
Abstract
Background
Creutzfeldt-Jacob disease (CJD) is a fatal neuro-degenerative disease, characterized by rapid and intense deterioration, mainly cognitive, leading to death. The typical onset of the disease is around the age of 67.
Purpose
To characterize the demographic and clinical features of the population of CJD patients with late-onset disease.
Methods
In this retrospective study, the Israeli national database of prion diseases was screened for CJD patients with disease age of onset > 80 years between 1960 and 2016. Patient’s demographic and clinical data were collected including sex, type of disease (sporadic/ genetic), clinical presentation, lab results including tau protein level, imaging, and EEG characteristics. Then, the clinical and demographic data of patients with late onset (> 80 years) (L) and patients with usual age of onset (< 80 years) (U) were compared.
Results
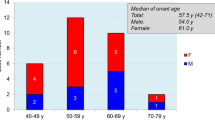
The study included 728 patients, 23 patients (3.3%) with late-onset disease (82.2.4±4 years, range 80–88) and 705 with usual disease onset (61.31 ± 9.47 years, range 34–80). Sporadic CJD was more common in the late-onset group (18/23 patients (78.2%) (L) vs. 256/705 patients (36.3%) (U)) (p = 0.0001, chi-square test). Classical EEG finding of periodic sharp wave activity were seen more often in the late-onset patients (55% (L) vs. 32.5% (U)) (p = 0.05, chi-square test). The rest of the demographic and clinical features were similar in both groups.
Conclusion
Late- and usual-onset diseases are similar in most of demographic and clinical features suggesting a common disease type with normal distribution of age of onset.

Similar content being viewed by others

References
Millhauser GL (2004) Copper binding in the prion protein. Acc Chem Res 37:79–85
Bremer J, Baumann F, Tiberi C et al (2010) Axonal prion protein is required for peripheral myelin maintenance. Nat Neurosci 13:310–318
Wulf MA, Senatore A, Aguzzi A (2017) The biological function of the cellular prion protein: an update. BMC Biol 15(1):34
Eggenberger E (2007) Prion disease. Neurol Clin 25:833–42.viii
Knight RS, Will RG (2004) Prion diseases. J Neurol Neurosurg Psychiatry 75:i36–i42
Goldfarb LG, Korczyn AD, Brown P, Chapman J, Gajdusek DC (1990) Mutation in codon 200 of scrapie amyloid precursor gene linked to Creutzfeldt-Jakob disease in Sephardic Jews of Libyan and non-Libyan origin. Lancet 336:637–638
Hsiao K, Meiner Z, Kahana E et al (1991) Mutation of the prion protein in Libyan Jews with Creutzfeldt-Jakob disease. N Engl J Med 324:1091–1097
Korczyn AD (1991) Creutzfeldt-Jakob disease among Libyan Jews. Eur J Epidemiol 7:490–493
Cali I, Cohen ML, Haik S et al (2018) Iatrogenic Creutzfeldt-Jakob disease with amyloid-β pathology: an international study. Acta Neuropathol Commun 6(1):5
Croes EA, van Duijn CM (2003) Variant Creutzfeldt-Jakob disease. Eur J Epidemiol 18:473–477
Lundberg PO (1998) Creutzfeldt-Jakob disease in Sweden. J Neurol Neurosurg Psychiatry 65:836–841
Sun Y, Fan LY, Huang CT et al (2021) Prognostic Features of Sporadic Creutzfeldt-Jakob Disease: an analysis of Taiwan’s Nationwide Surveillance. J Am Med Dir Assoc. 4:S1525-8610(21)00736–2. https://doi.org/10.1016/j.jamda.2021.08.010
Gambetti P, Kong Q, Zou W, Parchi P, Chen SG (2003) Sporadic and familial CJD: classification and characterisation. Br Med Bull 66:213–239
Minikel E, Vallabh S, Orseth M et al (2019) Age at onset in genetic prion disease and the design of preventive clinical trials. Neurology 93(2):e125–e134
Knight R (2002) Epidemiology of variant CJD. Dev Biol (Basel) 108:87–92
Corato M, Cereda C, Cova E, Ferrarese C, Ceroni M (2006) Young-onset CJD: age and disease phenotype in variant and sporadic forms. Funct Neurol 21:211–215
Boesenberg C, Schulz-Schaeffer WJ, Meissner B et al (2005) Clinical course in young patients with sporadic Creutzfeldt-Jakob disease. Ann Neurol 58:533–543
WHO (1998) Global surveillance, diagnosis and therapy of human transmissible spongiform encephalopathies: report of the WHO consultation. In: World health organization: emerging and other communicable diseases, surveillance and control, Geneva, pp 9–11
Hansen HC, Zschocke S, Sturenburg HJ, Kunze K (1998) Clinical changes and EEG patterns preceding the onset of periodic sharp wave complexes in Creutzfeldt-Jakob disease. Acta Neurol Scand 97:99–106
Caobelli F, Cobelli M, Pizzocaro C, Pavia M, Magnaldi S, Guerra UP (2015) The role of neuroimaging in evaluating patients affected by Creutzfeldt-Jakob disease: a systematic review of the literature. J Neuroimaging. 25:2–13
Nozaki I, Hamaguchi T, Sanjo N et al (2010) Prospective 10-year surveillance of human prion diseases in Japan. Brain 133:3043–3057
Appel SA, Chapman J, Kahana E et al (2010) Rapidly progressive Creutzfeldt-Jakob disease in patients with Familial Mediterranean Fever. Eur J Neurol 17:861–865
Funding
The work was partially supported by the National Institute of Health (NIH) Grant #NS043488.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethical approval
None.
Conflict of interest
The authors declare no competing interests.
Informed consent
None.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Trachtenbroit, I., Cohen, O.S., Chapman, J. et al. Epidemiological and clinical characteristics of patients with late-onset Creutzfeldt-Jakob disease. Neurol Sci 43, 4275–4279 (2022). https://doi.org/10.1007/s10072-022-05929-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10072-022-05929-9