
Abstract
18 (11 novel) Schiff bases, derivatives of salicylaldehyde, 2-hydroxyacetophenone, and 6-acetyl-, 8-acetyl-, and 8-formyl-7-hydroxy-4-methylcoumarin were synthesized and characterized by their spectral studies. 6-Acetyl-7-hydroxy-4-methylcoumarin was prepared by novel method under microwave assistance. These Schiff bases were evaluated for antibacterial activities against 12 bacterial and six fungi strains in vitro. N-(3,5-Dichloro-2-hydroxybenzylidene)-4-aminobenzenesulfonic acid sodium salt proved to be the most active against Staphylococcus aureus and MRSA strains (MIC 0.0194 µmol/cm3). The substitution pattern, two chlorine atoms in the salicylidene ring and the SO3Na group, is probably the most beneficial for the activity against Gram-(+) bacteria strains. All Schiff bases were evaluated for cancer efficacy against CFPAC-1 and HeLa cell cultures originating from human pancreas cancer or human cervical cancer. Schiff bases derived from salicylaldehyde are highly effective in pancreas and cervical cancer cells; however, they demonstrate also substantial toxicity towards NIH3T3 cells. Derivatives of coumarin contain three highly selective compounds: 7-hydroxy-8-[(4-methoxyphenylimino)methyl]-4-methyl-2H-chromen-2-one, N-[(7-hydroxy-4-methyl-2-oxo-2H-chromen-8-yl)methylene]-4-aminobenzenesulfonic acid sodium salt, and 7-hydroxy-8-[1-(4-hydroxyphenylimino)ethyl]-4-methyl-2H-chromen-2-one suggesting more promising potential of the second group of substances.
Graphical abstract

Similar content being viewed by others
Introduction
Schiff bases, formed in the reaction of aromatic or heteroaromatic substances’ primary amine with aldehyde or ketone, are also classified as imines or azomethines. They are a significant class of compounds in medicinal and pharmaceutical chemistry having a variety of biological applications that include antibacterial, antifungal, and antitumor activities. The imine group present in such compounds (of natural as well as synthetic origin) has been shown to be critical to their biological activities [1,2,3,4].
The increase in the mortality rate associated with infectious diseases is directly related to antimicrobial resistance to antibiotics [5]. New drugs to combat this problem are, therefore, in great demand [6].
Schiff bases have been shown to be interesting moieties for the design of antimicrobial agents [1,2,3,4]. Especially, several Schiff bases having dichloro or bromophenyl moieties have been proved to be biologically important [4]. The presence of halogen atom in the molecule increases the lipophilicity of the molecule and, therefore, affects the partitioning of a molecule into membranes and also facilitates hydrophobic interactions of the molecule with specific binding sites on either receptor or enzymes [7]. Salicylidene (2-hydroxybenzaldehyde-derived) Schiff bases (Fig. 1) are reported as promising antimicrobial drugs [4, 8, 9].

Salicylidene Schiff bases: N-salicylidene-2-hydroxyaniline (1) and derivatives of 5-chlorosalicylaldehyde (2)
De Souza reports the determination of the minimal inhibitory concentration (MIC) in vitro for M. tuberculosis H37Rv of a diverse group of compounds, including N-(salicylidene)-derived Schiff bases. The most effective compound among tested samples was N-(salicylidene)-2-hydroxyaniline (1) with a MIC of 8 µmol/dm3. Compound 1 showed a weak cytotoxicity to J774 macrophages and at 1000 µmol/dm3 80% of cells were viable indicating a higher selectivity of the drug to the pathogen than to the mammalian cells. The results for this compound are indicative of the promising antimycobacterial activity of this molecule [8].
A series of 26 Schiff bases were synthesized by reacting 5-chloroaldehyde and primary amines. The compounds were assayed for antibacterial (Bacillus subtilis, Escherichia coli, Pseudomonas fluorescens, and Staphylococcus aureus) and antifungal (Aspergillus niger, Candida albicans, and Trichophyton rubrum) activities by MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) method. Among the compounds tested, (E)-4-chloro-2-[(4-fluorobenzylimino)methyl]phenol (2b) showed the most favorable antimicrobial activity with MICs of 45.2, 1.6, 2.8, 3.4, and 47.5 µg/cm3 against B. subtilis, E. coli, P. fluorescens, S. aureus, and A. niger, respectively. Compound 2a showed significant activity against E. coli (3.1 μg/cm3, kanamycin: 3.9 µg/cm3). Compound 2c showed significant activity against P. fluorescens (3.1 µg/cm3, kanamycin 3.9 µg/cm3) and S. aureus (1.8 µg/cm3, kanamycin 1 µg/cm3, penicillin 2 μg/cm3).
The authors claimed that increased hydrophilicity and aromaticity, as well as the presence of heteroatoms resulted in an increase in the antimicrobial activity [9].
Hybrid Schiff bases containing salicylidene and sulfonamide moieties show promising antimicrobial activity [10]. 4-[(2-Hydroxybenzylidene)amino]-N-(pyrimidin-2-yl)benzenesulfonamides (3) (Fig. 2) were active against S. aureus, including a methicillin-resistant strain (MRSA) (MIC values within the range of 0.125–0.250 µmol/cm3 [10]. In general, dihalogenation of the salicylic moiety improved the antibacterial and antifungal activity but also increased the cytotoxicity, particularly with an increasing atomic mass.

4-[(2-Hydroxybenzylidene)amino]-N-(pyrimidin-2-yl)benzene sulfonamides 3
In the recent few years, the organic compounds containing coumarin Schiff bases as integral part of their structures, in particular, the transition metal complexes, have gained much attention not only because of their antimicrobial [11, 12] but also cytotoxic activity [13]. A variety of coumarin-derived Schiff bases have been identified or developed for treatment of different types of cancers [14, 15]. For example, they have been recently shown to be inhibitors of carbonic anhydrases (CA). Wang and coworkers have designed and synthesized a series of imine-linked sulfonamide–coumarin derivatives [16]. The leading compounds 4a and 4b (Fig. 3) were active in vitro against mouse melanoma B16-F10 and MCF7 cells, to obtain the IC50 values ≤ 0.19 μM. Both compounds inhibited the hCAs II (cytosolic, offtarget isoform)—the values of IC50 were 23 nM; and hCA IX (transmembrane, tumor associated enzyme)—24 nM, respectively.

