
Abstract
Epstein–Barr virus-positive gastric cancer [EBV (+) GC] is associated with EBV infection and is one of the GC subtypes defined by the Cancer Genome Atlas. EBV (+) GC has several distinct genomic or epigenomic features and clinicopathological characteristics compared with other molecular subtypes of GC. Here, we summarize the unique features of EBV (+) GC including the clinical and histopathological features, and discuss associated genetic and epigenetic aberrations. We also discuss noncoding RNAs [EBV-encoded RNAs and EBV-encoded microRNAs (miRNAs)] derived from EBV-infected cells, which have not been described in detail previously. These noncoding RNAs are defined by their roles; for example, EBV-encoded miRNAs play pivotal roles in oncogenesis and tumor progression in EBV (+) GC. We also discuss recent advances in therapeutic modalities for EBV (+) GC, as well as the potential of EBV infection as a predictive biomarker of the response to anti-PD-1 therapy with immune checkpoint inhibitors. We introduce our recent studies focusing on AT-rich interactive domain 1A gene mutations and programmed death ligand-1 overexpression/CD274 copy-number amplification, which are recurrently identified in EBV (+) GC. Finally, based on those findings, we propose potential therapeutic options using candidate-targeted therapies against EBV (+) GC.
Similar content being viewed by others


Introduction
A recent large-scale genome sequencing study conducted by The Cancer Genome Atlas (TCGA) revealed significant heterogeneity in gastric cancer (GC), resulting in the classification of GC into four molecular subtypes: microsatellite instability (MSI), genomically stable (GS), chromosomal instability (CIN), and Epstein–Barr virus-positive gastric cancer [EBV (+) GC] [1]. Among them, EBV (+) GC is defined by infection with EBV, which is a human herpes virus 4, and by several unique molecular features [2]. EBV acts as an oncogenic virus in GC as well as in Hodgkin’s lymphoma, Burkitt lymphoma, and nasopharyngeal carcinoma, and its presence is clinically demonstrated using EBV-encoded small RNA 1 (EBER-1) in situ hybridization (ISH) [2, 3]. Although EBV is associated with GC, the mechanisms of gastric carcinogenesis induced by EBV infection are not yet fully understood. EBV (+) GC is associated with the monoclonal proliferation of EBV-infected cells and shows several distinct genomic or epigenomic characteristics compared with other molecular subtypes of GC, suggesting that EBV infection contributes to the malignant transformation of normal cells in GC [1, 4,5,6]. Here, we summarize various features of EBV (+) GC and discuss the clinical and histopathological features of EBV (+) GC (Fig. 1). Furthermore, we describe in more detail the genetic and epigenetic modifications and unique characteristics of EBV (+) GC [1, 4, 6, 7]. We introduce several recent findings of genomic or epigenomic modifications in EBV (+) GC, focusing on AT-rich interactive domain 1A (ARID1A) gene mutations and programmed death ligand-1 (PD-L1) overexpression/CD274 copy-number amplification [8,9,10,11,12,13].

Summary of clinical features, macroscopic and microscopic features, and genetic and epigenetic features in Epstein–Barr virus-positive gastric cancer [EBV (+) GC]
Clinical and histopathological features of EBV (+) GC
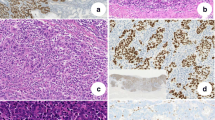
EBV (+) GC accounts for 2–20% of total GC cases, with a worldwide average of < 10% of total GC cases [1, 2, 11]. Geographical and environmental factors may affect the incidence rate, as EBV (+) GC is slightly more prevalent in Caucasians than in Asians [2]. EBV (+) GC predominantly occurs in men and at a relatively young age, and it localizes to the upper part of the stomach [2]. Consistent with previous studies, our analysis of a Japanese cohort, which included 401 GC tumors, identified 27 (6.7%) EBV (+) cases, which predominantly occurred in men, with a relatively early onset, and were located in the upper part of the stomach [11]. Typical macroscopic features of EBV (+) GC on endoscopic observation include superficial, depressed, ulcerated, or saucer-like tumors, as well as marked thickening of the gastric wall [5]. Typical microscopic features of EBV (+) GC include moderately-to-poorly differentiated adenocarcinoma with lymphocyte infiltration and a lace pattern within the mucosa [2, 5]. However, certain EBV (+) GCs manifest as conventional-type adenocarcinomas, namely, well-to-moderately differentiated adenocarcinoma without lymphocyte infiltration and a lace pattern. These cases are difficult to diagnose as EBV (+) GC without performing EBER-ISH.
