primers. Because extensive genomic contamination was made unlikely by the use of oligo-dT primers and confirmed not to be present by Bioanalyzer measurements, RNA samples were not treated with DNase. qPCR was performed using the SYBR Green Supermix reagent (Bio-Rad, Hercules, CA) on an iCycler system (Bio-Rad, Hercules, CA). To establish the sensitivity of our qPCR methodology, reactions were run with serial dilutions of amplified 18S cDNA of known concentrations to ensure that the correct product (i.e., by band size and melting temperature) could be amplified with only several copies in a well. Each reaction was run in triplicate, and the data was averaged for each cDNA sample and primer pair combination. Only those fold-changes with a 95% confidence interval that did not include the value 1 were considered significant. A fold-change of 1 between two conditions would indicate that the expression level is exactly the same in both conditions, the null hypothesis of this test. Therefore, one can conclude with 95% certainty that a fold-change interval that does not overlap with a value of 1 represents a real change in gene expression.
Gene set descriptions
GSEA was performed to gain insights into the functional relevance of tonotopic gene expression gradients using GSEA version 2 software (Subramanian et al. 2005). In a GSEA, genes are first ranked by their association with the class distinction of the comparison being made (e.g., HF versus LF). In other words, the genes most consistently overexpressed in the HF samples were ranked at one end of the list, and those most overexpressed in the LF samples were ranked at the other end. GSEA asks whether the genes that make up a particular gene set are overrepresented at either end of the list, rather than randomly distributed throughout. This question was answered by calculating for every gene set a running sum statistic known as the enrichment score (ES). The ES can be thought of as a running tally that is proportional to the number of genes in a set that are upregulated with a particular manipulation. Each ES was normalized to gene set size to produce a normalized enrichment score (NES). The statistical significance of each NES was determined by comparing that NES to the distribution of ESs generated by randomly permutating the genotype class labels. Both the p value and false discovery rate (FDR, q value) were calculated for each set. Gene sets whose NES had an associated FDR less than 0.25 and a p value less than 0.05 were considered significantly enriched.
The GeneSpring processed data from the 38,535 original probes was collapsed into 13,159 genes based on gene symbols. Some genes were assigned to one or multiple gene sets downloaded with the GSEA package. The mammalian gene set package included 837 gene sets, 50 of which were excluded by gene set size criteria (15–50), leaving 783 to be included in our analysis. One thousand one hundred ninety-seven out of the 1,892 curated gene sets also met this criterion (Table 1). The GSEA parameters used were as follows: metric = signal to noise; permutation number = 1,000; gene size minimum = 15; gene size maximum = 500; enrichment statistic = classic; permutation type = gene set.
Gene set descriptions
Two different gene set packages were used, both of which were downloaded directly from the Broad Institute Website (www.broad.mit.edu/gsea). To assess for tonotopic gradients in miRNA and transcription factor activity in the chicken gene expression data, a GSEA was performed using a gene set package called “c3.all.v2.5.symbols,” which was obtained from the BROAD Institute Website (Xie et al. 2005). This package includes gene sets defined by the presence of transcription factor motifs and predicted miRNA binding sites. Because the genes in each set of this package share regulatory motifs that are conserved across human, mouse, rat, and dog genomes, they will be referred to as the “mammalian gene sets.” The motifs used come from Xie et al. (2005) and the TRANSFAC database and include sets of genes sharing particular 3′-UTR miRNA binding motifs.
To assess the functional relevance of the pattern of differential gene expression, GSEA was performed using a gene set package called “c2.all.v2.5.symbols” which was also downloaded from the BROAD Institute Website. This package defines curated sets of genes based on specific experimental findings from experiments on human and animal model tissue. This package includes both canonical pathways and chemical/genetic perturbations based gene sets. These gene sets will be referred to as the “curated gene sets.” References for all curated gene sets are provided on the BROAD Website.
To ascertain whether any of the tonotopic gene expression gradient is attributable to protein kinase activation, two additional GSEAs were performed by generating gene sets from available data sets. To assess for tonotopic activity of protein kinase A (PKA), a set of genes putatively upregulated with increased PKA activity was used. Specifically, the data used was raw Affymetrix whole-genome microarray data from a previously reported experiment describing gene expression differences in the chicken BP after 24-h exposure to forskolin (100 μM), an adenylate cyclase activator which increases intracellular cAMP levels and therefore PKA activity (Frucht et al. 2010). These data were processed to generate a list of significantly (fold-change > 2, p value < 0.05) differentially expressed genes. The list of 39 affected genes was used for GSEA. A different data set was processed in the same fashion to produce a set of genes whose expression is putatively affected by protein kinase C (PKC) for use in a GSEA. This particular data set was from a previously reported experiment in which transcription was profiled with and without PMA-induced activation of PKC in human MM6 cells (Yu et al. 2008). The set of 3,456 genes that were differentially expressed (fold-change > 2, p value < 0.05) following PMA activation was used in a GSEA to look for evidence of tonotopic PKC activity.
