
Abstract
The light-gated dimeric cation channel channelrhodopsin-2 (ChR2) is one of the most important optogenetic tools. Upon light activation ChR2 undergoes conformational changes, the most prominent ones include a movement of transmembrane helix B. In the present work, we apply time resolved continuous wave EPR spectroscopy to follow spectral changes of a spin label bound to position C79 located in helix B. We observed an increase of the motional freedom of the spin label side chain in illuminated ChR2. The recovery of the underlying light-induced conformational change in the dark is correlated with the recovery of the P480 state of ChR2. The observed conformational changes might be thus key elements responsible for desensitizing the channel for cation conduction.
Similar content being viewed by others
1 Introduction
Retinal proteins like bacteriorhodopsin (BR), channelrhodopsins (ChR), sensory rhodopsins (SR) or animal rhodopsins have attracted increasing attention because of their structural relation with G protein coupled receptors and as key tools in optogenetics (for reviews see, e.g., [1, 2]). Animal rhodopsins are G protein coupled receptors, whereas microbial rhodopsins are active or passive ion transporters or act as sensors. These rhodopsins share a common architecture of seven transmembrane α-helices, which harbor a retinal bound to transmembrane helix 7 (TM7, helix G) of the protein by a protonated Schiff base. In microbal rhodopsins, the photoactivated retinal isomerisation initiates the so-called photocycle during which the protein performs its function, e.g., as an ion pump (BR), an ion channel (ChR) or as a light signaling device (SR). Prominent light-induced changes in the protein backbone conformation comprise outward tilts of the cytoplasmic end of TM6 (helix F), which are functionally significant for animal rhodopsin function [3, 4], for ion transport (BR) or for the interaction with transducers (SR) [1, 5]. The dynamics of the ion channel ChR seems to deviate from this pattern as a prominent conformational change upon illumination comprises significant movements of TM2 (helix B) [6,7,8].
Time-resolved continuous wave (cw) EPR spectroscopy has been a powerful tool for following such conformational changes. The first time-resolved cw EPR analyses on spin labeled BR was conducted in the Hubbell laboratory and revealed a light-induced transient outward movement of helix F during the BR photocycle [9]. Further time-resolved EPR investigations with spin labeled side chains introduced along the E–F loop and into the cytoplasmic ends of helices E and F as well as inter-spin distance measurements performed on stabilized photocycle intermediates revealed details of the time course of the conformational changes and showed an additional movement of helix G towards helix C [10,11,12,13,14]. A similar conformational change of helix F could be observed in SRII from Natronomonas pharaonis [5, 15]. In the present work, we use time-resolved cw EPR to follow light-induced conformational changes in channelrhodopsin-2 (ChR2) and to show its functional relevance.
ChR2 from the green alga Chlamydomonas reinhardtii forms a dimer and is a light-gated cation channel, which has been established as one of the most important optogenetic tools [16]. From a total of 737 amino acids, amino acids 1 to 315 form the transmembrane part consisting of seven almost parallel helices harboring the retinal [17, 18] (Fig. 1). The cytosolic domain (residues 316–737) does not play a crucial role for the channel function [19]. Helices C and D shape the dimerization sites of the ChR2 dimer [20, 21]. In the closed state [22], a cavity surrounded by helices A, B, C and G is found opened towards the extracellular side but occluded from the cytoplasmic side [21]. Residues within helices B and G seem to regulate the cation selectivity of the cation-conducting pathway [23]. A water-mediated hydrogen-bond between C128 (helix C) and D156 (helix D) acts as a putative gate (DC gate) with a strong influence on the closing kinetics of the channel [22, 24, 25].