4-Chloro-3-coumarinyl derived Schiff bases 4a and 4b
To the best of our knowledge, there are no data concerning the biological activity of Schiff bases having benzenesulfonic acid salt moiety combined with halogenated salicylidene or 7-hydroxycoumarin scaffolds. To fill this gap, in the present study, we report the synthesis, characterization, and in vitro cytotoxic and antimicrobial evaluation of a series of hybrid Schiff bases, derived from salicylaldehyde (structure 5a) or 7-hydroxycoumarin scaffold (structure 5b) and para-substituted aniline, with a view to explore their potency as better chemotherapeutic agents (Fig. 4).

Design of the novel Schiff bases—a series of compounds having salicylic moiety (5a) and coumarin moiety (5b). R = H, CH3; X and Y are substituents with various electronic effects
The aim of this study is evaluation of an impact of diverse moieties and substituents in the series of Schiff bases on antimicrobial and cytotoxic activity in vitro and finding preliminary conclusions about the structure–activity relationship. Furthermore, the objective of our work is to prepare a pool of compounds that can be used as ligands in the copper or silver complexes with increased biological activity [1].
Results and discussion
Chemistry
The classical synthesis of Schiff base involves the condensation of a carbonyl compound with primary amine. Various in situ methods for water elimination (for example, using dehydrating solvents or molecular sieves or carrying the reaction under azeotropic distillation) were developed. It is known, that efficiency of this synthesis is dependent on the use of highly electrophilic carbonyl components and strongly nucleophilic aromatic amines. If necessary, Brönsted–Lowry or Lewis acids were proposed as catalysts, to activate the carbonyl group of aldehydes or ketones and facilitate the nucleophilic attack by amines, then to eliminate a molecule of water. Recently, several new techniques have been reported. Among these innovations, microwave irradiation has been extensively used due to its operational simplicity, enhanced reaction rates, and great selectivity [4].
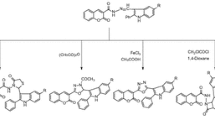
Compounds 8–13, derivatives of salicylaldehyde (Fig. 4, structure 5a), were obtained under conditions described in earlier reports [17,18,19]. Aldehydes 6a–6d were stirred with equimolar amount of appropriate aniline or sodium salt of sulfanilic acid (7a, 7e, 7f) at ambient temperature. Compound 14 was prepared by heating 2-hydroxyacetophenone and 4-methoxyaniline with the catalyst—lemon juice [20] in xylene at 100 °C (Scheme 1). The structures of Schiff bases 8–14 are listed in the Table 1.

8-Formyl-7-hydroxy-4-methylcoumarin (15) and 8-acetyl-7-hydroxy-4-methylcoumarin (17) were prepared according to earlier reports [21, 22]. As the yield of synthesis of 6-acetyl-7-hydroxy-4-methylcoumarin (16c) is low via Fries migration, we have synthesized it by novel method under microwave assistance in about 40%. Heating 7-hydroxy-4-methylcoumarin (16a) with acetic anhydride and anhydrous alumina in methanesulfonic acid resulted in the mixture of 7-acetoxy-4-methylcoumarin (16b) and the only product of Fries rearrangement: 6-acetyl-7-hydroxy-4-methylcoumarin (16c). Isolation by column chromatography on silica gel yielded the required compound 16c (Scheme 2). The synthesis of Schiff bases, derivatives of coumarin 18–28 is depicted in Scheme 3.


Schiff bases 18–22, derivatives of coumarin 15, were prepared by traditional but efficient method refluxing the ethanolic mixture of compound 15, substituted aniline 7a, 7b, 7d, 7e, and 7f, and catalytic amount of glacial acetic acid [4]. Coumarin 16 was more reactive, compared to the isomeric compound 17. The reaction of compound 16 with 4-methoxyaniline (7f) in xylene at 100 °C yielded Schiff base 23. Heating of the coumarin 17 with the corresponding aniline 7a–7g under catalytic amount of lemon juice in xylene resulted in the Schiff bases 24–28. The structures of Schiff bases 15–28 are listed in Table 2.
Microbiology
Compounds 8–28 were screened in vitro for their antimicrobial activity. The panel of pathogens involved Gram-(+) bacteria strains: Micrococcus luteus, Bacillus subtilis, Staphylococcus aureus, the MRSA strains and Enterococcus hirae; Gram-(−) bacteria strains: Escherichia coli, Pseudomonas aeruginosa; fungi: Candida albicans, Candida parapsilosis, Saccharomyces cerevisiae, Aspergillus brasiliensis. The MIC values of the active compounds are summarized in Table 3.
Salicylidene-derived Schiff bases (compounds 8, 12, 14), coumarins 15, 16a–16c, and 17 as well as coumarin-derived Schiff bases (18–23, 25–28) showed no significant antimicrobial activity. N-(2-hydroxybenzylidene)-4-aminobenzenesulfonic acid sodium salt (9) caused only some antifungal activity. N-(3,5-dichloro-2-hydroxybenzylidene)-4-aminobenzenesulfonic acid sodium salt (10) proved to be the most active against S. aureus and MRSA strains (MIC 0.0194 µmol/cm3). According to the Table 2C from Clinical and Laboratory Standards Institute [23], the susceptibility of Staphylococcus sp. to linezolid, an antibiotic used for the treatment of infections caused by Gram-positive bacteria, is defined as < 0.012–0.024 µmol/cm3. Compared with linezolid, the novel compound 10 is a promising structure for further research towards potential medicines active against S. aureus strains, including MRSA.
Two other halogenated Schiff bases 11 and 12 did not possess significant activity against these bacterial strains—replacement of the sulfonic acid salt substituent present in the compound 10 by hydroxy (compound 11) or methoxy (compound 12) groups results in reducing the antibacterial activity. The 3-methoxy-5-bromo substitution pattern and presence of the sulfonic substituent in compound 13 does not provide a beneficial effect on its activity against S. aureus and MRSA strains. We conclude that the substitution pattern, two chlorine atoms in the salicylidene ring and the SO3Na group, is probably the most beneficial for the activity against Gram-(+) bacteria strains.
Compounds 10 and 13 show antifungal activity against C. albicans strains (MIC: 0.0776 and 0.0704 µmol/cm3). Compared with the susceptibility of Candida sp. to fluconazole, an antifungal medication used for a number of fungal infections which are defined as < 0.026–0.11 µmol/cm3 [24], the MICs of compounds 10 and 13 look promising. Unfortunately, the use of coumarin scaffold instead of salicylidene for the synthesis of Schiff bases 22 and 24 causes a significant reduction in antimicrobial activity.
Cytotoxicity
Compounds 8–14 and 18–28 were examined in tumor and non-tumor cells to estimate theirs antitumor activity and selectivity. In experiments, we used following cell lines: CFPAC-1 and HeLa originating from human pancreas cancer or human cervical cancer, respectively, as well as mouse NIH-3T3 fibroblasts as non-tumor cells.