Results
Gene expression analysis statistics
We report here a systematic comparison of genome-wide expression along the tonotopic axis of the chicken auditory epithelium using Affymetrix whole-genome chicken microarrays. Zero-day-old chickens were used because this developmental time point succeeds the onset of hearing (Gottlieb 1965). The complete data set (included as Supplemental Data) should therefore be of interest to those studying expression and functional gradients along the tonotopic axis of the inner ear.
To examine gene expression gradients along the tonotopic axis of BPs, cochlear ducts were explanted and sensory epithelia were immediately isolated, microdissected, and sectioned into HF, MF, and LF segments (Fig. 1). Out of the 38,535 probe sets on the Affymetrix chicken array, 2,663 were differentially expressed between the HF and LF segments; 303 were differentially expressed between the HF and MF segments, and 1,306 genes were differentially expressed between the MF and LF segments. The complete list of differentially expressed genes for each of the three comparisons (i.e., HF vs. LF, HF vs. MF, MF vs. LF) are included as Supplemental Data. A proportional Venn diagram clearly indicates that far more genes are differentially expressed between the two extreme ends of the epithelium than between the MF segment and either of the two extremes (Fig. 1). This is consistent with the notion that most genes are being expressed differentially along the tonotopic axis in an apical-to-basal gradient. Gene expression data for all genes on the array (Tables S1, S2, and S3) as well as only the subset of genes identified as differentially expressed (Tables S4, S5, and S6) are included as supplementary material.
qPCR confirms microarray data
qPCR was performed on select genes in order to validate the microarray data. Primers were designed for 15 genes, six of which were upregulated in the HF segment and nine of which were upregulated in the LF segment (Table 2). These genes were selected to span a range of fold-changes in both directions based on their high level of expression in at least one segment of the BP. Some genes were selected because they are specifically known to have an important role in the inner ear and are known to be tonotopically expressed in the BP (e.g., KCNMA1). However, most of the genes chosen were selected because their expression pattern in the BP was previously uncharacterized. Out of the 15 genes examined, qPCR confirmed the directionality of all 15. The fold-changes of 12 of these 15 genes (80%) were significantly different from a value of 1 (Table 2), confirming their differential expression as detected by microarray. These findings validate the microarray results, so further analyses were performed on the gene expression data set.
Slo (KCNMA1) and its alternatively spliced STREX exon were tested by qPCR in the HF, MF, and LF segments of the BP (Fig. 2). Both showed a clear tonotopic expression gradient with higher expression in the LF than in the MF or HF segments (one-way ANOVA, p < 0.05). Slo expression at the HF end was 34.1% of that at the LF end, whereas HF expression of STREX was 10.7% of that at the LF end. These results show a steeper tonotopic gradient for STREX than for Slo, suggesting that the proportion of Slo transcripts including the STREX exon decreases going from the LF to HF segments.

Slo and the Slo exon STREX are transcribed tonotopically. Shown are the relative expression levels of both Slo and its alternatively spliced STREX exon in the low- (LF, n = 3), middle- (MF, n = 3), and high-frequency (HF, n = 4) segments of the post-hatch chicken basilar papilla as determined by qPCR. Expression levels were normalized first to 18S expression, then respectively to Slo and STREX expression levels in the LF segment. A clear tonotopic gradient can be appreciated for both Slo and STREX with expression that significantly decreased from the LF region to the HF region (one-way ANOVA, p < 0.05). The fold-change differences of LF versus MF and LF versus HF were significantly different between STREX and Slo, indicating a steeper gradient for STREX than for Slo (t tests, p < 0.05).
Tonotopic expression of structural proteins
Among the 2,663 genes found to be differentially expressed between the HF and LF segments were some genes known to have important functions in the inner ear, as well as many other genes whose functional significance is unknown (Table 3). As shown in Table 3, there was differential expression of the actin cross-linkers plastin 1 (up in LF) and plastin 2 (up in HF). Interestingly, some myosin transcripts were also tonotopically expressed. Myosin XVa expression was higher in the LF end of the BP while myosin IIIa transcription was higher in the HF end.