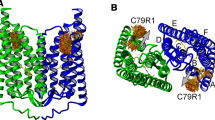
Structural model of the ChR2 dimer in ribbon diagram representation [PDB: 6EID] The view parallel (A) and perpendicular (B) to the membrane plane shows the arrangement of the two protomers (green, blue). The two corresponding retinal chromophores are depicted in yellow. The seven transmembrane helices are labeled in one of the monomers from A to G (B). Spin label rotamers (orange, nitroxide oxygen atoms in red, stick representation) had been calculated for positions 79 using MtsslWizard [26] and show the possible orientations of the spin label side chains C79R1 [7]. The observed movement of helix B (see text) is indicated by the grey arrows (modified from [7]) (Color figure online)
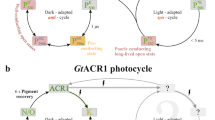
The channel function of ChR2 is linked to its photocycle which is initiated by the photoactivated isomerization of the retinal [27,28,29,30]. In the dark-adapted state, the retinal adopts an all-trans, C = N-anti conformation with a protonated Schiff base. Light activation leads to a branched photocycle with a 13-cis, C = N-anti or a 13-cis, C = N-syn retinal conformation [31]. The first resulting intermediate of the anti-cycle, PK500, decays to the blue-shifted intermediate PM390 in the µs time range [32]. During this transition the retinal Schiff base becomes deprotonated. Its reprotonation occurs during the transition to the intermediate PN520 which represents the conductive state of the protein [28]. The recovery of the H-bonding network of the DC gate and the protonation states of E90 and D156 lead to inhibition of the cation permeation after about 10 ms [32]. Extended photocycle models (e.g., [31, 33,34,35,36,37]) with parallel branches include the intermediate P480. It describes a light-adapted state of ChR2 with 13-cis, C = N-syn retinal conformation [31, 37] and represents the closed state of the syn-cycle, the so-called desensitized state. Photoexcitation of P480 comprises a slowly decaying second open state of high proton selectivity but low overall ion conductance [31]. P480 is accumulated during continuous illumination [31] and was shown to be stabilized by freeze trapping at 245 K [36]. Recently, we showed using double electron–electron resonance (DEER) that the thermal relaxation patterns of the light-induced distance changes between spin labels bound to C79 (C79R1) at helix B (Fig. 1) are in agreement with the rise and decay of the P480 state [7].
Channel opening and closing require conformational changes. IR spectroscopy revealed movements of the key residue E90 during the early formation of the ion conducting pore [38] which induces a tilt of helix B. DEER measurements of spin labeled ChR2 variants showed a movement of helix B [6,7,8] upon illumination accompanied by smaller structural changes of helix F in case of the C128T mutant, which has a more than 100 times slower transition from the open to the closed state [8]. Cw EPR measurements of a natural anion ChR labeled at the A–B loop supported the helix B movement upon light activation [39]. Cryo-electron microscopy measurements of two-dimensional crystals of the ChR2-C128T mutant confirmed a movement of helix F and a loss of order of helix B and indicated additional structural changes of helix G [40]. Electrophysiological analyses and all-atom molecular dynamics simulations also revealed movements of helices B, F and G [41]. Our data presented in the present work show an increase of the motional freedom of the spin label side chains C79R1 in illuminated ChR2. The recovery of the light-induced mobility changes to the initial value is correlated with the recovery of the P480 state of ChR2.
2 Materials and Methods
2.1 Expression and Purification
The ChR2 variant ChR2-C79R1 was prepared according to published protocols [6, 7, 24]. In brief, the expression of Chop2 (coding for amino acids residues 1 to 315 of GenBank accession number AF461397) with a C-terminal 9 × His-tag in Pichia pastoris was induced in the presence of all-trans retinal by methanol added three times over a period of 30 h. The cells were harvested by centrifugation and resuspended to 30% wet weight in 20 mM sodium phosphate (pH 7.4), 100 mM NaCl, 2 mM EDTA, 1 mM PMSF and 5% (v/v) glycerol. Cell lysis was induced mechanically by glass beads (0.5 mm) in a bead-beater (BioSpec Products). The suspension was centrifuged for 15 min at 5000 g. The supernatant was centrifuged for 1 h at 100,000 g. The membranes were collected and homogenized in 20 mM HEPES/NaOH (pH 7.4), 100 mM NaCl. Protein was solubilized with 1% [m/v] b-decyl maltoside and purified with a Ni–NTA sepharose column. The purity factor of the prepared samples was calculated to be 2.8 indicating that ~ 75% of the protomers bound a retinal [7].