Latent EBV infection
When EBV infects normal gastric epithelial cells, EBV-infected cells grow clonally, resulting in the development of EBV (+) GC. During the development of EBV-associated cancers, EBV infection is maintained in a latent form, and three types of latency states have been identified [2, 14]. EBV (+) GC belongs to latency I and II, in which EBERs (EBER-1 and EBER-2), EBV-determined nuclear antigen 1 (EBNA-1), BamHI-A rightward transcripts (BARTs), and BART microRNAs (miRNAs) are expressed and significantly associated with gastric tumorigenesis.
EBERs are the most abundantly expressed small noncoding RNAs in latent EBV-infected cells, and their involvement in malignant processes was reported previously [15, 16]. EBERs stimulate Tool-like receptor 3 (TLR3) and RIG-I in the TLR and RIG-I signaling pathways, respectively, including TANK-binding kinase 1 (TBK1) [14]. TBK1 is a serine/threonine protein kinase that plays essential roles in innate immunity, regulating inflammatory responses, and oncogenesis through the activation of the interferon regulatory factor 3 (IRF3) and IRF7 transcription factors. IRF is a family of transcriptional factors activating IFNs and regulating a differentiation and maturation of T cells, B cells, and plasma cells [17]. While IRF3 mediates immune response against viral infections, IRF3 is a key downstream transcriptional effector involved in inflammation and immunity in EBV-infected tumors. It is known that, in addition to TLR and RIG-I signaling pathways, TBK1 and IRF3 are involved in the cyclic GMP-AMP (cGAMP) synthase (cGAS)-STING signaling pathway, which is associated with the innate immune system. We recently identified a novel mechanism by which phosphorylated IRF3 induces PD-L1 overexpression in EBV (+) GC (discussed later) [11]. EBER-1, a highly expressed latency gene in EBV-infected tumors, can be used for the diagnosis of EBV infection by EBER-1-ISH.
Genetic aberrations in EBV (+) GC
Common genetic features of EBV (+) GC include frequent mutations in phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha (PIK3CA) and ARID1A and copy-number amplifications of Janus kinase 2 (JAK2) and CD274/PDCD1LG2.
Somatic mutations
ARID1A, a subunit of the Switch/Sucrose Non-fermentable (SWI/SNF) chromatin remodeling complex, is frequently mutated in GC [1, 9, 18]. ARID1A is a tumor suppressor gene and driver oncogene in GC, and most of the identified ARID1A mutations are truncating mutations that result in loss of ARID1A protein expression [1]. We and others reported that ARID1A protein expression is a useful prognostic indicator in GC or undifferentiated GC, and negative expression of ARID1A is associated with worse overall survival [8, 12, 19, 20]. According to a TCGA study, ARID1A is recurrently mutated in EBV (+) and MSI GC [1]. Analysis of the relation between ARID1A gene mutations and protein expression shows that most of the cases with ARID1A protein loss harbor ARID1A truncating mutations in MSI GC [18]. By contrast, cases with ARID1A protein loss despite the absence of ARID1A truncating mutations are detected in EBV (+) GC. These findings suggest that epigenetic modifications contribute to the loss of ARID1A protein expression in EBV (+) GC (discussed later).
PIK3CA, a p110α catalytic subunit of PI3K, is an oncogene in various cancers including GC [21, 22]. According to TCGA data, nonsynonymous mutations of PIK3CA are detected in 12% of total GC and are preferentially detected in 80% of EBV (+) GC [1]. PIK3CA mutations in EBV (+) GC are mainly localized in the kinase domain (exon 20), suggesting that the efficacy of PI3K inhibitors should be higher in patients with EBV (+) GC.
