
Abstract
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the causative agent of coronavirus disease 2019 (COVID-19), has caused more than 760 million cases and over 6.8 million deaths as of March 2023. Vaccination has been the main strategy used to contain the spread of the virus and to prevent hospitalizations and deaths. Currently, two mRNA-based vaccines and one adenovirus-vectored vaccine have been approved and are available for use in the U.S. population. The versatility, low cost, and rapid production of DNA vaccines provide important advantages over other platforms. Additionally, DNA vaccines efficiently induce both B- and T-cell responses by expressing the antigen within transfected host cells, and the antigen, after being processed into peptides, can associate with MHC class I or II of antigen-presenting cells (APCs) to stimulate different T cell responses. However, the efficiency of DNA vaccination needs to be improved for use in humans. Importantly, in vivo DNA delivery combined with electroporation (EP) has been used successfully in the field of veterinary oncology, resulting in high rates of response after electrochemotherapy. Here, we evaluate the safety, immunogenicity, and protective efficacy of a novel linear SARS-CoV-2 DNA vaccine candidate delivered by intramuscular injection followed by electroporation (Vet-ePorator™) in ferrets. The linear SARS-CoV-2 DNA vaccine candidate did not cause unexpected side effects. Additionally, the vaccine elicited neutralizing antibodies and T cell responses on day 42 post-immunization using a low dose of the linear DNA construct in a prime-boost regimen. Most importantly, vaccination significantly reduced shedding of infectious SARS-CoV-2 through oral and nasal secretions in a ferret model.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is the viral agent of one of the deadliest pandemics of the last 100 years. By March 2023, the World Health Organization (WHO) officially registered more than 760 million confirmed cases and over 6.8 million deaths worldwide caused by the coronavirus disease 2019 (COVID-19) pandemic (https://covid19.who.int). The earliest reports of COVID-19 were linked to the Huanan Seafood Wholesale Market in Wuhan, China, which sold aquatic animals, live poultry, and several wild animal species [1, 2]. Following the initial reports, human-to-human transmission was confirmed as individuals began to contract the infection without ever having been at the seafood market [3, 4]. The betacoronavirus RaTG13, isolated from the bat species Rhinolophus affinis, was found to be the animal coronavirus most closely related to SARS-CoV-2, sharing over 96% identity at the whole-genome level [5]. However, the genetic distance of approximately 4% (~ 1,150 mutations) between RaTG13 and SARS-CoV-2 isolate Wuhan-Hu-1 indicates that transmission likely did not occur directly from bats to humans and further suggests that a yet-unidentified animal species may have served as an intermediate host prior to spillover of the virus into humans [6, 7].
SARS-CoV-2 belongs to the subgenus Sarbecovirus, genus Betacoronavirus of the family Coronaviridae and is an enveloped positive-strand RNA virus [8]. The virus encodes a major surface glycoprotein, the spike (S) protein, which mediates receptor binding and is the main target of host immune responses [9]. The S protein mediates SARS-CoV-2 entry into target cells by initially binding to angiotensin-converting enzyme 2 (ACE2) on the host cell surface (via the S1 domain) and subsequently fusing (via the S2 domain) the viral membrane with a host membrane [9,10,11]. The virus-host receptor interaction occurs between the main functional motif of S within the receptor-binding domain (RBD), known as the receptor-binding motif (RBM), which is present at the tip of the trimer of the viral S protein, and the host cell receptor, ACE2 [12, 13]. Given its critical role in virus entry, the S RBD is also a major target of host responses to SARS-CoV-2, thus representing a good target for subunit vaccine development.
As of July 15, 2022, there are two mRNA vaccines (Pfizer/BNT162b2 and Moderna/mRNA-1273) and one recombinant-adenovirus-vectored vaccine (Janssen vaccine/Ad26.COV2.S) approved by the U.S. Food and Drug Administration (FDA) for human use in the U.S. [14,15,16]. Additionally, other 40 vaccine platforms have been approved for human use in at least one country. The list includes other non-replicating viral vector vaccines, such as Oxford/AstraZeneca/AZD1222, protein subunit vaccines, including Serum Institute of India/Novavax/COVOVAX and Novavax/Nuvaxovid, one virus-like particle (VLP) vaccine by Medicago/Covifenz, inactivated virus vaccines, such as Sinovac/CoronaVac and Sinopharm (Beijing)/Covilo, and one DNA vaccine, Zydus Cadila/ZyCoV-D in India [17, 18]. The last of these contains a circular plasmid DNA, which enters the nucleus of a host cell to be transcribed into messenger RNA (mRNA). An experimental DNA vaccine against Japanese encephalitis virus has shown promising results when tested in mice and pigs [19]. Before that, other DNA vaccines were approved for animal infectious diseases, including a horse vaccine against West Nile virus (WNV) and an Atlantic salmon vaccine against infectious hematopoietic necrosis virus (IHNV) [20].
Overall, DNA vaccination offers many advantages over other vaccine platforms. DNA vaccines can be manufactured quickly and are versatile, relatively simple, inexpensive, and safe compared to other vaccine types. Moreover, the DNA stability at room temperature facilitates their storage and transport, especially in countries or regions with restricted cold-chain resources [21]. Similar to live viruses, DNA vaccines are able to engage both the MHC-I and MHC-II pathways, inducing CD8+ and CD4+ T cells [22, 23]. In contrast to adenovirus/viral-vector-based vaccines, DNA vaccines do not induce anti-vector immunity, making them suitable for vaccine regimens that require boosters [21]. However, the efficiency of DNA vaccination needs to be improved for use in humans. Several approaches have been explored to improve efficiency of DNA vaccines, including the use of improved in vivo delivery methods using electroporation (EP) to optimize the uptake of exogenous DNA into cells [21, 24, 25]. The EP method applies brief electric pulses to induce reversible cell permeabilization through formation of transient pores, through which macromolecules such as DNA can translocate into the intracellular space and deliver the nucleic acid encoding the gene(s) of interest [21].