Within cytotoxicity assay, inhibitory concentrations required to inhibit 50% or 90% of control cell growth were calculated for each cell line and are displayed in the Table 4. According to this, the examined compounds are considered as not effective (IC50 values in the range 499 µM–100 µM), little effective (11 µM < IC50 < 99 µM) and effective IC50 ≤ 10 µM. All experiments were done three times independently. SD value was in range ± 2–15 for all substances and was omitted for more descriptive results. For better orientation in this table, values that are lower than those for non-tumor fibroblasts are indicated bold.
Coumarins 15, 16a, 16b, 16c, and 17 showed no significant cytotoxic activity. Most of synthesized Schiff bases showed moderate activity against tumor cells and can be categorized into the “little effective group” defined above. In particular, for CFPAC-1 cells compounds: 10–13, 18, 19, 21, 22, and 24 were little effective, whilst in HeLa cells compounds 10, 11, 13, 18–21, and 23–27 were little effective. Compound 20 for CFPAC-1 cells turned out highly effective. Compounds 8 and 9 in used concentrations were not effective for both tumor cell lines. HeLa cells turned out to be resistant against compound 22, whereas CFPAC-1 cells were little sensitive to 22 (Table 4). However, the IC50 value was relatively high for all tested compounds showing little cytotoxicity compared to known and well-established chemotherapeutics working in vitro in nanomolar concentrations, such as bortezomib. For instance, published so far data with most active imine-linked sulfonamide—coumarin derivatives—demonstrated the IC50 values ≤ 0.19 μM [16]. In our experiments except for only compound 14 against NIH3T3 cells (IC50 = 4 μM) and compound 20 against CFPAC-1 cells (IC50 = 10 μM), all other substances were less cytotoxic.
The ideal antitumor drug candidate should selectively target tumor cells and be relatively harmless for normal non-tumor cells. For assessment of tumor cell selectivity, selectivity index (SI) was calculated and is displayed in Table 5.
According to the definition, selective agents have the SI higher than one [16]. Among novel Schiff bases selective for CFPAC-1 cell line are agents 10, 11, 13, 18–22, and 24, whereas selective for HeLa cells are compounds 11, 20, 21, 23, 24, and 26. It is worth to emphasize that three compounds turned out to be highly selective: 20, 22, and 24. The most selective compound 24 has SI yielding approximately 6.5 for CFPAC-1, and 13.5 for HeLa cell line. It has to be admitted that this good selectivity of compound 24 is only resulted from insensitivity of NIH3T3 cells to those substances, in this context relatively high IC50 76 μM and 37 μM (in CFPAC1 and HeLa cells, respectively), in denominator, gave such a good selectivity index. Compound 24 is little cytotoxic for tested tumor cell lines and this should be improved to find out it as a potential antitumor drug.
It seems that benzene ring substituted with OH group in para position added to the coumarin moiety (17) gives the best tumor cell selectivity; however, IC50 for compound 24 should drop to nanomolar concentrations to talk about the antitumor activity of 24.
When we compare two chemically different group of compounds, namely derivatives of salicylaldehyde (10–13) and coumarin derivatives (18–28) in context of their anticancer activity, we can take some general conclusions. Schiff bases derived from salicylaldehyde are effective in pancreas and cervical cancer cells; however, they demonstrate also substantial toxicity towards NIH3T3 cells. Derivatives of coumarin represented by compounds 18–28 contain three highly selective compounds (20–22, 24) suggesting more promising potential of the second group of substances.
Conclusion
The present work comprises the new, microwave-assisted method of synthesis of 6-acetyl-7-hydroxy-4-methylcoumarin as well as the preparation of 18 (11 novel) Schiff bases, derivatives of salicylaldehyde, 2-hydroxyacetophenone, and 6-acetyl-, 8-acetyl-, and 8-formyl- 7-hydroxy-4-methylcoumarin. All these compounds were assayed for antimicrobial and cytotoxic activity. Several interesting structure–activity relationships in some Schiff bases were found. The novel compound N-(3,5-dichloro-2-hydroxybenzylidene)-4-aminobenzenesulfonic acid sodium salt (10) shows the highest antibacterial activity. It is noteworthy that compound 10 is soluble in water what potentially makes possible transport of the compound to the interior of the cell or interferences in the structure of the microbial cell membrane.
All but only three compounds (8, 9, 22 on HeLa cells) were active against cancer cells in micromolar concentrations ranging from ca 300 to 10 µM. Coumarin derivatives turned out to be more selective against cancer cells than derivatives of salicylaldehyde. The most selective compounds are 20 and 24. Compound 20 is the Schiff base derived from 8-formyl-7-hydroxy-4-methylcoumarin and 4-methoxyaniline; compound 24 is derived from 8-acetyl-7-hydroxy-4-methylcoumarin and 4-aminophenol. Both compounds are structural isomers differing in the positioning of the methyl group in the molecule. For these molecules, the desirable action is separated from an unwanted toxicity, thus constituting a promising hit for further structure optimization and development of potential anticancer agents. Especially, they are intended to be used as ligands in the copper or silver complexes with potentially increased biological activity.
Experimental
All reagents were obtained from Sigma-Aldrich and solvents from CHEMPUR S.A. or Polskie Odczynniki Chemiczne S.A. and were used without purification. 100% lemon juice LEMONDOR from Giancarlo Polenghi was used as a catalyst. All reaction mixtures were monitored using thin-layer chromatography (TLC) on 60F254 silica gel TLC plates (Merck) (developing system; chloroform–ethyl acetate 20:1). Column chromatography was carried out with silica gel (230–400 mesh) from Merck. Visualization was made with the use of UV laboratory lamp (λ = 254, 365 nm). High-resolution accurate mass determinations (HRMS) were recorded in the Faculty of Chemistry the University of Warsaw, on MAS Quattro LCT (TOF) mass spectrometer equipped with electrospray ionization (ESI). Melting points were determined on a ElectroThermal 9001 Digital Melting Point Melting Point apparatus. Nuclear magnetic resonance (1H NMR and 13C NMR, HMBC and HSQC) spectra were recorded on a Varian NMRS-300 NMR spectrometer with TMS as an internal standard. Atoms’ numbering is presented in Scheme 3. The chemical shifts were reported in parts per million (ppm); the coupling constants (J) were expressed in hertz (Hz). IR spectra (KBr or Nujol) were recorded on Shimadzu FTIR 8300 spectrophotometer in the Department of Technology of Drugs and Pharmaceutical Technology, Medical University of Warsaw or FTIR Nicolet iS5 in The Institute of Physics PAN in Warsaw. Elemental analysis (C, H, N, S) was conducted using the Elemental Analyser Vario EL III (Elementar) in the University of Wrocław, and their results were found to be in good agreement (± 0.3%) with the calculated values.