Another genetic feature of EBV (+) GC is lack of TP53 mutations. The TP53 tumor suppressor is the most recurrently mutated gene in GC; however, TP53 mutations are not frequent in EBV (+) GC among the GC subtypes classified by TCGA [1, 23, 24].
Somatic copy-number alterations
Somatic copy-number alterations are observed in various cancers and have critical roles via activating oncogenes and in inactivating tumor suppressors [25, 26]. Unique copy-number alterations with commonly amplified loci, such as 9p24.1, 17q12, and 11p13, or deleted loci, such as 16q23.1, 7q31.1, 9p24.1, 4q22.1, 10q23.31, and 20p12.1, are detected in EBV (+) GC [1, 27]. The most remarkable copy-number aberration in EBV (+) GC is focal chromosome 9p24.1 amplification, which includes JAK2, CD274, and PDCD1LG2, leading to constitutive expression of JAK2, PD-L1, and PD-L2, respectively [1]. Among these, we focused on elucidating the mechanism underlying PD-L1/CD274 overexpression in EBV (+) GC. The results showed that higher PD-L1 overexpression in EBV (+) GC tumor cells is due to high levels of CD274 focal amplification, whereas CD274 copy-number alteration is not observed in EBV (+) or EBV (−) GC tumor cells with lower PD-L1 overexpression [11]. These findings are in line with our previous report that focal and high-level amplification of CD274 results in higher PD-L1 overexpression in a small subset of small cell lung cancers, which is related to the mechanism inducing PD-L1 overexpression in EBV (+) GC. Detailed analysis of infiltrating immune cells in EBV (+) GC showed that, in contrast to EBV (+) GC tumors with higher PD-L1 overexpression due to focal CD274 amplification, those with lower PD-L1 overexpression are associated with the presence of CD8 T cells [11]. In addition, we elucidated a unique mechanism of PD-L1 overexpression in EBV (+) GC by which EBV infection activates IRF3, driving PD-L1 overexpression via interferon-γ (IFN-γ) [11, 28, 29]. TCGA data confirmed that EBV (+) GC tumors with focal CD274 amplification show higher PD-L1 overexpression, whereas those without CD274 copy-number aberrations show lower PD-L1 overexpression without infiltration of CD8 + lymphocytes [11]. These results indicate that copy-number alterations related to PD-L1 overexpression are associated with complex inflammatory signals in the tumor microenvironment of EBV (+) GC, suggesting the activation of unique innate antiviral immune responses to EBV infection.
Epigenetic aberrations in EBV (+) GC
Common epigenetic features of EBV (+) GC include extensive DNA promoter hypermethylation and altered expression of EBV-encoded miRNAs.
Methylation
DNA methylation has also known to have critical roles in tumorigenesis in the control of certain tissue-specific gene activity [30, 31]. Aberrant CpG hypermethylation in promoter and non-promoter CpG islands is a unique epigenetic aberration in EBV (+) GC. DNA hypermethylation of CDKN2A, but not the MLH1 promoter, is a distinct epigenetic feature of EBV (+) GC [1]. Many genes involved in cell cycle regulation, DNA repair, and apoptosis show hypermethylation in the CpG DNA promoter, which suppresses their expression in EBV (+) GC [2, 7]. A TCGA study identified several methylation-related silencing genes (RCOR2, RHOF, TMEM52, CLDN3, and HOXA10) in EBV (+) GC [27]. Recent work from our group identified markedly hypermethylated genes (ACE, SLC7A9, and TUBA8) in EBV (+) GC using the microarray dataset GSE31789, including CpG site methylation data for EBV (+) GC and EBV (−) GC [7, 10]. Consistently, EBV infection of MKN7 GC cells induces hypermethylation of EBV (+) marker genes and suppresses their expression, confirming that EBV infection-induced epigenetic modifications play a pivotal role in the oncogenesis and tumor progression of EBV (+) GC [7].