Tonotopic expression of sodium, potassium, and calcium channels
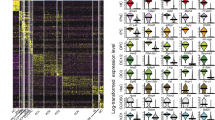
Many sodium, potassium, and calcium channels were differentially transcribed between the HF and LF ends (Table 4). An expression heat map shows that most of these genes are expressed in a gradient along the tonotopic axis (Fig. 3). Interestingly, the expression patterns of most of these channels have not been previously described. Specifically, multiple calcium-gated (i.e., KCNMA1, KCNMB4), voltage-gated (i.e., KCNQ2, KCNG4, KCNF1, KCND2, KCNAB1, KCNAB2), and inward-rectifying (i.e., KCNJ2, KCNJ15) potassium channels were all upregulated in the LF end. There was also differential expression of voltage-gated calcium channels (i.e., CACNA1B was up in the LF end; CACNA2D1 was up in the HF end) between the two ends of the BP. Additionally, there were sodium channels found by microarray differentially expressed between the HF and LF segments; SCN2A (voltage-gated) and SCNN1A (nonvoltage-gated) were up in the HF end of the BP, whereas SCN3B (voltage-gated) was up in the LF end. However, SCN2A was found by qPCR to not be differentially expressed. Finally, three transient receptor potential (TRP) channels were found to be tonotopically expressed. TRPC1 transcription was higher at the LF end, a finding confirmed by qPCR (Table 2). TRPC3 and TRPM3 were found by microarray to be upregulated in the HF end, but were undetectable by qPCR. These observations suggest important functional roles for some of these channels in the chicken inner ear.

Many ion channels are expressed in a gradient along the tonotopic axis of the chicken basilar papilla. Shown is a heat map showing normalized expression levels in the high- (HF), middle- (MF), and low-frequency (LF) segments of the basilar papilla for 15 ion channel genes. The depicted expression values are all normalized relative to the average for each row (i.e., the average expression for a particular gene across all three segments). As shown in the color key, light rectangles represent higher expression levels than dark rectangles. Most of the genes shown appear to be expressed in a gradient along the tonotopic axis.
Gene set enrichment analysis results
In order to determine the large-scale functional significance of differential gene expression, a GSEA was performed using manually curated gene sets downloaded from the Broad Institute Website. This package contains gene sets comprised of genes found by individual experiments to be associated with specific phenotypes in a variety of systems. Of the 1,197 gene sets included in this package, 293 were significantly enriched in the HF end whereas 82 were enriched in the LF end (Table 1). Interestingly, many gene sets comprised of genes associated with hematopoietic stem cells (HSC) are significantly enriched in the HF end of the BP, while only one HSC gene set is enriched in the LF end (Table 5). This finding is suggestive of a subpopulation of cells with HSC-like gene expression patterns in the HF end of the chick BP. GSEA statistics for all curated gene sets are included as supplemental material (Tables S7, S8, and S9).
To determine whether any miRNAs or transcription factors are differentially active between the HF and LF segments, an additional GSEA was performed using a gene set package with gene sets comprised of genes containing particular transcription factor and miRNA recognition sites. Fifty-six out of 783 of the gene sets in this set are enriched in the HF end, whereas 18 of the gene sets are significantly upregulated in the LF end (Table 1). Interestingly, many of the differentially expressed gene sets are miRNA (“MIR”) gene sets (Table 6). These results suggest tonotopic expression and therefore some functional relevance of these particular miRNAs in the chicken inner ear. GSEA statistics for all of these motif-based gene sets are included as supplemental material (Tables S10, S11, and S12).
GSEA was also performed using a PKC-based gene set and a PKA-based gene set as described in the Methods. A set of genes found to be differentially expressed following activation of PKC using PMA in human MM6 cells was significantly enriched in the HF end of the BP (Table 5; FDR < 0.001). Additionally, a set of genes differentially expressed in the chicken BP after 24 h of forskolin-induced PKA activation were significantly enriched in the LF end (Table 5; FDR = 0.002). These results suggest differential activation and/or expression of these kinases themselves along the tonotopic axis of the chicken BP.