2-[(4-Methoxyphenylimino)methyl]phenol (8)
Equimolar amounts of salicylaldehyde and 4-methoxyaniline dissolved in ethanol were heated to reflux for 1 h. Then the precipitate was filtered off and dried. Light green solid; yield 62%; m.p.: 81.5–82.6 °C (lit. 81 °C [17]).
N-(2-Hydroxybenzylidene)-4-aminobenzenesulfonic acid sodium salt (9)
Sodium salt of compound 9 was prepared according to [18]. Sulfanilic acid (2.5 g, 14.3 mmol) was suspended in 25 cm3 methanol and heated to reflux with 0.58 g powdered sodium hydroxide (14.4 mmol) for 1 h. Then the reaction mixture (white suspension) was left for 10 min at ambient temperature and salicylaldehyde was added dropwise with stirring. Change of color to yellow and precipitation of yellow solid were observed. Stirring was continued for 2 h. The precipitate was filtered off and washed with chloroform. Yellow solid; yield 98%; m.p.: > 300 °C; NMR spectra were found to be identical with the ones described in Ref. [18].
N-(3,5-Dichloro-2-hydroxybenzylidene)-4-aminobenzenesulfonic acid sodium salt (10, C13H9Cl2NO4S)
This product was prepared according to the procedure given for compound 9. Orange solid; yield 74%; m.p.: > 300 °C; 1H NMR (300 MHz, DMSO-d6): δ = 7.44 (d, J = 8.4 Hz 2H, H-9,13), 7.70 (d, J = 8.4 Hz, 2H, H-10,12), 7.74 (s, 2H, H-4,6), 9.05 (s, 1H, H-7) ppm; 13C NMR (75 MHz, DMSO-d6): δ = 120.44 (C-1), 120.84 (C-9,13), 121.67 (C-3), 121.94 (C-5), 126.86 (C-10,12), 130.71 (C-6), 132.26 (C-4), 145.89 (C-11), 147.85 (C-8), 156.02 (C-7), 162.42 (C-2) ppm; IR (KBr): \( \bar{\nu } \) = 3455 (OH), 1620 (C=N), 1320, 1045 (sulfone), 1175 (sulfonate salt), 850 (substitution para di) cm−1; HR-MS (TOF, ESI-): m/z calcd for C13H8Cl2NO4S ([M−H]−) 343.9551, found 343.9546.
4,6-Dichloro-2-[(4-hydroxyphenylimino)methyl]phenol (11)
3,5-Dichloro-2-hydroxybenzaldehyde (0.955 g, 5 mmol) and 0.546 g 4-aminophenol (5 mmol) were dissolved in 23 cm3 methanol. The mixture was heated to reflux for 1 h and concentrated; then it was left at ambient temperature. The precipitate was filtered off and purified by crystallization (ethanol). Red solid; yield 76%; m.p.: 192.9–193.5 °C (lit. 224 °C [19]).
4,6-Dichloro-2-[(4-methoxyphenylimino)methyl]phenol (12)
3,5-Dichloro-2-hydroxybenzaldehyde (0.907 g, 4.75 mmol) and 0.585 g 4-methoxyaniline (4.75 mmol) were dissolved in 13 cm3 methanol. The mixture was left at ambient temperature for 0.5 h. The precipitate was purified by crystallization (ethanol). Orange solid; yield 55%; m.p.: 114–115 °C (lit. 116 °C [19]).
N-(5-Bromo-2-hydroxy-3-methoxybenzylidene)-4-aminobenzenesulfonic acid sodium salt (13, C14H12BrNO5S)
This product was prepared according to the procedure given for compound 9. Red solid; yield 50%; m.p.: > 300 °C; 1H NMR (300 MHz, DMSO-d6): δ = 3.85 (s, 3H, OCH3), 7.26 (d, J = 2.4 Hz, H-4), 7.35 (d, J = 8.4 Hz, 2H, H-9,13), 7.47 (d, J = 2.4 Hz, H-6), 7.67 (d, J = 8.4 Hz, 2H, H-10,12), 8.95 (s, 1H, H-7) ppm; 13C NMR (75 MHz, DMSO-d6): δ = 55.88 (OCH3), 117.74 (C-2), 120.30 (C-4), 120.67 (C-9,13), 125.13 (C-6), 126.80 (C-1), 126.86 (C-10,12), 147.14 (C-8), 149.14 (C-3), 150.07 (C-7) ppm; IR (KBr): \( \bar{\nu } \) = 3700-3400 (OH), 1612 (C=N), 1320, 1045 (sulfone), 1182 (sulfonate salt), 851 (substitution para di) cm−1; HR-MS (TOF, ESI-): m/z calcd for C14H11BrNO5S ([M−H]−) 383.9541, found 383.9534.
2-[1-(4-Methoxyphenylimino)ethyl]phenol (14)
2-Hydroxyacetophenone (1.365 g, 10 mmol) and 1.60 g 4-methoxyaniline (13 mmol) were dissolved in 5 cm3 xylene. Then the catalyst 1.4 cm3 lemon juice was added. The reaction mixture was stirred magnetically and heated in the oil bath at 100 °C. The progress of reaction was monitored by TLC (chloroform–ethyl acetate 20:1). After 8 h the mixture was allowed to reach ambient temperature, then it was left in refrigerator for 24 h. Chloroform (10 cm3) and anhydrous MgSO4 were added to remove water. Then the filtrate was concentrated and the resulting precipitate was filtered off. The crude compound was purified by crystallization (methanol). Yellow solid; yield 23%; m.p.: 101–103 °C (lit. 98–99 °C [25]).
8-Formyl-7-hydroxy-4-methyl-2H-chromen-2-one (15)
Off-white solid; yield 11%; m.p.: 182–184 °C (lit. 174–176 °C [21]).