EBV-encoded miRNAs
EBV is the first virus shown to encode its own miRNAs. MiRNAs are small noncoding RNA molecules of approximately 21–25 nucleotides in length that interact with the 3′ untranslated region (UTR) of target mRNAs. MiRNAs thus regulate target gene expression at the post-transcriptional level through translational repression or by inducing the degradation of the target mRNA [32,33,34]. EBV-encoded miRNAs are divided into two major clusters, BamHI fragment H rightward open reading flame 1 (BHRF1) and BART miRNAs [2, 35]. Several predicted targets of EBV-encoded miRNAs were detected by computational prediction, and some of these were functionally investigated using an in vitro assay [36]. BHRF1 miRNAs, which encode three pri-miRNAs, show almost undetectable expression in EBV (+) GC; thus, their functional roles in lytic infection, in regulating host cell cycle and viral infection, and in progeny production, were revealed in various types of malignancy rather than in GC [37,38,39,40,41,42,43,44,45]. In GC specifically, BART miRNAs encoding 44 mature miRNAs may disrupt genes involved in apoptosis and cell cycle regulation, as confirmed in EBV (+) GC (Table 1) [2, 10, 36, 45,46,47,48,49,50,51,52,53,54,55,56,57,58,59]. It is currently accepted that EBV-encoded miRNAs play important regulatory roles in EBV-mediated gastric carcinogenesis.
We recently investigated EBV-encoded miRNAs that can regulate ARID1A expression in EBV (+) GC based on the identification of cases with ARID1A protein loss despite the lack of ARID1A truncating mutations in EBV (+) GC [18]. We first investigated epigenetic modifications such as DNA promoter hypermethylation; however, we found that ARID1A is not hypermethylated in EBV (+) GC tumors and EBV-infected GC cells [4, 10]. We therefore conducted in silico analysis to identify EBV-encoded miRNAs that may target ARID1A, and confirmed that miR-BART11-3p and miR-BART12 directly target ARID1A, downregulating ARID1A protein levels in EBV (+) GC harboring ARID1A-WT [10].
Therapeutic responses of EBV (+) GC patients in precision medicine
In this review, we summarized the clinical and histopathological features of EBV (+) GC and the genetic and epigenetic aberrations that underlie EBV-associated gastric carcinogenesis. Studies that contribute to our understanding of the molecular characteristics of these aberrations may improve the therapeutic efficacy of precision medicine for EBV (+) GC (Fig. 2). Because immune checkpoint inhibitors targeting the PD-1 axis are used for the treatment of GC, it was reported that EBV (+) tumors as well as MSI-high or a high PD-L1 combined positive score are used as predictive biomarkers of the response to anti-PD-1 mAbs in metastatic GC patients [60]. In fact, several clinical trials of immune checkpoint inhibitors in EBV (+) GC are currently underway [6]. However, tumors with PD-L1 overexpression associated with high levels of CD274 focal amplification without lymphocyte infiltration may not show a favorable response to anti-PD-1 mAbs [11]. In addition, because ARID1A is one of the most frequently mutated genes in EBV (+) GC, targeted therapies against ARID1A mutations could provide important therapeutic benefits to patients. However, because the ARID1A tumor suppressor gene is frequently inactivated, ARID1A is a poor therapeutic target by itself. Furthermore, potential candidate targets or pathways underlying ARID1A deficiency have not been identified, suggesting that specific inhibitors targeting genes downstream of ARID1A deficiency may not provide a therapeutic benefit [8]. Synthetic lethal approaches to the treatment of ARID1A-deficient tumors were recently proposed using specific inhibitors against enhancer of zeste homolog 2 (EZH2). These strategies demonstrate the selective sensitivity of EZH2 inhibitors against ARID1A-deficient GC cells, and suggest the potential efficacy of targeted therapy using a synthetic lethal approach for ARID1A-deficient EBV (+) GC [9]. Because treatment strategies based on synthetic lethality are a valid approach to the treatment of various cancers, inhibitors of poly ADP-ribose polymerase (PARP) or AKT are expected to show efficacy in EBV (+) GC harboring ARID1A mutations [61, 62]. These breakthrough treatments against ARID1A-mutated/-deficient or PD-L1 overexpressing tumors may yield positive results in EBV (+) GC.