Discussion
Reported here are 2,663 genes that are differentially expressed between the HF (i.e., proximal) and LF (i.e., distal) thirds of the BP. Among the list of genes found to be differentially expressed are many that would be predicted to show this pattern, as well as many with a previously uncharacterized pattern of expression or functional activity in the BP. Our data revealed that two inward-rectifying potassium channels (KCNJ2, KCNJ15) are transcribed at higher levels at the apical end, an interesting observation given that previous work has shown preferential expression of inward-rectifying potassium channels at the LF end of the BP (Fuchs et al. 1990; Fuchs 1992; Navaratnam et al. 1995; Wu et al. 1996). Also suggested by our microarray data is relative upregulation of a number of voltage-gated potassium channels in the LF end (Table 4), confirming physiological data on the presence of voltage-gated potassium currents preferentially found in LF hair cells of the turtle BP (Fuchs et al. 1990; Goodman and Art 1996). KCNMA1 was identified by our microarray data to be upregulated in the LF end of the BP, confirming recently reported results (Miranda-Rottmann et al. 2010). Similarly, there were increased amounts of the sodium channel transcript SCN3B in the LF end and of SCNN1A in the HF end. While sodium channels have not been observed in the chicken, hair cells from the LF end of alligator BP were found to contain TTX-sensitive currents (Evans and Fuchs 1987). Actin was detected by our array but found not to be tonotopically expressed (data not shown), a finding that is intuitive given that the total amount of actin per hair cell does not vary tonotopically in chickens (Tilney and Tilney 1988). The calcium binding protein calbindin was previously found to be expressed at higher levels in basal than in apical hair cells (Navaratnam et al. 1995; Navaratnam et al. 1997; Hiel et al. 2002), a finding that was not confirmed by our microarray data. However, this observation is likely the result of the 3′ bias of the Affymetrix chicken array. The NCBI Module Maker lists all chicken calbindin mRNAs (Gallus gallus Build 2.1, Chromosome: 2, Contig: NW_001471651.1) including ESTs, the vast majority of which (24/25) do not contain the sequence targeted by the calbindin probeset on this array. In contrast, previous work showing tonotopicity of calbindin expression used qPCR primers designed to amplify a portion of the transcript that is present in all 25 variants, and these results are therefore much more reliable (Navaratnam et al. 1995, 1997; Hiel et al. 2002). That calbindin was not found by microarray to be tonotopically expressed therefore likely reflects the spatial distribution of an exceedingly rare splice variant. Despite the known limitations of microarrays, these observations and the good correlation with qPCR serve to instill confidence that this data set generally reflects actual gene expression levels.
Among the genes examined here are genes whose products are known components of stereocilia tip-links. For example, the stereocilia tip-link protein protocadherin-15 was found by microarray to be upregulated in the LF end of the BP. However, protocadherin-15 was not detectable by qPCR (data not shown), suggesting that its identification by microarray was likely a false-positive result. Previous work has also shown a temporal but no appreciable spatial gradient of protocadherin-15 in the postnatal mouse organ of Corti (Lelli et al. 2009). The same study showed that protocadherin-15 expression is relatively high in embryonic mice, but drops to extremely low levels after birth. Our data suggest that protocadherin-15 is expressed at extremely low levels in the post-hatch chicken BP as well. Further work should aim to further characterize the spatiotemporal expression gradient of protocadherin-15 in BPs from animals of different ages to determine whether the pattern mirrors that seen in mammals.
KCNMA1, the transcript encoding the BK calcium-activated potassium channel, was upregulated in the LF end of the BP, confirming recently reported data (Miranda-Rottmann et al. 2010). However, the results from both studies are in apparent disagreement with both protein expression (Samaranayake et al. 2004) and electrophysiological data (Art et al. 1986, 1995) suggesting higher BK protein expression in the HF end of the BP. This discrepancy is likely due to post-transcriptional control of KCNMA1 (Bai et al. 2010, under review). There is precedent for this observation, as previous work has shown dissociation between mRNA expression and protein levels (Gygi et al. 1999; Ghaemmaghami et al. 2003). We have since established that Slo delivery to the surface of the cell is inhibited by beta-1 and beta-4 subunits (Bai et al. 2010, under review), which are expressed at higher levels in LF hair cells (Ramanathan et al. 1999, 2000; Bai et al. 2010, under review).
The TRP channel TRPC1 was found by qPCR to be upregulated in the LF end of the BP by both microarray and qPCR. Differential expression of TRP channels along the tonotopic axis is of particular interest given that they are among a small subset of channel families thought to possess the characteristics required of the as yet unidentified mechanoelectrical transduction (MET) channel in the inner ear (reviewed in Fettiplace 2009). Members of the TRP family are known to act as mechanically gated ion channels in a variety of species (reviewed in Christensen and Corey 2007). TRP proteins are capable of forming heteromultimeric channels with conductances that vary with channel composition (Bai et al. 2008; Köttgen et al. 2008). Channel composition may therefore partly give rise to the tonotopic gradient in MET channel conductance that is seen along the BP (Ricci et al. 2003). The present findings suggest closer examination of TRPC1 as a potential component of the transduction apparatus, especially in light of this protein’s known expression in the organ of Corti (Cuajungco et al. 2007).