2.2 Site-Directed Spin Labeling
Spin labeling was conducted as published [7]. In brief, the protein was bound to a Ni–NTA column and the cysteines were reduced with 5 mM DTT. After removal of DTT, the protein samples were incubated with 1 mM spin label ((1-oxyl-2,2,5,5-tetramethyl-3-pyrroline-3-methyl) methanethiosulfonate; MTSSL) for 12–20 h at 4 °C in darkness. Unbound spin label was removed by buffer exchange and the proteins were eluted with 1 M imidazole, before they were purified on a Superdex 200 column. Samples were concentrated by centrifugal filtration (Ultra-15, 10 kDa, Amicon). Spin labeling efficiencies of ChR2-C79R1 amounted to ~ 90% [7].
2.3 Reconstitution
ChR2-C79R1 was reconstituted in liposomes of POPC/POPG/cholesterol (8:1:1). The lipids were mixed in 20 mM HEPES (pH 7.4), vesicles were formed by vortexing and adding 10% cholat. To remove aggregates the lipids were centrifuged (3 min, 100 g). Protein and lipids were mixed in a ratio of 1:4 (w/w). After incubation for 10 min at room temperature, the mixture was subjected to pre-washed SM-2 Bio-Beads (Bio-Rad, Hercules, CA, US) to remove detergent.
2.4 EPR Spectroscopy and Light Activation of ChR2
Cw EPR spectra were recorded with a home-built X-band EPR spectrometer equipped with a dielectric resonator, MD5 (Bruker), using a microwave power of 0.5 mW and a magnetic field modulation of 0.15 mT. The magnetic field was controlled with an ER 032 controller (Bruker), the temperature of the cavity was held constant by a liquid flow cryostat. 20 μL of the sample were filled into EPR glass capillaries (0.9 mm inner diameter) with a final concentration between 80 and 440 μM of protein.
To light-activate ChR2, the sample was illuminated inside the resonator by a blue LED (470 nm, bandwidth 20 nm (M470F3, Thorlabs)) between 0.5 and 20 s. The changes of the EPR signals during and after light activation were followed for a total of 60–120 s. Light triggered EPR transients were recorded at different magnetic field values. 100 time traces were averaged for each magnetic field value to achieve sufficient signal to noise ratio. For these experiments, the illumination period was limited to 0.5 s to minimize bleaching of the sample. The recovery of the initial EPR signal upon light activation was fitted by a mono-exponential decay function with the amplitude factor and the time constant as fitting parameters. The plot of the fitted amplitude factors versus the magnetic field provided the so-called light–dark difference spectra [9].
The cw EPR spectra were simulated using 'Multi Component EPR Fitting' (written by C. Altenbach, University of California, Los Angeles, CA, in LabVIEW (National Instruments) available from http://www.chemistry.ucla.edu/directory/hubbell-wayne-l). For a given motional model with a number of spectral components, their respective A- and g-tensors, reorientational diffusion rates and line widths, the program solves the stochastic Liouville equation numerically. The deviation between the simulated and experimental spectra was minimized using the Levenberg–Marquardt algorithm. A simple motional model for solvent-exposed spin label side chains is the model of isotropic reorientational motion characterized by an isotropic rotational diffusion rate. For the present case this model with two spectral components provided reasonable agreement between simulation and experiment. Fitting parameters were the logarithm of the rotational diffusion rates (from which the reorientational correlation times were calculated), the line widths, and the fractions of the spectral components [42], all other parameters were fixed (gxx = 2.0085, gyy = 2.0061, gzz = 2.0022; Axx = 0.58 T, Ayy = 0.42 T, Azz,sm = 3.7 T, Azz,im = 3.5 T).