Treatment options for a patient with EBV (+) gastric cancer in precision medicine. Possible treatments focused on specific inhibitors against ARID1A aberrations and PD-L1. Immune checkpoint inhibitors may not show efficacy for tumors with higher PD-L1 overexpression associated with CD274 focal amplification without lymphocyte infiltration (“COLD” tumor) (break line)
Conclusion
The molecular features of EBV (+) GC have been elucidated in detail including genetic and epigenetic regulation. Because of the distinct molecular regulatory pattern of EBV (+) GC, several possible therapies ranging from immune checkpoint inhibitors to synthetic lethal approaches to the ARID1A axis can be selected as precision medicine strategies.
References
Cancer Genome Atlas Research. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513:202–9.
Shinozaki-Ushiku A, Kunita A, Fukayama M. Update on Epstein-Barr virus and gastric cancer (review). Int J Oncol. 2015;46:1421–34.
Xu M, Yao Y, Chen H, Zhang S, Cao SM, Zhang Z, et al. Genome sequencing analysis identifies Epstein-Barr virus subtypes associated with high risk of nasopharyngeal carcinoma. Nat Genet. 2019;51:1131–6.
Kaneda A, Matsusaka K, Aburatani H, Fukayama M. Epstein-Barr virus infection as an epigenetic driver of tumorigenesis. Cancer Res. 2012;72:3445–50.
Fukayama M, Abe H, Kunita A, Shinozaki-Ushiku A, Matsusaka K, Ushiku T, et al. Thirty years of Epstein-Barr virus-associated gastric carcinoma. Virchows Arch. 2020;476:353–65.
Ignatova E, Seriak D, Fedyanin M, Tryakin A, Pokataev I, Menshikova S, et al. Epstein-Barr virus-associated gastric cancer: disease that requires special approach. Gastric Cancer Off J Int Gastric Cancer Assoc Jpn Gastric Cancer Assoc. 2020;23:951–60.
Matsusaka K, Kaneda A, Nagae G, Ushiku T, Kikuchi Y, Hino R, et al. Classification of Epstein-Barr virus-positive gastric cancers by definition of DNA methylation epigenotypes. Cancer Res. 2011;71:7187–97.
Ashizawa M, Saito M, Min AKT, Ujiie D, Saito K, Sato T, et al. Prognostic role of ARID1A negative expression in gastric cancer. Sci Rep. 2019;9:6769.
Yamada L, Saito M, Thar Min AK, Saito K, Ashizawa M, Kase K, et al. Selective sensitivity of EZH2 inhibitors based on synthetic lethality in ARID1A-deficient gastric cancer. Gastric Cancer Off J Int Gastric Cancer Assoc Jpn Gastric Cancer Assoc. 2021;24:60–71.
Kase K, Saito M, Nakajima S, Takayanagi D, Saito K, Yamada L, et al. ARID1A deficiency in EBV-positive gastric cancer is partially regulated by EBV-encoded miRNAs, but not by DNA promotor hypermethylation. Carcinogenesis. 2021;42:21–30.
Nakano H, Saito M, Nakajima S, Saito K, Nakayama Y, Kase K, et al. PD-L1 overexpression in EBV-positive gastric cancer is caused by unique genomic or epigenomic mechanisms. Sci Rep. 2021;11:1982.
Saito M, Kohno T, Kono K. Heterogeneity of ARID1A expression in gastric cancer may affect patient survival and therapeutic efficacy. Hum Pathol. 2020;101:80–1.
Yamada L, Saito M, Thar Min AK, Saito K, Kase K, Onozawa H, et al. Explore the correlation between ARID1A and ANXA1 expressions in gastric cancer. Ann Cancer Res Ther. 2019;27:46–51.
Jangra S, Yuen KS, Botelho MG, Jin DY. Epstein-Barr virus and innate immunity: friends or foes? Microorganisms. 2019;7:183.