Given the limitations of microarray data sets, GSEA was performed to gain more meaningful insight into the spatial patterns of gene expression in the BP. Previous work has confirmed the utility of using GSEA to identify biologically important players in the BP (Frucht et al. 2010). One advantage of this approach is that gene sets found to be significantly enriched can be interpreted with confidence as they are not systematically biased by false-positives and false-negatives, which serve only to increase background noise in the data and decrease the sensitivity of this computational approach. Therefore, some genes sets that are actually enriched may be missed, but positive GSEA results are likely to reflect real gene expression patterns.
The GSEA results described here shed new light on the intricacy of tonotopicity in the post-hatch chicken BP. Interestingly, many sets of genes associated with HSC were enriched in the HF end of the BP. This finding suggests that there may exist a subpopulation of cells in the BP with properties similar to those of HSCs in the BP. HSCs, much like non-mammalian vertebrate hair cells, are quiescent until stimulated by external cues to divide and differentiate into new hair cells (Ryals and Rubel 1988; Jude et al. 2008; Narbonne and Roy 2008). The tonopocity of this pattern of gene expression is interesting, given that the HF end of the BP is more susceptible than the LF end to death following ototoxic insult (Matz et al. 1965; Johnstone and Boyle 1967). It may be more than merely coincidental that HSC genes are enriched in the HF end of the BP, which is the end of the BP that is most susceptible to damage by ototoxins. In birds, following ototoxic or traumatic injury, the entire auditory sensory epithelium regenerates so that morphology and architecture are almost completely restored (reviewed in Stone and Cotanche 2007). The present results therefore beg the question, are there as yet unobserved differences in regenerative mechanisms and/or capacity along the tonotopic axis of the BP? Further experimentation will be required to definitively answer this question.
In addition to the HSC gene sets, GSEA revealed that many sets of predicted targets of specific miRNAs were enriched in the HF end of the BP, suggesting upregulation of these miRNAs in the LF end. It is striking that all 22 miRNA gene sets that were significantly enriched showed this directionality. Myriad miRNA have already been shown not only to be expressed in the inner ear (Weston et al. 2006; Sacheli et al. 2009; Wang et al. 2010), but also to play an important role in inner ear development (Friedman et al. 2009; Soukup 2009; Soukup et al. 2009), function (Lewis et al. 2009; Mencía et al. 2009), and hair cell regeneration (Frucht et al. 2010). The present results suggest a tonotopic gradient in activity and accordingly expression in the chicken BP. Tonopocity of miRNA activity in the post-hearing onset chicken suggest a role for these specific miRNAs in inner ear function.
GSEA of tonotopic data were consistent with increased activity of PKA at the low-frequency end of the papilla and PKC activity at the high-frequency end of the BP. These findings have implications for electrical tuning. The primary determinant of changing oscillatory frequency in membrane potential is the changing kinetic properties of the BK channel (Art et al. 1986, 1995). Specifically, deactivation times in response to a step voltage decreases with increase in characteristic frequency (Art et al. 1986, 1995). Thus, the inclusion of the STREX exon, which is preferentially expressed in LF hair cells, prolongs BK channel deactivation times consistent with native BK channels at this location (Art et al. 1986, 1995; Xie and McCobb 1998). Similarly, the presence of increased PKA activity in low-frequency hair cells is consistent with the longer deactivation times produced by addition of PKA (with the STREX exon and the beta-4 subunit) in heterologous expression systems (Petrik and Brenner 2007). The inference that PKC is increased in high-frequency hair cells corroborates the physiological finding of shorter open times in higher-frequency hair cells (Art et al. 1995). PKC has been shown to shorten BK channel open time (Zhou et al. 2010).
It is worth emphasizing that all of these experiments were performed on tissue from 0-day-old chickens. This age was selected for study because, unlike in mammals, there is much evidence that, in chickens, this time point is preceded by both acquisition and maturation of hearing. This observation is confirmed by behavioral (i.e., whole animal), physiological, and molecular evidence. For example, not only do chickens at embryonic day 19 have evoked cochlear nuclei responses (Saunders et al. 1973, 1974), but the frequency tuning and threshold levels of embryonic chickens resemble those of adult animals (Rebillard and Rubel 1981). Furthermore, acquisition of BK currents necessary for electrical tuning is measurable in embryonic chickens and is co-incident with hearing onset (Sokolowski et al. 1993). Interestingly, recent work suggests that part of this maturation process may involve developmental switching in the splice variants of BK channel subunit transcripts (Kim et al. 2010). Moreover, work in our lab has confirmed an identical spatial expression pattern of transcripts encoding KCNMA1 and its related proteins between 0-day-old and 14-day-old chickens (Bai and Navaratnam, unpublished observations). It therefore seems unlikely that genes found to be differentially expressed here represent transient developmental changes rather than true stable tonotopic expression, but this possibility cannot be definitively excluded. Future work should therefore aim to characterize these expression gradients in more mature BPs as well. An additional point that requires consideration is that, as was likely the case with calbindin, the microarray probesets may only be detecting a specific transcript variant when in reality there may be many additional variants in this tissue, perhaps some of which are tonotopically expressed. Therefore, in addition to the well-known methodological limitations of microarrays, there are also biological implications to the sensitivity of the approach.