3 Results
We used a ChR2 mutant that has seven cysteines replaced, C34S/C36S/C87S/C179L/C183L/C208A/C259L, with two native cysteines remaining, C79 and C128 (Mut2C) [6]. Incubation of Mut2C with (1-oxyl-2,2,5,5-tetramethyl-pyrroline-3-methyl) methanethiosulfonate spin label (MTSSL) leads to a spin label side chain at position C79, C79R1, because C128 was found to be inaccessible for MTSSL for intact ChR2 [6]. Electrophysiological and spectroscopic experiments supported the conclusion that spin labeled Mut2C shows wildtype characteristics [6]. DEER spectroscopy of spin labeled Mut2C uncovered light-induced inter-spin distance changes between positions C79R1 and C79R1’ of the ChR2 dimer which were interpreted as movements of helix B upon light activation [6, 8]. A DEER study of trapped intermediates provided strong evidence that this movement is correlated with the presence of the P480 state [7]. In the present work, time resolved continuous wave (cw) EPR measurements were performed to follow possible EPR signal changes of C79R1 upon light activation.
3.1 Cw EPR Spectra of ChR2-C79R1
In general, a cw EPR spectrum of a spin labeled protein reflects the mobility of the spin label side chain, R1, which is strongly influenced by its local environment. Sharp lines reveal a high mobility of R1, whereas a broad spectrum points to sterical restrictions of the spin label side chain reorientational motion caused by van-der-Waals interaction of R1 with neighboring side chain or backbone atoms. The cw EPR spectra of solubilized and reconstituted ChR2-C79R1 (Fig. 2) both show broad spectral lines revealing two components, an immobile (im) and a semi-mobile (sm) component. The reorientational motion of R1 for the sample reconstituted into liposomes seems to be less restricted compared to the solubilized sample because the immobile component is less prominent. For a more quantitative analysis simulated EPR spectra were fitted to the experimental ones. We used a very simple model with two spectral components characterized by different rotational correlation times for the assumed isotropic reorientational motion. Fitting parameters were the fractions of the components and the two rotation correlation times, τsm and τim (see Table 1). For both samples, the values of τsm were found to be similar (4.0–4.5 ns), whereas the value of τim of the reconstituted sample exceeds that of the solubilized sample (170 ± 30 ns vs. 105 ± 10 ns). The difference in τim for solubilized and reconstituted C79R1 might be due to the reduced protein overall rotational diffusion in liposomes compared to micelles. Interestingly, the fractions for the two components were significantly different (Table 1) with the semi-mobile component accounting for more than 70% of the reconstituted sample. Thus C79R1 is more mobile in the reconstituted form of ChR2.

The experimental EPR spectra (continuous line) of A solubilized and B reconstituted ChR2-C79R1 can be simulated by a superposition of two spectral components, which characterize an immobile (im) and a semi-mobile (sm) spin label side chain R1 (inset). Fitting of these simulated spectra (dashed) to the experimental ones reveal the reorientational correlation times and the fractions of these components (see Table 1)
3.2 Time-Resolved cw EPR Upon Light Activation of ChR2-C79R1
Changes of the EPR signal upon light activation were recorded at fixed magnetic field values. Depending on the field position the EPR signals increase or decrease during illumination and recover to their initial values in the dark (Fig. 3). The recovery of the initial signal could be fitted by a single exponential with a time constant of ~ 20 s (for averaged time constants see below). This time constant does neither depend on the duration of the light activation nor on the field position. In contrast, the amplitudes of the transient signals show a significant dependence on the field position (Fig. 3a) and increase with increasing duration of illumination (Fig. 3b) due to the accumulation of activated molecules. The slope of the signal rise decreases for longer illumination times (> ~ 10 s) where the number of molecules being activated by light (per time interval) and of those relaxing to the initial state (per time interval) fall into the same order of magnitude. With the present light intensity, a delay between light activation and EPR signal change could not be detected, thus the time course of the rise of the signal could not be resolved with the present setup.

