
Abstract
Background
Polymorphisms in inflammation-related genes have been associated with a risk of gastric carcinoma (GC). However, the biological mechanisms underlying these associations are still elusive. Our objective was to determine whether chronic inflammation-associated IL1Β signalling, as seen in the context of Helicobacter pylori infection, could be linked to gastric carcinogenesis by modulating the behaviour of gastric epithelial cells.
Methods
The effect of IL1B was assessed by studying the expression and activation status of the IL1Β-activated transcription factors C/EBPβ and CREB in GC cell lines. Interaction between CREB and C/EBPβ was explored through interference RNA, chromatin immunoprecipitation and chemical inhibition. CREB and C/EBPβ expression was analysed in 66 samples of primary GC and in normal gastric mucosa. GC cell growth was analysed in vitro by BrdU incorporation and in vivo employing a chicken embryo chorioallantoic membrane model.
Results
We found that IL1B regulates the expression/activation status of both C/EBPβ and CREB in GC cells through an ERK1/2-dependent mechanism. Our results show that CREB is a direct transactivator of CEBPB, acting as an upstream effector in this regulatory mechanism. Furthermore, we found CREB to be overexpressed in 94 % of GC samples and significantly associated with C/EBPβ expression (P < 0.05). Finally, we demonstrated both in vitro and in vivo that CREB can mediate IL1B-induced GC cell proliferation.
Conclusions
Our results support the hypothesis that the effect of chronic inflammation on gastric carcinogenesis, as seen in the context of genetically susceptible individuals infected with Helicobacter pylori, includes the modulation of signalling pathways that regulate survival mechanisms in epithelial cells.
Summary
IL1B is able to increase the expression/activation status of CREB and its target gene C/EBPβ, which are mandatory for GC cell survival. Our results may help inform new strategies for the prevention and treatment of GC, including the control of chronic inflammation.
Similar content being viewed by others
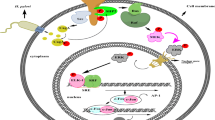


Introduction
Gastric carcinoma (GC) is the second most common cause of cancer-related death in the world. The main risk factor for the onset of GC is life-time infection with Helicobacter pylori (H. pylori), a stomach-colonising bacterium [1]. Infection with H. pylori leads to chronic gastritis that may progress to gastric atrophy, intestinal metaplasia, dysplasia and finally GC [2, 3].
The risk of developing GC depends both on environmental factors and host-related factors [4, 5]. In this model, gene polymorphisms that increase the production of pro-inflammatory mediators lead to an enhanced chronic inflammatory response to H. pylori infection and to an increased risk of progression towards GC [5, 6]. There are numerous studies demonstrating that polymorphisms in genes such as IL1Β, IL1RN, TNFA and IFNGR1 are associated with a risk of developing GC [7–11]. Moreover, these polymorphisms have been shown to be associated with increased gene expression, both in vitro and in vivo [12]. Perhaps the most striking evidence favouring this model comes from a transgenic mouse model showing that overexpression of the IL1Β gene in gastric mucosa leads to an increased risk of developing gastric disease, including dysplasia and GC, even in the absence of H. pylori infection [13].
According to the prevailing model, the link between enhanced chronic inflammation and GC depends essentially on the “destructive” effects of inflammation over the gastric epithelium, resulting in atrophy of the gastric mucosa and increased cell turnover [14, 15]. However, it is well demonstrated that inflammatory mediators, and other growth factors secreted by inflammatory cells, can act directly on other cell types, such as epithelial cells. Therefore, in addition to the mucosal destruction and repair effect, enhanced chronic inflammation could also play a role in gastric carcinogenesis by providing gastric epithelial cells with a survival stimulus through the secretion of growth factors [16, 17]. Coupled with mutagenic events, this could ultimately lead to an increased risk of cell transformation and GC development.
In this regard, IL1Β is particularly interesting since polymorphisms in its promoter region have been shown to be associated with an increased risk of GC [7–9, 18]. IL1Β is a powerful pro-inflammatory cytokine that activates different transcription factors [19], some of which are also activated by H. pylori infection [20, 21]. One of the IL1Β-activated transcription factors is CCAAT/enhancer-binding protein beta (C/EBPβ) [22]. We previously reported that C/EBPβ is overexpressed in pre-malignant lesions and in GC, suggesting that this protein may facilitate gastric carcinogenesis by inducing the expression of COX-2 [23]. Furthermore, C/EBPβ expression in GC was significantly associated with loss of expression of the putative gastric tumour-suppressor TFF1 [24, 25].