Shinozaki A, Sakatani T, Ushiku T, Hino R, Isogai M, Ishikawa S, et al. Downregulation of microRNA-200 in EBV-associated gastric carcinoma. Cancer Res. 2010;70:4719–27.
Banerjee AS, Pal AD, Banerjee S. Epstein-Barr virus-encoded small non-coding RNAs induce cancer cell chemoresistance and migration. Virology. 2013;443:294–305.
Zhao GN, Jiang DS, Li H. Interferon regulatory factors: at the crossroads of immunity, metabolism, and disease. Biochim Biophys Acta. 2015;1852:365–78.
Wang K, Kan J, Yuen ST, Shi ST, Chu KM, Law S, et al. Exome sequencing identifies frequent mutation of ARID1A in molecular subtypes of gastric cancer. Nat Genet. 2011;43:1219–23.
Tober JM, Halske C, Behrens HM, Kruger S, Rocken C. Intratumoral heterogeneity and loss of ARID1A expression in gastric cancer correlates with increased PD-L1 expression in Western patients. Hum Pathol. 2019;94:98–109.
Kim YB, Ahn JM, Bae WJ, Sung CO, Lee D. Functional loss of ARID1A is tightly associated with high PD-L1 expression in gastric cancer. Int J Cancer. 2019;145:916–26.
Takenaka M, Saito M, Iwakawa R, Yanaihara N, Saito M, Kato M, et al. Profiling of actionable gene alterations in ovarian cancer by targeted deep sequencing. Int J Oncol. 2015;46:2389–98.
Liu Y, Sethi NS, Hinoue T, Schneider BG, Cherniack AD, Sanchez-Vega F, et al. Comparative molecular analysis of gastrointestinal adenocarcinomas. Cancer Cell. 2018;33:721–35.
Cai H, Jing C, Chang X, Ding D, Han T, Yang J, et al. Mutational landscape of gastric cancer and clinical application of genomic profiling based on target next-generation sequencing. J Transl Med. 2019;17:189.
Miyabe K, Saito M, Koyama K, Umakoshi M, Ito Y, Yoshida M, et al. Collision of Epstein-Barr virus-positive and -negative gastric cancer, diagnosed by molecular analysis: a case report. BMC Gastroenterol. 2021;21:97.
Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899–905.
Zack TI, Schumacher SE, Carter SL, Cherniack AD, Saksena G, Tabak B, et al. Pan-cancer patterns of somatic copy number alteration. Nat Genet. 2013;45:1134–40.
Gulley ML. Genomic assays for Epstein-Barr virus-positive gastric adenocarcinoma. Exp Mol Med. 2015;47:e134.
Mimura K, Kua LF, Shiraishi K, Kee Siang L, Shabbir A, Komachi M, et al. Inhibition of mitogen-activated protein kinase pathway can induce upregulation of human leukocyte antigen class I without PD-L1-upregulation in contrast to interferon-gamma treatment. Cancer Sci. 2014;105:1236–44.
Mimura K, Teh JL, Okayama H, Shiraishi K, Kua LF, Koh V, et al. PD-L1 expression is mainly regulated by interferon gamma associated with JAK-STAT pathway in gastric cancer. Cancer Sci. 2018;109:43–53.
Esteller M. Epigenetics in cancer. N Engl J Med. 2008;358:1148–59.
Koch A, Joosten SC, Feng Z, de Ruijter TC, Draht MX, Melotte V, et al. Analysis of DNA methylation in cancer: location revisited. Nat Rev Clin Oncol. 2018;15:459–66.
Anastasiadou E, Jacob LS, Slack FJ. Non-coding RNA networks in cancer. Nat Rev Cancer. 2018;18:5–18.
Slack FJ, Chinnaiyan AM. The role of non-coding RNAs in oncology. Cell. 2019;179:1033–55.
Saito M, Schetter AJ, Mollerup S, Kohno T, Skaug V, Bowman ED, et al. The association of microRNA expression with prognosis and progression in early-stage, non-small cell lung adenocarcinoma: a retrospective analysis of three cohorts. Clin Cancer Res. 2011;17:1875–82 (Epub 2011/02/26).