EPR signal changes of the reconstituted sample during illumination and their relaxation in the dark. A Depending on the magnetic field positions (inset, arrows) the EPR signal rises (green) or falls (blue) during illumination with blue light (the illumination period of 20 s is indicated by the horizontal bars). In the dark the EPR signal recovers to its initial value following a mono-exponential decay (black). B The absolute amplitudes of single transients (squares) recorded at the B-field position indicated by the blue arrow in A increase with the length of the illumination period (illumination periods of 2 s (black), 10 s (blue) and 20 s (red) are indicated by the horizontal bars). From averaged transients a mono-exponential decay function with a relaxation time of 21 s was determined which fits the depicted signal relaxation in the dark (starting at time point 0) reasonably well (continuous lines) (Color figure online)
The amplitude factors of the fitted exponentials plotted as function of the magnetic field (histograms in Fig. 4) constitute the so-called light–dark difference spectra and provide information about mobility change of R1 during light activation. The observed spectral changes are in reasonable agreement with calculated difference spectra where the component fractions are shifted from the immobile to the semi-mobile component (cyan area in Fig. 4). Thus, for both samples, solubilized and reconstituted ChR2-C79R1, the fractions of the semi-mobile components increase during light activation.

The cw EPR spectra (black) of ChR2-C79R1 (A solubilized, B reconstituted) inform about the spin label side chain mobility in the dark. The amplitude factors of a mono-exponential decay function fitted to the EPR transients upon light activation of 0.5 s duration, recorded at different magnetic fields, represent the light minus dark difference EPR spectra (histograms, multiplied by 150, dark green). They reflect the light induced mobility changes. Spectra calculated from the difference of the spin normalized single fractions (semi-mobile minus immobile) are shown for comparison (cyan area). Averaged transients with fittings of a mono-exponential decay (red) are shown in the insets (Color figure online)
Upon inversion of negative transients all transient time traces were averaged and the signal recovery in the dark was fitted with a mono-exponential function (Fig. 4, insets). The relaxation times of solubilized and reconstituted ChR2-C79R1 were found to be (23.5 ± 1.5) s and (21.1 ± 1.3) s (T = 298 K), respectively, and are thus identical within experimental error.
In general, mobility changes of the R1 side chain indicate changes of its environment and/or alterations of the associated backbone and its dynamics. The observed results for C79R1 thus reflect light-induced conformational changes of the protein in the vicinity of C79R1 and/or changes of the orientation or dynamics of helix B. DEER experiments had revealed that the distance between C79R1 nitroxides in the ChR2 dimer increase upon light activation [6,7,8]. These distance changes were attributed to movements of helix B. Trapping of intermediates enabled to identify the P480 state as the state with the most prominent distance increase [7]. The time constant for the recovery to the C79R1 initial state observed in the present work is similar to the time constant reported for the decay of the P480 state (~ 28 s at 298 K [32], 40 s at 288 K [31]). Hence, these data are strong evidence that the mobility changes of C79R1 upon light activation are related to the rise and decay of the P480 state.
4 Discussion
ChR2 has emerged as an important tool in the field of optogenetics. To rationally design ChR2 variants for advanced optogenetic approaches with high ion selectivity and improved photocurrent inactivation, a better understanding of the molecular basis for the transition between the two open states with their different ion selectivity is required. In this respect the functional role and importance of the observed helix B movement is still under discussion. Here, we performed cw and time resolved cw EPR spectroscopy of solubilized and reconstituted ChR2-C79R1 to follow this movement and to relate the corresponding conformational change to ChR2 photocycle transitions.
The cw EPR spectra of solubilized and reconstituted ChR2-C79R1 considerably differ in their component composition with the immobile component being less populated in the reconstituted sample. Such effects of the lipid environment on retinal protein conformations have been observed previously. The structural properties of sensory rhodopsin, particularly at the lipid–water interface, are influenced by the substitution of the lipid bilayer with detergent [43]. This interface region seems to play a crucial role in constraining protein secondary structure. C79R1 is located at the membrane-water interface. Thus, reconstitution into liposomes promotes the more native conformation of the cytoplasmic end of helix B and of the A-B loop that allows for greater freedom of motion for the spin label. Upon light activation the fractions of the more mobile components in the EPR spectra of both the solubilized and reconstituted samples transiently increase indicating an increased dynamics of the cytoplasmic end of helix B. This is in line with density changes of helix B observed by cryo-electron microscopy of light activated ChR2-C128T which were interpreted as partial unwinding or loss of order of the cytoplasmic part of helix B [40].