Another important IL1Β-activated transcription factor is cAMP response element-binding protein (CREB), which has been described as a major player in inflammation [19, 26]. In non-small cell lung cancer, IL1Β induces the activation of CREB through ERK1/2 signalling, resulting in the expression of a set of pro-angiogenic cytokines that are crucial factors in tumour progression [27]. Furthermore, CREB was recently described to play an important pro-oncogenic role in both cancer development and progression; it was found to be overexpressed in several cancer types [28–31]. It has been demonstrated, both in hepatocytes [32] and in pre-adipocytes [33], that CREB is able to regulate the transcription of the CEBPB gene by directly interacting with its promoter.
The main objective of this study was to determine whether chronic inflammation-associated IL1Β signalling, as seen in the context of H. pylori infection, could be linked to gastric carcinogenesis by modulating the behaviour of gastric epithelial cells. We addressed this objective by showing that CREB and C/EBPβ transcription factors can be activated by IL1Β signalling in the GC context. We also demonstrated that CREB acts upstream of C/EBPβ in GC cell lines. Finally, we showed in vitro and in vivo that this signalling mechanism promotes GC cell survival.
Results
IL1B increases C/EBPβ and CREB expression in an ERK1/2-dependent manner
To evaluate the effect of IL1B on the expression and activation status of C/EBPβ and CREB, we incubated GC cell lines AGS and GP202 with IL1B for 24 h. In both AGS and GP202 cells, incubation with IL1B led to an increase in the expression of all isoforms (LAP*, LAP and LIP) of C/EBPβ (Fig. 1). Regarding CREB, we observed an increase in both expression and phosphorylation levels in both cell lines (Fig. 1).

Effects of IL1B stimulation and ERK1/2 inhibition on CREB, pCREB, and C/EBPβ protein levels. Both AGS and GP202 cells, when treated with 10 ng/mL of IL1B for 24 h, exhibited an increase in activated ERK1/2 (pERK1/2). In parallel, the expression levels of CREB, pCREB and C/EBPβ also increased. The ERK1/2 chemical inhibitor U0126 (25 μM) reverted the effect of IL1B on CREB, pCREB and C/EBPβ protein levels
Since ERK1/2 has been previously implicated in the regulation of C/EBPβ and CREB, we investigated whether it could mediate the effect of IL1B over those two transcription factors. Incubation of AGS and GP202 cells with the ERK1/2 inhibitor U0126 led to a decrease in the expression of C/EBPβ and CREB and to a decrease in phosphorylation levels of CREB (Fig. 1). The level of ERK1/2 phosphorylation in both cell lines was measured as a control for the efficacy of inhibition with U0126 (Fig. 1). These results demonstrate that IL1B is able to regulate the expression/activation status of both C/EBPβ and CREB in GC cells.
CREB is a transcriptional regulator of C/EBPβ in GC cells
Knowing that CREB is a transcriptional regulator of the CEBPB gene in other cell types, we decided to investigate whether the same regulatory mechanism could be at work in GC cells. Since both AGS and GP202 cells yielded the same results and the IL1B-stimulatory effect was more pronounced in AGS cells, we decided to perform the next set of experiments only in the AGS cells. Using both small interfering RNA (siRNA) and short-hairpin RNA (shRNA), we found that knocking down CREB results in downregulation of C/EBPβ expression (Fig. 2a). Conversely, silencing C/EBPβ using siRNA had no impact on CREB expression (Fig. 2a). These results show that CREB acts upstream of C/EBPβ in this regulatory mechanism.

Effect of CREB downregulation on C/EBPβ protein levels. a CREB silencing (CREB-specific bands are indicated by arrows; the upper bands on the CREB blots represent unspecific binding of the CREB antibody) was followed by a downregulation of C/EBPβ protein levels, whereas the silencing of C/EBPβ had no impact on CREB protein. b Schematic representation of the CEBPB promoter (2663 bp) showing the three CRE-binding sites (BS1, BS2 and BS3) and a control region (CR) located at the 3′ end of CEBPB. c CREB interacts with all three CRE-binding motifs present on the CEBPB promoter. No Ab no antibody used, Input 1/100 of the sheared initial chromatin, CREB chromatin immunoprecipitated using an anti-CREB antibody, IgG chromatin immunoprecipitated with an unspecific antibody of the same family as the anti-CREB antibody
To check whether CREB acts directly on the CEBPB gene, we analysed the CEBPB promoter (2663 base pairs) in order to find putative cAMP response element (CRE)-binding motifs. We employed a previously described [32] nucleotide position numbering scheme. The analysis revealed the presence of three CRE-binding sites, ranging from nucleotides −2174 to −2171 (BS1), from −959 to −956 (BS2), and from −66 to −63 (BS3) (Fig. 2b). Using chromatin immunoprecipitation (ChIP), we observed that CREB binds all three CRE-binding sites on the CEBPB promoter (Fig. 2c).