In summary, the present results suggest that 2,663 genes are differentially expressed between the HF and LF thirds of the post-hatch chicken BP. While many of these genes likely reflect tonotopic differences in hair cells, which are known to have varying properties, some of these genes may reflect tonotopic differences in non-sensory supporting cells as well. Among those genes identified as differentially expressed were many potassium, calcium, and sodium channels, the inner ear expression patterns of which have not been previously described. Given the large number of genes found to be differentially expressed between the HF and LF ends of the BP, there must be gene expression regulatory mechanisms underlying this expression pattern. The present results point to specific miRNA and transcription factors that may play an important role in the maintenance of these gene expression gradients. Because the frequency selectivity properties of hair cells in non-mammalian vertebrates seem to be attributable more to the physiological properties of individual cells than to physical properties of the basilar membrane, it would be interesting to eventually determine whether transfection with pre-miRNAs and/or transcription factors can alter the tonotopic axis.
References
Affymetrix I (2004) Eukaryotic sample and array processing. GeneChip expression analysis technical manual. Santa Clara, CA, Affymetrix, Inc. 701021 Rev. 5:2.1.3–2.3.18
Affymetrix I (2009) GeneChip. Expression analysis technical manual.
Art JJ, Crawford AC et al (1986) Electrical resonance and membrane currents in turtle cochlear hair cells. Hear Res 22:31–36
Art JJ, Wu YC et al (1995) The calcium-activated potassium channels of turtle hair cells. J Gen Physiol 105(1):49–72
Bai C-X, Giamarchi A et al (2008) Formation of a new receptor-operated channel by heteromeric assembly of TRPP2 and TRPC1 subunits. EMBO Rep 9(5):472–479
Bai JP, Surguchev A et al (2010) The beta-4 subunit increases Slo responsiveness to physiological Ca2+ concentrations and together with beta-1 reduces surface expression of Slo in hair cells. Am J Physiol Cell Physiol under review.
Christensen AP, Corey DP (2007) TRP channels in mechanosensation: direct or indirect activation? Nat Rev Neurosci 8(7):510–521
Crawford AC, Fettiplace R (1981) An electrical tuning mechanism in turtle cochlear hair cells. J Physiol (Lond) 312:377–412
Cuajungco MP, Grimm C et al (2007) TRP channels as candidates for hearing and balance abnormalities in vertebrates. Biochim Biophys Acta 1772(8):1022–1027
Evans MG, Fuchs PA (1987) Tetrodotoxin-sensitive, voltage-dependent sodium currents in hair cells from the alligator cochlea. Biophys J 52(4):649–652
Fettiplace R (2009) Defining features of the hair cell mechanoelectrical transducer channel. Pflugers Arch - Eur J Physiol 458(6):1115–1123
Friedman LM, Dror AA et al (2009) MicroRNAs are essential for development and function of inner ear hair cells in vertebrates. Proc Natl Acad Sci USA 106(19):7915–7920
Frucht CS, Uduman M et al (2010) Gene expression analysis of forskolin treated basilar papillae identifies MicroRNA181a as a mediator of proliferation. PLoS ONE 5(7):e11502
Fuchs PA (1992) Ionic currents in cochlear hair cells. Prog Neurobiol 39(5):493–505
Fuchs PA, Nagai T et al (1988) Electrical tuning in hair cells isolated from the chick cochlea. J Neurosci 8(7):2460–2467
Fuchs PA, Evans MG et al (1990) Calcium currents in hair cells isolated from the cochlea of the chick. J Physiol (Lond) 429:553–568
Ghaemmaghami S, Huh W-K et al (2003) Global analysis of protein expression in yeast. Nature 425(6959):737–741
Goodman MB, Art JJ (1996) Variations in the ensemble of potassium currents underlying resonance in turtle hair cells. J Physiol (Lond) 497(Pt 2):395–412
Gottlieb G (1965) Prenatal auditory sensitivity in chickens and ducks. Science 147:1596–1598
Gygi SP, Rochon Y et al (1999) Correlation between protein and mRNA abundance in yeast. Mol Cell Biol 19(3):1720–1730
Hiel H, Navaratnam DS et al (2002) Topological and developmental gradients of calbindin expression in the chick’s inner ear. J Assoc Res Otolaryngol 3(1):1–15