The current time-resolved EPR experiments reveal that the mobility changes of ChR2-C79R1, induced by illumination with long light pulses, relax with a time constant that agrees with the decay rate of the P480 state [31, 32]. Thus the transition between the initial state and the P480 state is connected with the observed conformational change of helix B. A unifying photocycle model [31] discriminates between two photocycles, the anti-cycle, which is responsible for the channel conductivity for cations and protons, and the syn-cycle, which characterizes the opening and closing process for only protons. In this model, the P480 state as a part of the syn-cycle is accumulated during continuous illumination, such an accumulation is observed in our experiments (Fig. 3b). The thermal relaxation patterns of the interspin distance changes between C79R1 and C79R1’ [6, 8] were also found to be in agreement with the rise and decay of the P480 state [7]. Together with the present results this implicates that structural changes of helix B occur, when the cation channel is closed, preferably under high light intensities and long illumination times. A functional model with two closed and two open states was introduced in 2009 [44]. The authors identified the deprotonation of E90 to take place only in the syn-cycle, which was confirmed to occur in the submicrosecond time range [38] and which has to be related to the rise of the P480 state. This rise is suggested here to be accompanied by the outward movement of helix B, which is too fast to be time resolved in the present experimental setup. It was shown earlier that the transitions between the P480 state and the initial state are correlated with the isomerization state of the retinal [37] and a rearrangement of the helical backbone of ChR2 [30]. Our data show that this rearrangement includes the movement of helix B with a change of the mobility of the C79R1 side chain.
Helix B plays a significant role in the ion selectivity for ChR2. There are three glutamate residues located on helix B (E90, E97, and E101) that are crucial to proton and sodium selectivity [45]. Furthermore, mutagenesis of V86 and K93 were shown to alter ChR2 ion conductance properties, including cation selectivity and the relative population of the two conductive states [23]. The amino acids E90, K93, E97, E101, and M107 are interconnected via a hydrogen-bond network with the retinal pocket, this network contributes to the stabilization of the helix [22]. Upon retinal isomerization this network is broken which may account for the observed helix movement. Deprotonation of E90 and the subsequent hydration of the pore mediated by this movement of helix B prepare the proton conductance of the open state in the syn-cycle [31].
The present results provide experimental evidence that the movement of helix B might serve as or reflect the function of a switch between the anti- and the syn-cycle as suggested in [23]. Continuous illumination leads to an accumulation of the P480 state [31] with helix B moved outwardly, thus diminishing the flux of cations. Since the rise time of the EPR signal change could not be resolved we cannot exclude that the movement of helix B occurs in the PN520 state in accordance with results on the ChR2-C128T mutant [8], and that it may be also important for ion permeation and selectivity.
Availability of Data and Materials
All data generated or analyzed during this study are included in this published article.