These results were validated by showing that 24-h chemical inhibition of the interaction between CREB and its co-activator CREB binding protein (CBP) led to a reduced binding of CREB to the CRE-binding sites on the CEBPB gene promoter (Fig. 3a). To confirm that the decrease in CREB binding to the CEBPB promoter actually leads to downregulation of transcription and protein synthesis, we evaluated the relative amount of C/EBPβ mRNA and protein after 48 h of treatment with the CBP–CREB interaction inhibitor. This experiment resulted in a significant reduction in both C/EBPβ transcript levels (Fig. 3b) and C/EBPβ protein levels (Fig. 3c).

Effects of CBP–CREB interaction inhibition on CREB activity and C/EBPβ. a ChIP performed on AGS cells after treatment with CBP–CREB interaction inhibitor (+) revealed a decrease in the amount of CREB linked to the CEBPB promoter compared to untreated cells (−). AGS cells treated with CREB–CBP interaction inhibitor (25 μM) for 48 h showed a significant decrease in C/EBPβ b transcript levels and c protein expression. Real-time PCR results represent the mean ± S.D. of three independent experiments. Significance: *(P < 0.05), **(P < 0.01) and ***(P < 0.001)
CREB and C/EBPβ proteins are co-expressed in normal gastric mucosa and in GC
To determine whether there is an association between the expression of CREB and C/EBPβ, we analysed the immunohistochemical (IHC) expression of these two proteins in a series of 66 cases of GC. In normal gastric mucosa, we found that both C/EBPβ and CREB are expressed in the nuclei of epithelial cells in the proliferative isthmus/neck zone (Fig. 4a, b). To confirm that CREB and C/EBPβ are expressed in the same cells, we performed double immunofluorescence in a tissue fragment of normal gastric mucosa (Fig. 4g, j). In GC, we observed that CREB and C/EBPβ were expressed in cell nuclei in 94 and 73 % of the cases, respectively. A comparison of the IHC results indicates that there is a statistically significant association (P = 0.04) between CREB and C/EBPβ expression in our series of GC. We also observed that GC cases with higher CREB expression scores were significantly associated with intestinal and mixed histological subtypes (P = 0.003) (Table 1). No associations were detected between the expression of CREB and other clinicopathological characteristics of the tumours.

Immunohistochemical expression of CREB and C/EBPβ in normal gastric mucosa and in GC cases. a CREB is expressed in epithelial cells located in the neck/isthmus region of normal gastric glands. b C/EBPβ is also expressed in neck/isthmus normal epithelial cells. c, e Examples of CREB-positive GC cases (scoring 3); d, f C/EBPβ is overexpressed in the same GC cases that are positive for CREB. Magnification 100×. g–j Double immunofluorescence staining for CREB and C/EBPβ in normal gastric mucosa, showing co-expression of the two proteins in gastric epithelial cells: g DAPI nuclear staining, h C/EBPβ staining, i CREB staining, j merged image for DAPI, C/EBPβ and CREB staining. Magnification 400×
CREB modulates IL1B-induced proliferation of GC cells
To determine whether IL1B is able to increase the survival of GC cells, we measured BrdU incorporation and performed TUNEL assays in the AGS GC cell line after incubating cells with IL1B. In parallel, we determined whether any of the effects of IL1B are mediated by CREB. Our control experiments showed that CREB expression is downregulated by the shRNA used (Fig. 5a). Figure 5b shows that IL1B is able to significantly increase cellular proliferation, and that this effect can be reverted by downregulating CREB levels. IL1B had no significant effect on the rate of apoptosis (data not shown).

CREB modulates both IL1B-induced and basal cell proliferation. a Downregulation of CREB expression using shRNA. b AGS cells expressing normal levels of CREB (transfected with scrambled shRNA) showed an increase in cell proliferation after 24 h of treatment with 10 ng/mL IL1B, while CREB downregulation (transfected with anti-CREB shRNA) was responsible for a significant decrease in IL1B-induced cell proliferation. sc shRNA control scrambled shRNA, shRNA CREB anti-CREB shRNA. c AGS, d GP202, and e MKN28 cells were treated for 24 and 48 h to assess the time-dependent impact of treatment on GC cell proliferation; the protein levels of cyclin D1 were checked in f AGS, g GP202 and h MKN28 cells after 24 and 48 h of CBP–CREB interaction inhibitor treatment. BrdU results represent the mean ± S.D. of three independent experiments. Significance: *P < 0.05, **P < 0.01 and ***P < 0.001
To explore the role of CREB in GC cell proliferation, we evaluated the expression of the cell-cycle regulator cyclin D1. After performing a dose–response experiment to determine the effect of the CBP–CREB interaction inhibitor on cell proliferation, we selected a concentration of 25 μM. In the AGS cell line, CREB inhibition had a significant inhibitory effect on cell proliferation (Fig. 5c). The effect was more pronounced after 48 h of treatment. Moreover, this effect was also confirmed by measuring the expression level of cyclin D1 after CREB inhibition after 24 and 48 h of treatment (Fig. 5f). The effect was also seen in the GP202 GC cell line (Fig. 5d, g) and in the intestinal-type GC cell line MKN28 (Fig. 5e, h). Overall, these results further support the notion that CREB plays an important role in GC cell survival, in both diffuse and intestinal histological types, by regulating the proliferation of GC cells.