Johnstone BM, Boyle AJ (1967) Basilar membrane vibration examined with the Mössbauer technique. Science 158(799):389–390
Jude CD, Gaudet JJ et al (2008) Leukemia and hematopoietic stem cells: balancing proliferation and quiescence. Cell Cycle 7(5):586–591
Kim J-M, Beyer R et al (2010) Expression of BK-type calcium-activated potassium channel splice variants during chick cochlear development. J Comp Neurol 518(13):2554–2569
Köttgen M, Buchholz B et al (2008) TRPP2 and TRPV4 form a polymodal sensory channel complex. J Cell Biol 182(3):437–447
Lelli A, Asai Y et al (2009) Tonotopic gradient in the developmental acquisition of sensory transduction in outer hair cells of the mouse cochlea. J Neurophysiol 101(6):2961–2973
Lewis MA, Quint E et al (2009) An ENU-induced mutation of miR-96 associated with progressive hearing loss in mice. Nat Genet 41(5):614–618
Matz GJ, Wallace TH et al (1965) The ototoxicity of kanamycin. A comparative histopathological study. The Laryngoscope 75(11):1690–1698
Mencía A, Modamio-Høybjør S et al (2009) Mutations in the seed region of human miR-96 are responsible for nonsyndromic progressive hearing loss. Nat Genet 41(5):609–613
Miranda-Rottmann S, Kozlov AS et al (2010) Highly specific alternative splicing of transcripts encoding BK channels in the chicken’s cochlea is a minor determinant of the tonotopic gradient. Mol Cell Biol 1–45
Narbonne P, Roy R (2008) Genes that affect both cell growth and polarity mediate stem cell quiescence. Front Biosci 13:995–1002
Navaratnam DS, Escobar L et al (1995) Permeation properties and differential expression across the auditory receptor epithelium of an inward rectifier K+ channel cloned from the chick inner ear. J Biol Chem 270(33):19238–19245
Navaratnam DS, Bell TJ et al (1997) Differential distribution of Ca2+-activated K+ channel splice variants among hair cells along the tonotopic axis of the chick cochlea. Neuron 19(5):1077–1085
Petrik D, Brenner R (2007) Regulation of STREX exon large conductance, calcium-activated potassium channels by the beta4 accessory subunit. Neuroscience 149(4):789–803
Ramanathan K, Michael TH et al (1999) A molecular mechanism for electrical tuning of cochlear hair cells. Science 283(5399):215–217
Ramanathan K, Michael TH et al (2000) beta subunits modulate alternatively spliced, large conductance, calcium-activated potassium channels of avian hair cells. J Neurosci 20(5):1675–1684
Rebillard G, Rubel EW (1981) Electrophysiological study of the maturation of auditory responses from the inner ear of the chick. Brain Res 229(1):15–23
Ricci AJ, Crawford AC et al (2003) Tonotopic variation in the conductance of the hair cell mechanotransducer channel. Neuron 40(5):983–990
Rosenblatt KP, Sun ZP et al (1997) Distribution of Ca2+-activated K+ channel isoforms along the tonotopic gradient of the chicken’s cochlea. Neuron 19(5):1061–1075
Rozen S, Skaletsky HJ (eds) (2000) Primer3 on the WWW for general users and for biologist programmers. Bioinformatics methods and protocols: methods in molecular biology. Humana Press, Totowa, NJ
Ryals BM, Rubel EW (1988) Hair cell regeneration after acoustic trauma in adult Coturnix quail. Science 240(4860):1774–1776
Sacheli R, Nguyen L et al (2009) Expression patterns of miR-96, miR-182 and miR-183 in the developing inner ear. Gene Expression Patterns 9(5):364–370
Samaranayake H, Saunders JC et al (2004) Ca(2+) and K(+) (BK) channels in chick hair cells are clustered and colocalized with apical-basal and tonotopic gradients. J Physiol (Lond) 560(Pt 1):13–20
Saunders JC, Coles RB et al (1973) The development of auditory evoked responses in the cochlea and cochlear nuclei of the chick. Brain Res 63:59–74
Saunders JC, Gates GR et al (1974) Brain-stem evoked responses as an index of hearing thresholds in one-day-chicks and ducklings. J Comp Physiol Psychol 86(3):426–431
Schmittgen TD, Livak KJ (2008) Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 3(6):1101–1108
Schroeder A, Mueller O et al (2006) The RIN: an RNA integrity number for assigning integrity values to RNA measurements. BMC Mol Biol 7:3