Kim DN, Chae HS, Oh ST, Kang JH, Park CH, Park WS, et al. Expression of viral microRNAs in Epstein-Barr virus-associated gastric carcinoma. J Virol. 2007;81:1033–6.
Lagana A, Forte S, Russo F, Giugno R, Pulvirenti A, Ferro A. Prediction of human targets for viral-encoded microRNAs by thermodynamics and empirical constraints. J RNAi Gene Silenc. 2010;6:379–85.
Xia T, O’Hara A, Araujo I, Barreto J, Carvalho E, Sapucaia JB, et al. EBV microRNAs in primary lymphomas and targeting of CXCL-11 by ebv-mir-BHRF1-3. Cancer Res. 2008;68:1436–42.
Xu DM, Kong YL, Wang L, Zhu HY, Wu JZ, Xia Y, et al. EBV-miR-BHRF1-1 targets p53 gene: potential role in Epstein-Barr virus associated chronic lymphocytic leukemia. Cancer Res Treat Off J Korean Cancer Assoc. 2020;52:492–504.
Cristino AS, Nourse J, West RA, Sabdia MB, Law SC, Gunawardana J, et al. EBV microRNA-BHRF1-2-5p targets the 3’UTR of immune checkpoint ligands PD-L1 and PD-L2. Blood. 2019;134:2261–70.
Wang YF, He DD, Liang HW, Yang D, Yue H, Zhang XM, et al. The identification of up-regulated ebv-miR-BHRF1-2-5p targeting MALT1 and ebv-miR-BHRF1-3 in the circulation of patients with multiple sclerosis. Clin Exp Immunol. 2017;189:120–6.
Poling BC, Price AM, Luftig MA, Cullen BR. The Epstein-Barr virus miR-BHRF1 microRNAs regulate viral gene expression in cis. Virology. 2017;512:113–23.
Li J, Callegari S, Masucci MG. The Epstein-Barr virus miR-BHRF1–1 targets RNF4 during productive infection to promote the accumulation of SUMO conjugates and the release of infectious virus. PLoS Pathog. 2017;13:e1006338.
Ma J, Nie K, Redmond D, Liu Y, Elemento O, Knowles DM, et al. EBV-miR-BHRF1-2 targets PRDM1/Blimp1: potential role in EBV lymphomagenesis. Leukemia. 2016;30:594–604.
Skalsky RL, Corcoran DL, Gottwein E, Frank CL, Kang D, Hafner M, et al. The viral and cellular microRNA targetome in lymphoblastoid cell lines. PLoS Pathog. 2012;8:e1002484.
Yoon CJ, Chang MS, Kim DH, Kim W, Koo BK, Yun SC, et al. Epstein-Barr virus-encoded miR-BART5-5p upregulates PD-L1 through PIAS3/pSTAT3 modulation, worsening clinical outcomes of PD-L1-positive gastric carcinomas. Gastric Cancer Off J Int Gastric Cancer Assoc Jpn Gastric Cancer Assoc. 2020;23:780–95.
Song Y, Li X, Zeng Z, Li Q, Gong Z, Liao Q, et al. Epstein-Barr virus encoded miR-BART11 promotes inflammation-induced carcinogenesis by targeting FOXP1. Oncotarget. 2016;7:36783–99.
Min K, Kim JY, Lee SK. Epstein-Barr virus miR-BART1-3p suppresses apoptosis and promotes migration of gastric carcinoma cells by targeting DAB2. Int J Biol Sci. 2020;16:694–707.
Liu J, Zhang Y, Liu W, Zhang Q, Xiao H, Song H, et al. MiR-BART1-5p targets core 2beta-1,6-acetylglucosaminyltransferase GCNT3 to inhibit cell proliferation and migration in EBV-associated gastric cancer. Virology. 2020;541:63–74.
Wang J, Zheng X, Qin Z, Wei L, Lu Y, Peng Q, et al. Epstein-Barr virus miR-BART3-3p promotes tumorigenesis by regulating the senescence pathway in gastric cancer. J Biol Chem. 2019;294:4854–66.