CREB inhibition decreases GC cell growth in vivo
To evaluate the effect of CREB on tumour growth, we used the chicken embryo chorioallantoic membrane (CAM) model. The CAM effectively supports the growth of inoculated human cancer cells due to the immunodeficiency of the chick during its early developmental stages. Before CAM inoculation, we confirmed the knockdown of CREB protein on AGS cells transfected with anti-CREB shRNA (Fig. 6a). To avoid inter-animal differences in the results, scrambled shRNA and anti-CREB shRNA transfected AGS cells were inoculated in distinct areas of the CAM of the same egg and allowed to proliferate for 6 days. At this end point, the tumour area was quantified. As can be seen in Fig. 6b, inhibition of CREB led to reduced growth of the inoculated cells. Figure 6c shows that, on average, the tumour growth area was significantly smaller in cells with CREB inhibition. These results demonstrate that CREB-mediated signalling is important for GC cell growth in vivo.

CREB inhibition reduces in vivo cell growth. a Before inoculating AGS cells in the chorioallantoic membrane (CAM), CREB protein knockdown was checked by western blotting. b AGS cells transfected with shRNA against CREB give rise to small-sized tumours compared with scramble-transfected cells; c Images representing the different sizes (delimited by red dashed lines) of scramble and shRNA CREB tumours in two paired CAM experiments (#1 and #2). Values of P < 0.05 were considered to be statistically significant
Discussion
Our results demonstrate that IL1B is able to activate CREB and C/EBPβ in GC cells. This process is mediated by ERK1/2, since its inhibition by U0126 reverted the effects induced by IL1B. This is in agreement with previously reported results showing that IL1B is able to induce GC cell proliferation in an ERK1/2-dependent manner [16]. We also demonstrated that CREB is able to transactivate C/EBPβ in GC cells.
Our in vitro observations were reinforced by the analysis of the expression of CREB and C/EBPβ in a series of GC samples and normal gastric mucosa. We showed that in normal gastric mucosa, CREB is expressed in the proliferative neck/isthmus region of the gastric glands; in GC, it is overexpressed in the majority of tumour samples. These results are in accordance with those published by Chen et al. [34], showing that CREB mRNA levels are upregulated in GC samples when compared with adjacent normal mucosa.
CREB expression was also significantly associated with the expression of C/EBPβ in GC. The observation that in normal gastric mucosa CREB and C/EBPβ are expressed in a cellular compartment that includes progenitor cells suggests that CREB and C/EBPβ may be involved in maintaining a proliferative phenotype in gastric epithelial cells. This would be in accordance with the observed pattern of overexpression of both proteins in the majority of the GC cases included in the present study. These results are also in keeping with our previous demonstration that C/EBPβ is overexpressed in pre-malignant lesions and in GC, suggesting that this protein might facilitate the transformation of gastric epithelial cells by inducing the expression of COX-2 [23] and by inhibiting the expression of the putative gastric tumor suppressor gene TFF1 [24, 25].
In order to complement the aforementioned observations with a biological readout, we evaluated the role of CREB in mediating IL1B-induced changes in cell proliferation and apoptosis. Although no significant effect was observed in relation to apoptosis, our results show that CREB is an effector of IL1B-induced cell proliferation, since downregulation of CREB impairs the pro-mitogenic action of IL1B on GC cells in vitro. These results were further supported by the CAM assays showing that inhibition of CREB reduces the ability of GC cells to survive in this in vivo model.
Infection with H. pylori leads to chronic inflammation and an increased risk of developing GC. Our results support the hypothesis that the effect of chronic inflammation on tumourigenesis includes modulation of critical signalling pathways that regulate survival in epithelial cells. In this scenario, H. pylori infection leads to overexpression of IL1B which, in turn, activates CREB and C/EBPβ. This effect may be more pronounced in individuals that carry genetic susceptibility polymorphisms that have been demonstrated to be associated with enhanced chronic inflammation, such as those in the IL1B gene promoter. Coupling cell survival with an increased likelihood of accumulating genetic mutations may help explain why individuals with pro-inflammatory genetic polymorphisms have an increased risk of developing GC.
Materials and methods
Tissue material
Surgical specimens from 66 GCs were resected and diagnosed at Hospital S. João, Porto, Portugal. Tissue fragments were fixed in 10 % formaldehyde followed by paraffin embedding. Tumour-representative areas of each GC were selected to create a tissue microarray (TMA) block. Serial sections of 3 μm were obtained from the TMA block and used for routine staining with haematoxylin and eosin and immunohistochemistry. This study was performed in accordance with institutional ethical standards. All of the samples enrolled in this study were unidentified.