Sokolowski BH, Stahl LM et al (1993) Morphological and physiological development of vestibular hair cells in the organ-cultured otocyst of the chick. Dev Biol 155(1):134–146
Soukup GA (2009) Little but loud: small RNAs have a resounding affect on ear development. Brain Res 1277:104–114
Soukup GA, Fritzsch B et al (2009) Residual microRNA expression dictates the extent of inner ear development in conditional Dicer knockout mice. Dev Biol 328(2):328–341
Stone J, Cotanche D (2007) Hair cell regeneration in the avian auditory epithelium. Int J Dev Biol 51(6–7):633–647
Subramanian A, Tamayo P et al (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102(43):15545–15550
Tilney LG, Tilney MS (1988) The actin filament content of hair cells of the bird cochlea is nearly constant even though the length, width, and number of stereocilia vary depending on the hair cell location. J Cell Biol 107(6 Pt 2):2563–2574
Tucker T, Art JJ et al (1996) Routes of calcium entry and extrusion in turtle hair cells. Ann N Y Acad Sci 781:123–137
Wang X-R, Zhang X-M et al (2010) MicroRNA expression in the embryonic mouse inner ear. NeuroReport 21(9):611–617
Weston MD, Pierce ML et al (2006) MicroRNA gene expression in the mouse inner ear. Brain Res 1111(1):95–104
Wu YC, Art JJ et al (1995) A kinetic description of the calcium-activated potassium channel and its application to electrical tuning of hair cells. Prog Biophys Mol Biol 63(2):131–158
Wu YC, Tucker T et al (1996) A theoretical study of calcium microdomains in turtle hair cells. Biophys J 71(5):2256–2275
Xie J, McCobb DP (1998) Control of alternative splicing of potassium channels by stress hormones. Science 280(5362):443–446
Xie X, Lu J et al (2005) Systematic discovery of regulatory motifs in human promoters and 3' UTRs by comparison of several mammals. Nature 434(7031):338–345
Yu W, Ramakrishnan R et al (2008) Cyclin T1-dependent genes in activated CD4 T and macrophage cell lines appear enriched in HIV-1 co-factors. PLoS ONE 3(9):e3146
Zhou X-B, Wulfsen I et al (2010) Dual role of protein kinase C on BK channel regulation. Proc Natl Acad Sci USA 107(17):8005–8010
Acknowledgments
This work is part of a dissertation submitted to fulfill in part the requirements for the degree of Doctor of Philosophy at Yale University.
CSF was supported by NIH MSTP TG 2T32GM07205 and an Ohse Grant for Surgical Research at Yale School of Medicine. CSF, JSS, and DSN were supported by NIH/NIDCD grants DC 007894, DC 000273, and DC 008130. The authors would like to acknowledge the support of the National Organization for Hearing Research, which provided seed funding for this project to DSN. MU was supported by NIH Grant T15 LM07056 from the National Library of Medicine.
We thank Adam Thornberg and Shrikant Mane, PhD, Yale Keck Facility, for assistance with the RNA amplification and microarray hybridizations.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Table S1
HF vs. LF—fold-change and p values for all genes (XLS 5660 kb)
Table S2
HF vs. MF—fold-change and p values for all genes (XLS 5662 kb)
Table S3
MF vs. LF—fold-change and p values for all genes (XLS 5662 kb)
Table S4
Differentially expressed genes for HF vs. LF (XLS 469 kb)
Table S5
Differentially expressed genes for HF vs. MF (XLS 74 kb)
Table S6
Differentially expressed genes for MF vs. LF (XLS 239 kb)
Table S7
Results of GSEA using curated gene sets, HF vs. LF (XLS 210 kb)
Table S8
Results of GSEA using curated gene sets, HF vs. MF (XLS 210 kb)
Table S9
Results of GSEA using curated gene Sets, MF vs. LF (XLS 212 kb)
Table S10
Results of GSEA using mammalian gene sets, HF vs. LF (XLS 142 kb)
Table S11
Results of GSEA Using Mammalian Gene Sets, HF v. MF (XLS 142 kb)
Table S12
Results of GSEA using mammalian gene sets, MF vs. LF (XLS 142 kb)
Rights and permissions
About this article
Cite this article
Frucht, C.S., Uduman, M., Kleinstein, S.H. et al. Gene Expression Gradients along the Tonotopic Axis of the Chicken Auditory Epithelium. JARO 12, 423–435 (2011). https://doi.org/10.1007/s10162-011-0259-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10162-011-0259-2