6-Acetyl-7-hydroxy-4-methyl-2H-chromen-2-one (16c)
7-Hydroxy-4-methylcoumarin (16ª, 8.80 g, 50 mmol) was dissolved in 35 cm3 methanesulfonic acid (540 mmol). Then 35 cm3 acetic anhydride (370 mmol) and 40 g anhydrous alumina (390 mmol) were added. The reaction mixture was heated in microwave oven; conditions: two cycles of heating, time: 4 min (reflux), power: 500 W. Then the mixture was allowed to reach ambient temperature and filtered through silica gel under reduced pressure. The filtrate was poured to 60 g ice water and extracted with dichloromethane (3 × 60 cm3). The organic layer was washed with saturated aqueous solution of NaHCO3. Solvent was evaporated and the product 2 was isolated by column chromatography on silica gel 230–400 mesh (chloroform–ethyl acetate). Colorless solid; yield 42%; m.p.: 207–208 °C (lit. 210–211 °C [26]); 1H and 13C NMR spectra were found to be identical with the ones described in Ref. [26].
8-Acetyl-7-hydroxy-4-methyl-2H-chromen-2-one (17)
Colorless solid; yield 80%; m.p.: 174–175 °C (lit. 174–175 °C [26]); 1H and 13C NMR spectra were found to be identical with the ones described in Ref. [26].
8-Formyl-7-hydroxy-4-methyl-2H-chromen-2-one (15) derived Schiff bases—general procedure
Compound 15 (0.408 g, 2 mmol), appropriate aniline or the sodium salt of sulfanilic acid (2 mmol) were dissolved in 5 cm3 ethanol, one drop of glacial acetic acid was added, and the mixture was magnetically stirred and heated to reflux in the oil bath (1–4 h). The progress of reaction was monitored by TLC (chloroform–ethyl acetate 20:1). When the reaction was complete, the mixture was left at ambient temperature overnight. The precipitate was filtered off under reduced pressure, and purified by crystallization (ethanol).
7-Hydroxy-8-[(4-bromophenylimino)methyl]-4-methyl-2H-chromen-2-one (18, C17H12BrNO3)
This product was prepared according to the general procedure. Orange solid; yield 68%; m.p.: 222–223.5 °C; Rf= 0.68; 1H NMR (300 MHz, CDCl3): δ = 2.43 (d, J = 1.2 Hz, 3H, H-9), 6.16 (bq, J = 1.2 Hz, 1H, H-3), 6.95 (d, J = 9 Hz, 1H, H-6), 7.24–7.29 (m, 2H, H-12,16), 7.55–7.60 (m, 2H, H-13,15), 7.61 (d, J = 9 Hz, 1H, H-5), 9.36 (s, 1H, H-10) ppm; 13C NMR (75 MHz, CDCl3): δ = 19.18 (C-9), 107.18 (C-8), 111.44 (C-3), 111.63 (C-4a), 114.87 (C-6), 121.52 (C-14), 123.25 (C-12,16), 129.68 (C-5), 132.88 (C-13,15), 153.41 (C-4), 157.28 (C-8a,10), 160.36 (C-2), 166.06 (C-7) ppm; IR (Nujol): \( \bar{\nu } \) = 1710 (C=O), 1620 (C=N) cm−1; HR-MS (TOF, ESI-): m/z calcd for C17H11NO3Br ([M−H]−) 355.9922, found 355.9915.
7-Hydroxy-8-[(4-hydroxyphenylimino)methyl]-4-methyl-2H-chromen-2-one (19, C17H13NO4)
This product was prepared according to the general procedure. Orange solid; yield 60%; m.p.: 253 °C (decomposition); Rf = 0.65; 1H NMR (300 MHz, DMSO-d6): δ = 2.42 (d, J = 0.9 Hz, 3H, H-9), 6.24 (bq, J = 0.9 Hz, 1H, H-3), 6.85–6.90 (m, 2H, H-13,15), 6.91 (d, J = 9 Hz, 1H, H-6), 7.39–7.44 (m, 2H, H-12,16), 7.77 (d, J = 9 Hz, 1H, H-5), 9.19 (s, 1H, H-10), 9.81 (s, 1H, H-10) ppm; 13C NMR (75 MHz, DMSO-d6): δ = 18.40 (C-9), 106.22 (C-8), 110.18 (C-3), 110.69 (C-4a), 114.31 (C-6), 116.16 (C-13,15), 122.75 (C-12.16), 129.81 (C-5), 137.34 (C-11), 153.17 (C-10), 153.81 (C-8a), 154.06 (C-4), 157.54 (C-14), 159.25 (C-2), 165.67 (C-7) ppm; IR (Nujol): \( \bar{\nu } \) = 3250 (O–H), 1696 (C=O), 1622 (C=N) cm−1.
7-Hydroxy-8-[(4-methoxyphenylimino)methyl]-4-methyl-2H-chromen-2-one (20, C18H15NO4)
This product was prepared according to the general procedure. Orange solid; yield 69%; m.p.: 203.7–205.6 °C; Rf = 0.46; 1H NMR (300 MHz, CDCl3): δ = 2.42 (d, J = 0.9 Hz, 3H, H-9), 3.86 (s, 3H, C14-OCH3), 6.14 (bq, J = 0.9 Hz, 1H, H-3), 6.96 (d, J = 9 Hz, 1H, H-6) 6.95–7.00 (m, 2H, H-13,15), 7.40–7.56 (m, 2H, H-12,16), 7.58 (d, J = 9 Hz, 1H, H-5), 9.33 (s, 1H, H-10) ppm; 13C NMR (75 MHz, CDCl3): δ = 19.18 (C-9), 55.81 (C14-OCH3), 107.16 (C-8), 111.06 (C-3), 111.18 (C-4a), 114.99 (C-13,15), 115.21 (C-6), 122.75 (C-12.16), 129.09 (C-5), 139.45 (C-11), 153.54 (C-8a), 154.28 (C-4), 154.57 (C-10), 159.68 (C-2), 160.61 (C-14), 166.4 (C-7) ppm; IR (Nujol): \( \bar{\nu } \) = 1721 (C=O), 1602 (C=N), 1379 (C-O-Casym) cm−1; HR-MS (TOF, ESI-): m/z calcd for C18H14NO4 ([M−H]−) 308.0923, found 308.0924.
7-Hydroxy-8-[(4-methylphenylimino)methyl]-4-methyl-2H-chromen-2-one (21)
This product was prepared according to the general procedure. Orange solid; yield 57%; m.p.: 196.5–199.0 °C (lit.188–189 °C [27]).