Immunohistochemistry and immunofluorescence
Tissue sections from formalin-fixed paraffin-embedded (FFPE) tissues were first deparaffinised, hydrated, and then treated with 1× citrate buffer (pH 6.0) (Thermo Scientific, Waltham, MA, USA) for 45 min at 100 °C. All of the following steps were performed at room temperature (RT). Unspecific endogenous peroxidase activity was eliminated with 3 % hydrogen peroxidase in methanol for 15 min. To reduce nonspecific background staining, slides were blocked with Ultra V Block (Thermo Scientific) for 10 min. Slides were rinsed in PBS–0.1 % Tween20 and incubated for 1 h with the antibody anti-CREB [E306] (Abcam, Cambridge, UK) diluted 1:1000 and then overnight (ON) with anti-C/EBPβ (Abcam) diluted 1:1000 in UltraAB Diluent (Thermo Scientific). Slides were then incubated with Dako Real EnVision HRP Rabbit/Mouse solution (Dako, Glostrup, Denmark) for 30 min. Slides were washed, developed for 1–3 min with 2 % Dako REAL™ DAB+ Chromogen solution (Dako), counterstained with haematoxylin, dehydrated, and mounted with mounting medium (Thermo Scientific). All washing steps were performed in PBS–0.1 % Tween20 buffer. Normal gastric mucosa was used as a control, and negative controls were obtained by substituting the primary antibody with immunoglobulins of the same class and concentration. Slides were reviewed by a pathologist, and the percentage of positive cells was semiquantitatively scored as either 0 (corresponding to positivity in <5 % of the tumour cells), 1 (corresponding to positivity in >5 % and <50 % of the tumour cells), 2 (corresponding to positivity in >50 % and <75 % of the tumour cells), or 4 (corresponding to positivity in >75 % of the tumour cells).
For immunofluorescence, antigen retrieval was performed with 1× citrate buffer (pH 6,0). After unspecific protein blocking with Ultra V Block (Thermo Scientific) for 10 min, slides were incubated with the antibody anti-CREB [E306] diluted 1:1000 for 1 h, followed by the antibody anti-C/EBPβ diluted 1:1000 ON. After washing twice with PBS–Tween 0.021 % for 10 min, the slides were incubated with a mixture of two secondary antibodies raised in different species (with Texas Red conjugated against rabbit and FITC conjugated against mouse) for 45 min at room temperature and protected from light. To counterstain cell nuclei, slides were mounted with Vectashield Mounting Medium with DAPI (Vector Laboratories, Burlingame, CA, USA). Finally, slides were visualised and images were captured (via ApoTome) under a fluorescence microscope (Zeiss, Oberkochen, Germany).
Cell culture, chemical treatments and transfections
AGS, MKN28 and GP202 cell lines were maintained in RPMI medium (Gibco, Waltham, MA, USA), supplemented with 10 % foetal bovine serum (FBS) (PAA, Pasching, Austria), and 100 IU/mL penicillin and 100 μg/mL streptomycin (Gibco), in a humidified incubator under an atmosphere of 5 % CO2 at 37 °C. Cells were grown until 60–80 % confluence and treated with 10 ng/mL of IL1B (Sigma, St. Louis, MO, USA) and incubated for 24 h.
For ERK1/2 inhibition, cells were treated with 25 μM U0126 (Cayman, Ann Arbor, MI, USA) together with IL1B for 24 h. As U0126 was diluted in DMSO, an appropriate volume of DMSO was added to the cells as the control condition. For CBP–CREB interaction inhibition, cells were treated with 25 μM of the specific inhibitor (Merck–Millipore, Darmstadt, Germany) or DMSO vehicle (Sigma) for 24 and 48 h.
For silencing experiments, AGS cells grown until 60–80 % confluence were transfected with 1.0 μg of anti-CREB shRNA expression vector or 1.0 μg scrambled shRNA (Origene, Rockville, MD, USA) using Lipofectamine 2000 (Invitrogen, Waltham, MA, USA), according to the manufacturer’s specifications. At 48 h post-transfection, cells were selected by adding 1.0 μg/mL of puromycin (Sigma) to the culture medium. Individual puromycin-resistant colonies were isolated after 2 weeks of selection and expanded in the presence of puromycin (1.0 μg/mL). For the transient silencing of CREB, AGS cells were transfected either with 100 nM of siRNA against CREB (Qiagen, Hilden, Germany) or with 100 nM of siRNA control (Qiagen), using Lipofectamine 2000 (Invitrogen) as a vehicle. In parallel, C/EBPβ silencing was performed by transfecting AGS cells with 150nM of siRNA against CEBPB or siRNA control (Qiagen). Protein downregulation after gene silencing was evaluated by western blotting following 72 h of culture.