References
O.P. Ernst, D.T. Lodowski, M. Elstner, P. Hegemann, L.S. Brown, H. Kandori, Chem. Rev. 114, 126 (2014)
H. Kandori, Biophys. Rev. 12, 355 (2020)
D.L. Farrens, C. Altenbach, K. Yang, W.L. Hubbell, H.G. Khorana, Science 274, 768 (1996)
C. Altenbach, A.K. Kusnetzow, O.P. Ernst, K.P. Hofmann, W.L. Hubbell, Proc. Natl. Acad. Sci. USA 105, 7439 (2008)
J.P. Klare, E. Bordignon, M. Engelhard, H.-J. Steinhoff, Photochem. Photobiol. Sci. 3, 543 (2004)
T. Sattig, C. Rickert, E. Bamberg, H.J. Steinhoff, C. Bamann, Angew. Chem. Int. Ed. Engl. 52, 9705 (2013)
M. Schumacher, J.P. Klare, C. Bamann, H.-J. Steinhoff, Appl. Magn. Reson. 53, 731 (2022)
N. Krause, C. Engelhard, J. Heberle, R. Schlesinger, R. Bittl, FEBS Lett. 587, 3309 (2013)
H.-J. Steinhoff, R. Mollaaghababa, C. Altenbach, K. Hideg, M. Krebs, H.G. Khorana, W.L. Hubbell, Science 266, 105 (1994)
T. Rink, M. Pfeiffer, D. Oesterhelt, K. Gerwert, H.J. Steinhoff, Biophys. J. 78, 1519 (2000)
N. Radzwill, K. Gerwert, H.J. Steinhoff, Biophys. J. 80, 2856 (2001)
H.J. Steinhoff, R. Mollaaghababa, C. Altenbach, H.G. Khorana, W.L. Hubbell, Biophys. Chem. 56, 89 (1995)
R. Mollaaghababa, H.J. Steinhoff, W.L. Hubbell, H.G. Khorana, Biochemistry 39, 1120 (2000)
T. Rink, J. Riesle, D. Oesterhelt, K. Gerwert, H.J. Steinhoff, Biophys. J. 73, 983 (1997)
A.A. Wegener, J.P. Klare, M. Engelhard, H.J. Steinhoff, Embo. J. 20, 5312 (2001)
O. Yizhar, L.E. Fenno, T.J. Davidson, M. Mogri, K. Deisseroth, Neuron 71, 9 (2011)
J.P. Klare, I. Chizhov, M. Engelhard, Bioenergetics (2007). https://doi.org/10.1007/400_2007_041
J. Pinhassi, E.F. DeLong, O. Béjà, J.M. González, C. Pedrós-Alió, Microbiol. Mol. Biol. Rev. 80, 929 (2016)
G. Nagel, T. Szellas, W. Huhn, S. Kateriya, N. Adeishvili, P. Berthold, D. Ollig, P. Hegemann, E. Bamberg, Proc. Natl. Acad. Sci. USA 100, 13940 (2003)
M. Muller, C. Bamann, E. Bamberg, W. Kuhlbrandt, J. Mol. Biol. 414, 86 (2011)
H.E. Kato, F. Zhang, O. Yizhar, C. Ramakrishnan, T. Nishizawa, K. Hirata, J. Ito, Y. Aita, T. Tsukazaki, S. Hayashi, P. Hegemann, A.D. Maturana, R. Ishitani, K. Deisseroth, O. Nureki, Nature 482, 369 (2012)
O. Volkov, K. Kovalev, V. Polovinkin, V. Borshchevskiy, C. Bamann, R. Astashkin, E. Marin, A. Popov, T. Balandin, D. Willbold, G. Buldt, E. Bamberg, V. Gordeliy, Science 358, eaan8862 (2017)
R. Richards, R.E. Dempski, J. Biol. Chem. 292, 7314 (2017)
C. Bamann, R. Gueta, S. Kleinlogel, G. Nagel, E. Bamberg, Biochemistry 49, 267 (2010)
M. Nack, I. Radu, M. Gossing, C. Bamann, E. Bamberg, G.F. von Mollard, J. Heberle, Photochem. Photobiol. Sci. 9, 194 (2010)
G. Hagelueken, R. Ward, J.H. Naismith, O. Schiemann, Appl. Magn. Reson. 42, 377 (2012)
J.L. Spudich, C.-S. Yang, K.-H. Jung, E.N. Spudich, Annu. Rev. Cell Dev. Biol. 16, 365 (2000)
C. Bamann, T. Kirsch, G. Nagel, E. Bamberg, J. Mol. Biol. 375, 686 (2008)
E. Ritter, K. Stehfest, A. Berndt, P. Hegemann, F.J. Bartl, J. Biol. Chem. 283, 35033 (2008)
I. Radu, C. Bamann, M. Nack, G. Nagel, E. Bamberg, J. Heberle, J. Am. Chem. Soc. 131, 7313 (2009)