N-[(7-Hydroxy-4-methyl-2-oxo-2H-chromen-8-yl)methylene]-4-aminobenzenesulfonic acid sodium salt (22, C17H13NO6S)
This product was prepared according to the general procedure. Orange solid; yield 90%; m.p.: > 300 °C; 1H NMR (300 MHz, DMSO-d6): δ = 2.43 (s, 3H, H-9), 6.26 (d, J = 0.9 Hz, 1H, H-3), 6.95 (d, J = 9 Hz, 1H, H-6,) 7.48 (d, J = 8.4 Hz, 2H, H-12,16), 7.71 (d, J = 8.4 Hz, 2H, H-13,15), 7.82 (d, J = 9.0 Hz, 1H, H-5), 9.28 (s, 1H, H-10) ppm; 13C NMR (75 MHz, DMSO-d6): δ = 18.37 (C-9), 106.20 (C-8), 110.38 (C-3), 110.88 (C-4a), 114.29 (C-6), 120.72 (C-12,16), 126.96 (C-13,15), 130.63 (C-5), 145.94 (C-14), 147.62 (C-11), 154.00 (C-8a), 154.12 (C-4), 156.89 (C-10), 159.11 (C-2), 165.62 (C-7) ppm; IR (Nujol): \( \bar{\nu } \) = 3260 (OH), 1728 (C=O), 1610 (C=N), 1319, 1050 (sulfone), 1180 (sulfonate salt), 849 (substitution para di) cm−1; HR-MS (TOF, ESI-): m/z calcd for C17H12NO6S ([M−H]−) 358.0385, found 358.0380.
7-Hydroxy-6-[1-(4-methoxyphenylimino)ethyl]-4-methyl-2H-chromen-2-one (23, C19H17NO4)
Coumarin 16c (10 mmol) and 1.60 g 4-methoxyaniline (13 mmol) were dissolved in 5 cm3 xylene. The reaction mixture was stirred magnetically and heated in the oil bath at 100 °C. The progress of reaction was monitored by TLC (chloroform–ethyl acetate 20:1). After 6 h the mixture was allowed to reach ambient temperature, then it was left in refrigerator for 24 h. Chloroform (10 cm3) and anhydrous MgSO4 were added to remove water. Then the filtrate was concentrated and the resulting precipitate was filtered off. The crude compound was purified by crystallization (methanol). Yellow solid; yield 83%; m.p.: 239–241 °C; Rf = 0.57; 1H NMR (300 MHz, acetone-d6): δ = 2.53 (d, 3H, J = 3.0 Hz, H-9), 2.77–2.80 (m, 3H, C10-CH3), 3.84 (s, 3H, C14-OCH3), 6.12 (bs, 1H, H-3), 6.75 (d, 1H, J = 4.2 Hz, H-8), 7.03 (m, 4H, H-12,13,15,16), 8.13 (s, 1H, H-5) ppm; 13C NMR (75.4 MHz, acetone-d6): δ = 18.60 (C-9), 55.88 (C14-OCH3), 105.02 (C-8), 112.20 (C-3), 112.32 (C-4a), 115.36 (C-13,15), 123.89 (C-6), 123.97 (12,16), 128.22 (C-5), 139.77 (C-11), 153.95 (C-4), 158.66 (C-8a), 160.57 (C-2), 167.14 (C-7), 172.81 (C-10) ppm; IR (KBr): \( \bar{\nu } \) = 2841, 1723 (C=O), 1612 (C=N), 1500 (C=C), 858 (substitution para di) cm−1; HR-MS (TOF, ESI +): m/z calcd for C19H17NNaO4 ([M + Na]+) 346.1055, found 346.1049.
8-Acetyl-7-hydroxy-4-methyl-2H-chromen-2-one (17) derived Schiff bases—general procedure
Coumarin 17 (10 mmol) and appropriate aniline (13 mmol) were dissolved in 5 cm3 xylene. Then the catalyst—1.4 cm3 lemon juice—was added. The reaction mixture was stirred magnetically and heated in the oil bath at 100 °C. The progress of reaction was monitored by TLC (chloroform–ethyl acetate 20:1). After 8 h the mixture was allowed to reach ambient temperature, then it was left in refrigerator for 24 h. Chloroform (10 cm3) and anhydrous MgSO4 were added to remove water. Then the filtrate was concentrated and the resulting precipitate was filtered off. The crude compound was purified by crystallization (methanol).
7-Hydroxy-8-[1-(4-hydroxyphenylimino)ethyl]-4-methyl-2H-chromen-2-one (24)
This product was prepared according to the general procedure. Yellow solid; yield 85%; m.p.: 256–257 °C (lit. 217 °C [28]); 1H NMR spectrum was found to be different from the one described in Ref. [28].
7-Hydroxy-8-[1-(4-methoxyphenylimino)ethyl]-4-methyl-2H-chromen-2-one (25, C19H17NO4)
This product was prepared according to the general procedure. Yellow solid; yield 87%; m.p.: 198–200 °C; Rf= 0.48; 1H NMR (300 MHz, acetone-d6): δ = 2.47 (d, 3H, J = 1.2 Hz, H-9), 2.68 (m, 3H, C10-CH3), 3.85 (s, 3H, C14-OCH3), 6.11 (s, 1H, H-3), 6.89 (dd, 1H, J = 9.0, 3.6 Hz, H-6), 7.03–7.12 (m, 4H, H-12,13,15,16), 7.75 (d, 1H, J = 9.0 Hz, H-5) ppm; 13C NMR (75.4 MHz, acetone-d6): δ = 19.15 (C-9), 33.99 (C10-CH3), 55.79 (C14-OCH3), 110.41 (C-4a), 111.68 (C-3), 114.96 (C-13,15), 115.37 (C-6), 116.34 (C-8), 121.43 (C-12,16), 129.81 (C-5), 133.01 (C-7), 146.12 (C-11), 154.56 (C-8a), 154. 89 (C-4), 159.55 (C-2), 160.13 (C-10,14) ppm; IR (KBr): \( \bar{\nu } \) = 3001, 2964, 1709 (C=O), 1597 (C=N), 1502 (C=C), 827 (substitution para di) cm−1; HR-MS (TOF, ESI-): m/z calcd for C19H16NO4 ([M−H]−) 322.1079, found 322.1073.