Western blotting (immunoblotting)
Cells were washed with 1× PBS (pH 7.4) and lysed in NP-40 buffer supplemented with phosphatase (Sigma) and protease inhibitors (Roche, Penzberg, Germany). After Bradford protein quantification, 40 μg of total protein were loaded into 12.5 % acrylamide gels, separated by SDS-PAGE under denaturing conditions and electrotransferred to a Hybond ECL Nitrocellulose Membrane (GE Healthcare, Little Chalfont, UK). After blocking, membranes were incubated for 1.5 h with the primary antibodies anti-ERK1/2 #9102 (Cell Signaling Technology, Danvers, MA, USA) diluted 1:1000, anti-pERK1/2 #9106 (Cell Signaling Technology) diluted 1:1000, anti-CREB [E306] diluted 1:500, anti-pCREB [E113] (Abcam) diluted 1:1000, anti-C/EBPβ [H-7] (Santa Cruz Biotechnology, Santa Cruz, CA, USA) diluted 1:500, anti-cyclin D1 (Santa Cruz Biotechnology) diluted 1:500, and anti-α-tubulin (Sigma) diluted 1:10000 in PBS-0.5 % Tween20 plus 5 % non-fat dried milk or 4 % BSA (bovine serum albumin). The blots were then washed with PBS–0.5 % Tween20 and incubated with horseradish peroxidase-conjugated secondary antibodies diluted 1:10000 in PBS–0.5 % Tween20 plus 0.5 % non-fat dried milk. Chemiluminescent bands were visualised using Western Blot ECL (GE Healthcare).
BrdU incorporation assay
Cells were allowed to reach 60–80 % confluence and 1× BrdU was added to the culture medium for 1 h. Cells were then washed with 1× PBS and fixed in freshly prepared 4 % (v/v) paraformaldehyde at RT for 30 min. The glass slides were removed from the 6-well plate, transferred to individual wells in a 12-well plate, and washed with 1xPBS. In order to denature the DNA and permeabilize cells, hydrochloric acid (HCl) 2 M was added to each slide for 20 min, followed by washing steps with PBS–0.5 % Tween20 plus 0.05 % BSA. Cells in glass slides were incubated for 1 h with mouse primary antibody against BrdU (Dako) diluted 1:10, washed twice with PBS–0.5 % Tween20 plus 0.05 % BSA, and incubated for 30 min with anti-mouse secondary antibody marked with Alexa Fluor 594 (Invitrogen) diluted 1:100. Glass slides were rinsed in PBS–0.5 % Tween20 plus 0.05 % BSA twice, mounted with Vectashield Mounting Medium with DAPI (Vector Laboratories), and finally visualised under fluorescent microscopy. For each experiment, the BrdU technique was performed in triplicate. In each assay, at least 1000 cells were counted and BrdU incorporation was expressed as the ratio of DAPI to BrdU-positive cells.
Promoter analysis and chromatin immunoprecipitation (ChIP)
The nucleotide sequence of human CEBPB promoter was obtained from the UCSC Genome Browser (http://genome.ucsc.edu/). The putative CRE-binding sites present on CEBPB promoter were identified using the Genomatix MatInspector software (http://www.genomatix.de/solutions/genomatix-genome-analyzer.html).
ChIP assay was performed using a Magna ChIP G Kit (Millipore, Billerica, MA, USA), according to the manufacturer’s protocol instructions. Briefly, 1 × 107 of AGS cells were crosslinked with 1 % formaldehyde for 10 min at RT, and the reaction was stopped by 1× glycine solution for 5 min. Cells were rinsed with 1× PBS, lysed in order to isolate nuclei, and then sonicated in Nuclear Lysis to shear the chromatin to sizes of 200–500 bp. Then, 50 μl of the supernatant were immunoprecipitated by adding 2 μg of rabbit antibody anti-CREB (Abcam) or 2 μg of control rabbit polyclonal anti-IgG antibody (Abcam), and the mixture was placed on a rotator at 4 °C ON in the presence of magnetic G beads. DNA–protein crosslinks were reversed by heating samples at 62 °C for 2 h on a shaking platform. To elute DNA, a series of wash steps followed by elution (50 μl) were performed in spin columns. PCR conditions: 95 °C for 15 min, 35 times (95 °C for 1 min, 58–60 °C for 1 min, 72 °C for 1 min). The reactions were carried out with HotStarTaq DNA Polymerase (Qiagen) as described by the manufacturer, using 2 μl of DNA template. The PCR products were analysed by electrophoresis on a 2 % agarose gel. PCR primer pairs flanking CREB-binding sites and for the control region (CR) were designed using the Primer 3 software.