J. Kuhne, J. Vierock, S.A. Tennigkeit, M.A. Dreier, J. Wietek, D. Petersen, K. Gavriljuk, S.F. El-Mashtoly, P. Hegemann, K. Gerwert, Proc. Natl. Acad. Sci. USA 116, 9380 (2019)
V.A. Lorenz-Fonfria, T. Resler, N. Krause, M. Nack, M. Gossing, G. Fischer von Mollard, C. Bamann, E. Bamberg, R. Schlesinger, J. Heberle, Proc. Natl. Acad. Sci. USA 110, E1273 (2013)
V.A. Lorenz-Fonfria, J. Heberle, Biochim. Biophys. Acta 1837, 626 (2014)
S. Bruun, D. Stoeppler, A. Keidel, U. Kuhlmann, M. Luck, A. Diehl, M.A. Geiger, D. Woodmansee, D. Trauner, P. Hegemann, H. Oschkinat, P. Hildebrandt, K. Stehfest, Biochemistry 54, 5389 (2015)
M. Saita, F. Pranga-Sellnau, T. Resler, R. Schlesinger, J. Heberle, V.A. Lorenz-Fonfria, J. Am. Chem. Soc. 140, 9899 (2018)
J. Becker-Baldus, C. Bamann, K. Saxena, H. Gustmann, L.J. Brown, R.C. Brown, C. Reiter, E. Bamberg, J. Wachtveitl, H. Schwalbe, C. Glaubitz, Proc. Natl. Acad. Sci. USA 112, 9896 (2015)
J. Becker-Baldus, A. Leeder, L.J. Brown, R.C.D. Brown, C. Bamann, C. Glaubitz, Angew. Chem. Int. Ed. Engl. 60, 16442 (2021)
J. Kuhne, K. Eisenhauer, E. Ritter, P. Hegemann, K. Gerwert, F. Bartl, Angew. Chem. Int. Ed. Engl. 54, 4953 (2015)
H. Li, O.A. Sineshchekov, G. Wu, J.L. Spudich, J. Biol. Chem. 291, 25319 (2016)
M. Muller, C. Bamann, E. Bamberg, W. Kuhlbrandt, J. Mol. Biol. 427, 341 (2015)
M. Takemoto, H.E. Kato, M. Koyama, J. Ito, M. Kamiya, S. Hayashi, A.D. Maturana, K. Deisseroth, R. Ishitani, O. Nureki, PLoS ONE 10, e0131094 (2015)
D.E. Budil, S. Lee, S. Saxena, J.H. Freed, J. Magn. Reson. Ser. A 120, 155 (1996)
J.P. Klare, E. Bordignon, M. Doebber, J. Fitter, J. Kriegsmann, I. Chizhov, H.-J. Steinhoff, M. Engelhard, J. Mol. Biol. 356, 1207 (2006)
K. Nikolic, N. Grossman, M.S. Grubb, J. Burrone, C. Toumazou, P. Degenaar, Photochem. Photobiol. 85, 400 (2009)
K. Ruffert, B. Himmel, D. Lall, C. Bamann, E. Bamberg, H. Betz, V. Eulenburg, Biochem. Biophys. Res. Commun. 410, 737 (2011)
Acknowledgements
We thank Ernst Bamberg (MPI of Biophysics) for continuous support, Heike Biehl for help with the ChR2 sample preparation and Johann P. Klare for fruitful discussions.
Funding
Open Access funding enabled and organized by Projekt DEAL. This work was financially supported by the Max Planck Society to CB and the Deutsche Forschungsgemeinschaft to HJS (DFG, SFB 944) and to CB (DFG, SFB 807-P12).
Author information
Authors and Affiliations
Contributions
Conceptualization: CB, HJS; Methodology: MS, CB, HJS; Formal analysis and investigation: MS, HJS; Writing—original draft preparation: MS; Writing—review and editing: MS, CB, HJS; Funding acquisition: CB, HJS; Resources: CB, HJS.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Ethical Approval
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Schumacher, M., Bamann, C. & Steinhoff, HJ. Time-Resolved EPR Spectroscopy of Channelrhodopsin-2 Helix B Movements. Appl Magn Reson 55, 207–218 (2024). https://doi.org/10.1007/s00723-023-01612-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00723-023-01612-0