7-Hydroxy-8-[1-(4-ethoxyphenylimino)ethyl]-4-methyl-2H-chromen-2-one (26, C20H19NO4)
This product was prepared according to the general procedure. Yellow solid; yield 76%; m.p.: 167–170 °C; Rf = 0.39; 1H NMR (300 MHz, acetone-d6): δ = 1.40 (t, 3H, J = 6.9 Hz, H-19), 2.47 (d, 3H, J = 0.9 Hz, H-9), 2.67, 2.69 (m, 3H, C14-OCH2CH3), 4.10 (q, 2H, J = 6.9 Hz, C14-OCH2CH3), 6.11 (s, 1H, H-3), 6.88 (d, 1H, J = 8.7 Hz, H-6), 7.02–7.11 (m, 4H, H-12,13,14,15,16), 7.75 (d, 1H, J = 9.0 Hz, H-5) ppm; 13C NMR (75.4 MHz, acetone-d6): δ = 15.25 (C14-OCH2CH3), 19.19 (C-9), 64.14 (C-11), 64.40 (C14-OCH2CH3), 110.40 (C-4a), 111.70 (C-3), 115.27 (C-13,15), 116.00 (C-6), 121.40 (12,16), 124.35 (C-11), 129.78 (C-5), 133.03 (C7), 154.53 (C-8a), 154.79 (C-4), 160.09 (C-2), 168.61 (C-14), 173.12 (C-10) ppm; IR (KBr): \( \bar{\nu } \) = 2984, 1722 (C=O), 1590 (C=N), 1507 (C=C), 832 (substitution para di) cm−1; HR-MS (TOF, ESI-): m/z calcd for C20H18NO4 ([M−H]−) 336.1236, found 336.1234.
7-Hydroxy-8-[1-(4-methylphenylimino)ethyl]-4-methyl-2H-chromen-2-one (27, C19H17NO3)
This product was prepared according to the general procedure. Yellow solid; yield 81%; m.p.: 149–150 °C; Rf= 0.54; 1H NMR (300 MHz, acetone-d6): δ = 2.37 (s, 3H, C14-CH3), 2.47 (d, 3H, J = 1.2 Hz, H-9), 2.65–2.80 (m, 3H, C10-CH3), 6.12 (br.s, 1H, H-3), 6.89 (d, 1H, J = 9.3 Hz, H-6), 7.15 (d, 2H, J = 9.0 Hz, H-13,15), 7.30 (d, 2H, J =7.8 Hz, H-14,16), 7.76 (d, 1H, J = 9.0 Hz, H-5) ppm; 13C NMR (75 MHz, acetone-d6): δ = 19.15 (C-9), 21.02 (C14-CH3), 34.00 (C-11), 111.67 (C-8), 113.10 (C-4a), 115.24 (C-6), 115.39 (C-3), 120.26 (C-12,16), 129.85 (C-5), 130.78 (C-13,15), 133.00 (C-14), 146.82 (C-11), 150.41 (C-4), 154.56 (C-8a), 154.80 (C-10), 159.60 (C-2), 160.09 (C-7) ppm; IR (KBr): \( \bar{\nu } \) = 3023 (OH), 2985, 1722 (C=O), 1603 (C=N), 1505 (C=C), 839 (substitution para di) cm−1; HR-MS (TOF, ESI +): m/z calcd for C19H17NNaO3 ([M + Na]+) 307.1208 found 307.1186.
7-Hydroxy-8-[1-(4-ethylphenylimino)ethyl]-4-methyl-2H-chromen-2-one (28, C20H19NO3)
This product was prepared according to the general procedure. Yellow solid; yield 61%; m.p.: 122–123 °C; Rf= 0.53; 1H NMR (300 MHz, acetone-d6): δ = 1.26 (t, 3H, J = 7.5 Hz, C14-CH2CH3), 2.47 (d, 3H, J = 0.9 Hz, H-9), 2.64–2.80 (m, 5H, C10-CH2CH3), 6.12 (br.s, 1H, H-3), 6.93 (d, 1H, J = 9.0 Hz, H-6), 7.04 (d, 2H, J = 8.1 Hz, H-13,15), 7.32 (d, 2H, J = 8.1 Hz, H-14,16), 7.76 (d, 1H, J = 8.7 Hz, H-5), 13.40 (s, 1H, C7-OH) ppm; 13C NMR (75 MHz, acetone-d6) δ =18.18 (C14-CH3), 33.01 (C-11, C14-CH2-CH3), 110.70 (C-8), 114.28 (C-3), 114.43 (C-4a), 119.31 (C-6), 119.42 (C-12,16), 128.13 (C-5), 132.04 (C-13,15), 134.00 (C-14), 146.62 (C-11), 151.41 (C-4), 156.46 (C-8a), 155.81 (C-10), 161.59 (C-2), 160.39 (C-7) ppm; IR (KBr): \( \bar{\nu } \) = 2962, 2930, 1667 (C=O), 1600 (C = N), 1514 (C=C), 835 (substitution para di) cm−1; HR-MS (TOF, ESI-): m/z calcd for C20H18NO3 ([M−H]−) 320.1289, found 320.1292.
In vitro antibacterial activity assay
Strains
The following microbial strains were chosen from American Type Culture Collection (ATCC): Gram-(+) bacteria strains: Micrococcus luteus 10240, Bacillus subtilis 6633, Staphylococcus aureus (6538, 6538P), Enterococcus hirae 10541; Gram-(−) bacteria strains: Escherichia coli 8739, Pseudomonas aeruginosa (9027, 15442); fungi (yeast strains): Candida albicans (10231, 2091), Candida parapsilosis 22019, Saccharomyces cerevisiae 9763; fungi (mold strain): Aspergillus brasiliensis 16404. The MRSA hospital strains (methicillin-resistant S. aureus) from Clinical Hospital Prof. Orlowski in Warsaw collection: A854 (isolated from throat mucosa, 30.03.2011) resistance: CIP, E, DA, AK and A876 (isolated from lower leg injury, 19.02.2012) resistance: E, DA, CIP, AK.
Antimicrobial activity: preliminary test
In the preliminary tests, antimicrobial activity was determined by a modified cylinder–plate method. Stainless steel cylinders of 6 mm diameter were put on a Mueller–Hinton two agar (for bacteria strains) or Sabouraud agar (for yeast strains) plate inoculated with one of the tested strains. An aliquot of 100 mm3 of each compound in concentration of 10 mg/cm3 (dissolved in 10% DMSO, in a 0.08 M phosphate buffer, pH 7.0) was placed into the cylinder. As a negative control 10% DMSO (in 0.08 M phosphate buffer, pH 7.0) without any compound was used. The plates with bacterial strains were incubated at 37°C for 24 h, whereas plates with yeast strains at 30°C for 48 h. The inhibition of bacterial growth was observed as a halo around the cylinder containing the tested compound. Size of inhibition zone reflected an antimicrobial activity of the compound.