RNA isolation, cDNA synthesis and quantification of CEBPB mRNA transcript
Total RNA was isolated from AGS cells grown in 6-well culture plates using the RNeasy Mini Kit (Qiagen). Reverse transcription was performed using a SuperScript II Reverse Transcriptase Kit (Invitrogen) with 1000 μg of total RNA in a 20-μl volume reaction, after treatment with DNase I (Invitrogen).
To determine the relative amount of CEBPB transcript, we performed quantitative target amplification, using cDNA as the template, with the SYBR Green PCR Kit (Qiagen), according to the manufacturer’s protocol. As an internal control, we quantified the expression levels of beta-actin transcript.
Analysis of in vivo tumour growth
The chicken embryo CAM model was used to evaluate the growth capability of AGS cells transfected with scramble or shCREB RNA (n = 16). Briefly, fertilised chick (Gallus gallus) eggs were incubated horizontally at 37.8 °C in a humidified atmosphere. On embryonic day 3 (E3), a square window was opened on the shell after removing 1.5–2 mL of albumin to allow detachment of the developing CAM. The window was sealed with a transparent adhesive tape and the eggs returned to the incubator. Cells, re-suspended in 10 µl of complete medium, were placed on top of E10 growing CAM, and 2 × 106 cells per embryo from each cell line (scrambled shRNA vs CREB shRNA) were placed into a 3-mm nylon ring under sterile conditions. The eggs were re-sealed and returned to the incubator for an additional 5 days. After removing the ring, the CAM was excised from the embryos, photographed ex ovo under a stereoscope at 20× magnification (SZX16 coupled with a DP71 camera, Olympus, Tokyo, Japan). The area of CAM tumour was determined using the Cell A program (Olympus).
Statistical analysis
The clinicopathological features of GC cases were compared using the χ 2 test. When two conditions were compared, Student’s t test was used, whereas ANOVA was employed when the comparison involved more than two conditions. The paired t test was used for tumour area comparisons. In order to accurately access putative differences in tumour areas between the two cell lines, only eggs bearing two tumours with areas ≥1 mm2 (n = 16) were considered, independently of the cell group. Values of P < 0.05 were considered to be statistically significant.
References
Parkin DM, Bray F, Ferlay J, et al. Global cancer statistics, 2002. CA Cancer J Clin. 2005;55:74–108.
Correa P. Human gastric carcinogenesis: a multistep and multifactorial process—First American Cancer Society Award Lecture on cancer epidemiology and prevention. Cancer Res. 1992;52:6735–40.
Crew KD, Neugut AI. Epidemiology of gastric cancer. World J Gastroenterol. 2006;12:354–62.
Milne AN, Carneiro F, O’Morain C, et al. Nature meets nurture: molecular genetics of gastric cancer. Hum Genet. 2009;126:615–28.
Resende C, Ristimäki A, Machado JC. Genetic and epigenetic alteration in gastric carcinogenesis. Helicobacter. 2010;15:34–9.
Amieva MR, El-Omar EM. Host-bacterial interactions in Helicobacter pylori infection. Gastroenterology. 2008;134:306–23.
El-Omar EM, Carrington M, Chow W-H, et al. Interleukin-1 polymorphisms associated with increased risk of gastric cancer. Nature. 2000;404:398–402.
Machado JC, Pharoah P, Sousa S, et al. Interleukin 1B and interleukin 1RN polymorphisms are associated with increased risk of gastric carcinoma. Gastroenterology. 2001;121:823–9.
Figueiredo C, Machado JC, Pharoah P, et al. Helicobacter pylori and interleukin 1 genotyping: an opportunity to identify high-risk individuals for gastric carcinoma. J Natl Cancer Inst. 2002;94:1680–7.
Machado JC, Figueiredo C, Canedo P, et al. A proinflammatory genetic profile increases the risk for chronic atrophic gastritis and gastric carcinoma. Gastroenterology. 2003;125:364–71.
Canedo P, Corso G, Pereira F, et al. The interferon gamma receptor 1 (IFNGR1) −56C/T gene polymorphism is associated with increased risk of early gastric carcinoma. Gut. 2008;57:1504–8.
Hwang I-R, Kodama T, Kikuchi S, et al. Effect of interleukin 1 polymorphisms on gastric mucosal interleukin 1β production in Helicobacter pylori infection. Gastroenterology. 2002;123:1793–803.
Tu S, Bhagat G, Cui G, et al. Overexpression of interleukin-1β induces gastric inflammation and cancer and mobilizes myeloid-derived suppressor cells in mice. Cancer Cell. 2008;14:408–19.
Beales ILP, Calam J. Interleukin 1β and tumour necrosis factor α inhibit acid secretion in cultured rabbit parietal cells by multiple pathways. Gut. 1998;42:227–34.