Zheng X, Wang J, Wei L, Peng Q, Gao Y, Fu Y, et al. Epstein-Barr Virus MicroRNA miR-BART5–3p Inhibits p53 Expression. J Virol. 2018. https://doi.org/10.1128/JVI.01022-18.
Wang D, Zeng Z, Zhang S, Xiong F, He B, Wu Y, et al. Epstein-Barr virus-encoded miR-BART6-3p inhibits cancer cell proliferation through the LOC553103-STMN1 axis. FASEB J. 2020;34:8012–27.
He B, Li W, Wu Y, Wei F, Gong Z, Bo H, et al. Epstein-Barr virus-encoded miR-BART6–3p inhibits cancer cell metastasis and invasion by targeting long non-coding RNA LOC553103. Cell Death Dis. 2016;7:e2353.
Dong M, Gong LP, Chen JN, Zhang XF, Zhang YW, Hui DY, et al. EBV-miR-BART10-3p and EBV-miR-BART22 promote metastasis of EBV-associated gastric carcinoma by activating the canonical Wnt signaling pathway. Cell Oncol (Dordr). 2020;43:901–13.
Min K, Lee SK. EBV miR-BART10-3p promotes cell proliferation and migration by targeting DKK1. Int J Biol Sci. 2019;15:657–67.
Song Y, Li Q, Liao S, Zhong K, Jin Y, Zeng T. Epstein-Barr virus-encoded miR-BART11 promotes tumor-associated macrophage-induced epithelial-mesenchymal transition via targeting FOXP1 in gastric cancer. Virology. 2020;548:6–16.
Wu Y, Wang D, Wei F, Xiong F, Zhang S, Gong Z, et al. EBV-miR-BART12 accelerates migration and invasion in EBV-associated cancer cells by targeting tubulin polymerization-promoting protein 1. FASEB J. 2020;34:16205–23.
Choi H, Lee SK. TAX1BP1 downregulation by EBV-miR-BART15-3p enhances chemosensitivity of gastric cancer cells to 5-FU. Adv Virol. 2017;162:369–77.
Yoon JH, Min K, Lee SK. Epstein-Barr Virus miR-BART17-5p promotes migration and anchorage-independent growth by targeting kruppel-like factor 2 in gastric cancer. Microorganisms. 2020;8:258.
Kim H, Choi H, Lee SK. Epstein-Barr virus miR-BART20-5p regulates cell proliferation and apoptosis by targeting BAD. Cancer Lett. 2015;356:733–42.
Kono K, Nakajima S, Mimura K. Current status of immune checkpoint inhibitors for gastric cancer. Gastric Cancer Off J Int Gastric Cancer Assoc Jpn Gastric Cancer Assoc. 2020;23:565–78.
Shen J, Peng Y, Wei L, Zhang W, Yang L, Lan L, et al. ARID1A deficiency impairs the DNA damage checkpoint and sensitizes cells to PARP inhibitors. Cancer Discov. 2015;5:752–67.
Williamson CT, Miller R, Pemberton HN, Jones SE, Campbell J, Konde A, et al. ATR inhibitors as a synthetic lethal therapy for tumours deficient in ARID1A. Nat Commun. 2016;7:13837.
Choy EY, Siu KL, Kok KH, Lung RW, Tsang CM, To KF, et al. An Epstein-Barr virus-encoded microRNA targets PUMA to promote host cell survival. J Exp Med. 2008;205(11):2551–60.
Choi H, Lee H, Kim SR, Gho YS, Lee SK. Epstein-Barr virus-encoded microRNA BART15-3p promotes cell apoptosis partially by targeting BRUCE. J Virol. 2013;87(14):8135–44.
Acknowledgements
This work was supported by JSPS KAKENHI Grant Numbers 20K09083 and 21K08675.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflicts of interest to declare.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Saito, M., Kono, K. Landscape of EBV-positive gastric cancer. Gastric Cancer 24, 983–989 (2021). https://doi.org/10.1007/s10120-021-01215-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10120-021-01215-3