Minimum inhibitory concentration
For the compounds, which showed some activity against any of the tested strains, a minimum inhibitory concentration (MIC) was determined based on M7-A9 method [23]. The stock solution was prepared by dissolving each compound in DMSO. A series of gradient solutions in the range from 1 to 0.0156 mg/cm3 was obtained by the double dilution in 0.08 M phosphate buffer (pH 7.0). The 250 mm3 aliquot of the successive dilution of tested compound was mixed with 4.75 cm3 of a liquefied Mueller–Hinton two or Sabouraud agar, precooled to 45°C. The suspension of the particular strain of density 0.5 unit (McFarland scale) was diluted ten times and aliquot of 2 mm3 was applied on agar plate surface. The lowest concentration of tested compound, which totally inhibited growth of the examined strain, was taken as MIC value. As a negative control the solution of the highest concentration of DMSO in 0.08 M phosphate buffer (pH 7.0) was used. All tests were performed in triplicates. In control samples, MIC values of ciprofloxacin ranging between 0.08 and 1.36 × 10−3 µmol/cm3 for bacterial strains.
In vitro antitumor activity assay
Cell culture
The influence of novel compounds on growth and proliferation of cells was studied in three established mammalian cell lines. We performed experiments in two cancer cell lines: CFPAC—human pancreatic cancer cell line and HeLa cells—human cervical cancer cell line. As a non-tumor cell line NIH3T3 fibroblasts were investigated. All cell lines were from ATCC. Cells were cultured exponentially in RPMI1640 (CFPAC) or in DMEM (HeLa and NIH3T3). The culture media containing stable glutamine were supplemented with 10% fetal bovine serum and standard antibiotics. All are from Biochrom, Germany. Cells were kept in a humidified atmosphere of 5% CO2, at 37 °C and passaged every 3 days.
Crystal violet dye elution (CVDE) assay
For estimation of cytostatic/cytotoxic effects of experimental compounds, cells were seeded into 96-well flat-bottom plates at a density of 105 cells/well in 100 mm3 of culture medium. After adhesion of cells, particular compounds were added in a range of concentrations 1–500 µM for 48 h. As a control, cells were incubated with solvent only. After 48 h of incubation with compounds, medium was removed and CVDE assay was performed as described in Reference [29]. According to the growth inhibition induced by particular drug, a dose–response curve was drawn and inhibitory concentrations, IC50 and IC90, respectively, were estimated.
References
Chaturvedi D, Kamboj M (2016) Chem Sci J 7:2
Kajal A, Bala S, Kamboj S, Sharma N, Saini V (2013) J Catalysts. https://doi.org/10.1155/2013/893512
Qin W, Long S, Panunzio M, Biondi S (2013) Molecules 18:12264
da Silva CM, da Silva DL, Modolo LV, Alves RB, de Resende MA, Martins CVB, de Fatima A (2011) J Adv Res 22:1
Cosgrove SA (2006) Clin Infect Dis 42:S82
https//amr-review.org//sites/default/files/securing new drugs for future generations final web_0.pdf. Accessed 24.09.2018
Karthikeyan MS, Prasad DJ, Poojary B, Bhat KS, Holla BS, Kumarib NS (2006) Bioorg Med Chem 14:7482
de Souza AO, Galetti FCS, Silva CL, Bicalho B, Parma MM, Fonseca SF, Marsaioli AJ, Trindade ACLB, Rossimíriam P, Gil F, Bezerra FS, Andrade-Neto M, de Oliveira MCF (2007) Quim Nova 30:1563
Shi L, Ge H-M, Tan S-H, Li H-Qi, Song Y-C, Zhu H-L, Tan R-Xi (2007) Eur J Med Chem 42:558
Krátký M, Dzurková M, Janoušek J, Koneˇcná K, Trejtnar F, Stolaˇríková J, Vinšová J (2017) Molecules 22:1573
Prabhakara CT, Patil SA, Toragalmath SS, Kinnal SM, Badami PS (2016) J Photochem Photobiol B 157:1
Satyanarayana VSV, Sreevani P, Sivakumar A, Vijayakumar V (2008) Arkivoc 2008(17):221–233
Utreja D, Sarbjit Singh V, Kaur M (2015) Curr Bioact Compd 11:215
Emami S, Dadashpour S (2015) Eur J Med Chem 102:611
Kaur M, Kohli S, Sandhu S, Bansal Y, Bansal G (2015) Anti-Cancer Agents Med Chem 15:1032
Wang Z-C, Qin Y-J, Wang P-F, Yang Y-A, Wen Q, Zhang X, Qiu H-Y, Duan Y-T, Wang Y-T, Sang Y-L, Zhu H-L (2013) Eur J Med Chem 66:1
Frischmann PD, Jiang J, Hui JKH, Grzybowski JJ, MacLachlan MJ (2008) Org Lett 10:1255
Robert F, Naik AD, Hidara F, Tinant B, Robiette R, Wouters J, Garcia Y (2010) Eur J Org Chem 4:621–637
Cronenberger L, Gaigé T, Pachéco H (1968) Bull Soc Chim Biol 50:929
Patil S, Jadhav SD, Patil UP (2012) Arch Appl Sci Res 4:1074
Al-Kawkabani A, Boutemeur-Kheddis B, Makhloufi-Chebli M, Hamdi M, Talhi O, Silva AMS (2013) Tetrahedron Lett 54:5111
Hejchman E, Maciejewska D, Wolska I (2008) Monatsh Chem 139:1337
Clinical and Laboratory Standards Institute (2012) Performance standards for antimicrobial susceptibility testing; Approved standard M7-A9, Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically, 9th edn. CLSI, Wayne
Clancy CJ, Yu VL, Morris AJ, Snydman DR, Nguyen MH (2005) Antimicrob Agents Chemother 49:3171
Hothi HS, Makkar JR, Sharma JR, Manrao MR (2006) Eur J Med Chem 41:253
Yadav P, Satapathi S, Kumaria M, Chaturvedi A, Li L, Samuelson LA, Kumarb J, Sharma SK (2014) J Photochem Photobiol A 280:39
Traven VF, Ivanov IV, Panov AV, Safronova OB, Chibisova TA (2008) Russ Chem Bull 57:1989
Balaji PN, Seleena SKP, Gummadi V, Tummala H, Bodhanapu N, Thummala L (2013) Res J Pharm Biol Chem Sci 4:9
Młynarczuk-Biały I, Roeckmann H, Kuckelkorn U, Schmidt B, Umbreen S, Gołąb J (2006) J Cancer Res 66:7598
Acknowledgements
This project was supported by Medical University of Warsaw, Faculty of Pharmacy, FW24/NM1/17.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Hejchman, E., Kruszewska, H., Maciejewska, D. et al. Design, synthesis, and biological activity of Schiff bases bearing salicyl and 7-hydroxycoumarinyl moieties. Monatsh Chem 150, 255–266 (2019). https://doi.org/10.1007/s00706-018-2325-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00706-018-2325-5