Konturek P, Kania J, Konturek J, et al. H. pylori infection, atrophic gastritis, cytokines, gastrin, COX-2, PPAR gamma and impaired apoptosis in gastric carcinogenesis. Med Sci Monit 2003;9:SR53–66.
Beales I. Effect of interleukin-1beta on proliferation of gastric epithelial cells in culture. BMC Gastroenterol. 2002;2:7.
Yasunaga Y, Shinomura Y, Kanayama S, et al. Increased production of interleukin 1 beta and hepatocyte growth factor may contribute to foveolar hyperplasia in enlarged fold gastritis. Gut. 1996;39:787–94.
El-Omar EM, Rabkin CS, Gammon MD, et al. Increased risk of noncardia gastric cancer associated with proinflammatory cytokine gene polymorphisms. Gastroenterology. 2003;124:1193–201.
Newton K, Dixit VM. Signaling in innate immunity and inflammation. Cold Spring Harbor Perspect Biol. 2012;4:3.
Naumann M, Crabtree JE. Helicobacter pylori-induced epithelial cell signalling in gastric carcinogenesis. Trends Microbiol. 2004;12:29–36.
Kudo T, Lu H, Wu JY, et al. Pattern of transcription factor activation in Helicobacter pylori—infected Mongolian gerbils. Gastroenterology. 2007;132:1024–38.
Giltiay NV, Karakashian AA, Alimov AP, et al. Ceramide- and ERK-dependent pathway for the activation of CCAAT/enhancer binding protein by interleukin-1β in hepatocytes. J Lipid Res. 2005;46:2497–505.
Regalo G, Canedo P, Suriano G, et al. C/EBPbeta is over-expressed in gastric carcinogenesis and is associated with COX-2 expression. J Pathol. 2006;210:398–404.
Sankpal NV, Mayo MW, Powell SM. Transcriptional repression of TFF1 in gastric epithelial cells by CCAAT/enhancer binding protein-β. Biochim Biophys Acta 2005;1728:1–10.
Sankpal NV, Moskaluk CA, Hampton GM, et al. Overexpression of CEBPbeta correlates with decreased TFF1 in gastric cancer. Oncogene. 2006;25:643–9.
Zhao D. Protein kinase Cδ-mediated CREB activation regulates ghrelin-induced cyclooxygenase-2 expression and prostaglandin E2 production in human colonic epithelial cells. J Cell Biochem. 2007;102:1245–55.
Sun H, Chung WC, Ryu SH, et al. Cyclic AMP-responsive element binding protein- and nuclear factor-kappaB-regulated CXC chemokine gene expression in lung carcinogenesis. Cancer Prev Res (Phila). 2008;1:316–28.
Shankar DB, Cheng JC, Kinjo K, et al. The role of CREB as a proto-oncogene in hematopoiesis and in acute myeloid leukemia. Cancer Cell. 2005;7:351–62.
Seo HS, Liu DD, Bekele BN, et al. Cyclic AMP response element-binding protein overexpression: a feature associated with negative prognosis in never smokers with non-small cell lung cancer. Cancer Res. 2008;68:6065–73.
Aggarwal S, Kim SW, Ryu SH, et al. Growth suppression of lung cancer cells by targeting cyclic AMP response element-binding protein. Cancer Res. 2008;68:981–8.
Chhabra A, Fernando H, Watkins G, et al. Expression of transcription factor CREB1 in human breast cancer and its correlation with prognosis. Oncol Rep. 2007;18:953–8.
Niehof M, Manns MP, Trautwein C. CREB controls LAP/C/EBP beta transcription. Mol Cell Biol. 1997;17:3600–13.
Zhang JW, Klemm DJ, Vinson C, et al. Role of CREB in transcriptional regulation of CCAAT/enhancer-binding protein beta gene during adipogenesis. J Biol Chem. 2004;279:4471–8.
Chen L, Yang Q, Kong WQ, et al. MicroRNA-181b targets cAMP responsive element binding protein 1 in gastric adenocarcinomas. IUBMB Life. 2012;64:628–35.
Acknowledgments
This work was supported by Fundação para a Ciência e Tecnologia (SFRH/BD/46504/2008; ERA-PTG/0001/2010), FEDER, National Funds through the FCT—Foundation for Science and Technology, under the projects “PEst-C/SAU/LA0003/2013” and Programa Operacional Factores de Competitividade—COMPETE. IPATIMUP integrates the i3S Research Unit, which is partially supported by FCT.
Confict of interest
None.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Resende, C., Regalo, G., Durães, C. et al. Interleukin-1B signalling leads to increased survival of gastric carcinoma cells through a CREB-C/EBPβ-associated mechanism. Gastric Cancer 19, 74–84 (2016). https://doi.org/10.1007/s10120-014-0448-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10120-014-0448-x