In the present study we assessed the safety, tolerance, immunogenicity, and protective efficacy of a novel linear SARS-CoV-2 DNA vaccine candidate delivered by intramuscular injection followed by electroporation (Vet-ePorator™), using a ferret model of SARS-CoV-2 infection.
Materials and methods
Linear DNA vaccine
Codon-optimized complementary DNA (cDNA) encoding the RBD region of the SARS-CoV-2 S protein was designed as described previously [26] and chemically synthesized by Genscript, Nanjing, Jiangsu, China. Briefly, the synthetic codon-optimized RBD-encoding construct was designed considering codon usage bias, GC content, CpG dinucleotide content, mRNA secondary structure, cryptic splicing sites, premature polyA sites, internal chi sites and ribosome-binding sites, negative CpG islands, RNA instability motifs (ARE), repeat sequences (direct repeats, inverted repeats, and dyad repeats), and restriction sites that may interfere with cloning. In addition, to improve translational initiation efficiency and performance, Kozak and Shine-Dalgarno sequences were inserted into the synthetic gene. To increase the efficiency of translational termination, two consecutive stop codons were inserted at the end of the cDNA. For the construction of the RBD-encoding DNA plasmid, the cDNA was amplified via PCR using sequence-specific primers and directionally cloned into a linearized pTK1A-TPA vector cleaved by the restriction enzymes PacI and NotI. The linear DNA amplicon construct encoding the RBD was synthesized as described previously [26]. For phosphorothioate-modified amplicons, a sulfur atom was substituted for the non-bridging oxygen in the phosphate backbone of the oligonucleotide. In addition, the five terminal bases of both the forward and reverse primers were modified to increase DNA amplicon stability. The DNA was amplified using a large-scale PCR system with Q5 polymerase from New England Biolabs, USA, resulting in 2780-bp linear DNA (linDNA) amplicon expression cassettes. For purification, the amplicon was first concentrated by ethanol precipitation and then purified on an Akta Pure 150 FPLC instrument (GE Healthcare, Chicago, IL, USA) with a GE HiPrep 26/60 Sephacryl S-500 High Resolution size exclusion column and 0.3 M NaCl running buffer. The purified DNA was ethanol precipitated again and resuspended in phosphate-buffered saline (PBS) to a final concentration of 1 mg/ml− 1 ± 10% and sterile filtered using a 0.22-µm polyethersulfone membrane. Further analytical characterization of the linear DNA construct was performed using a NanoDrop spectrophotometer (Thermo Fisher, Waltham, MA, USA), a 2100 Bioanalyzer, and an Alliance HPLC System (Waters, Milford, MA USA). Sanger sequencing of the linear DNA construct showed no sequence errors when compared to the plasmid DNA template sequence. The linear DNA construct was then lyophilized using a VirTis Genesis Pilot system. All removable internal components of the system were autoclaved. All interior surfaces were wipe-sterilized with Actril and swabbed for confirmation of sterility by microtesting (plating). The purified sterile linear DNA solution was aseptically dispensed into sample vials (2 ml amber glass, 15 × 32 mm with a 13-mm crimp) at a predetermined volume, and a 13-mm 2-leg stopper was inserted into the mouth of each vial. Stoppered vials were then placed in an autoclaved bag and stored in a -20 ± 5°C freezer for 18 hours before lyophilization. After the lyophilization program was run, the vials were stoppered under vacuum, and West 13-mm smooth vial caps were applied and crimped manually. Prior to use in the study, the lyophilized linear DNA constructs were resuspended in 1 ml of sterile water (Hospira Inc., Lake Forest, IL USA). The expression level of SARS-CoV-2 DNA vaccine construct was assessed in vitro, and the immunogenicity was assessed in BALB/c mice, K18-hACE2 rats, and ferrets as described previously [26].
Ethics statement
The viral isolate used in this study was obtained from residual de-identified diagnostic human nasopharyngeal samples tested at Weill Cornell Medicine, kindly provided to us by Dr. Melissa Cushing. The protocols and procedures for transfer of de-identified diagnostic samples were reviewed and approved by the Cornell University Institutional Review Boards (IRB approval numbers 2101010049). The ferrets were handled in accordance with the Animal Welfare Act. The study procedures were reviewed and approved by the Institutional Animal Care and Use Committee of Cornell University (IACUC approval number 2021-0052).
Animal studies
A total of 15 12- to 16-month-old ferrets (Mustela putorius furo; three males and two females [n = 5] per group were obtained from a commercial breeder (Triple F, Gillett, PA, USA). The ferrets were divided into three groups: sham-immunized (sterile water) (G1), 0.25 mg single dose (G2), and 0.25 mg prime + booster dose (G3). All animals were housed in the animal biosafety level 2 (ABSL-2) facility at the East Campus Research Facility (ECRF) of Cornell University during the immunization phase. After a 72-h acclimation period in the ABSL-2 facility, the animals were vaccinated intramuscularly, under inhalation anesthesia with isoflurane, with 1 ml of the linear DNA vaccine (dose of 0.25 mg for G2 and G3) or sterile water (G1, the sham-vaccinated group), followed by intramuscular electroporation (Vet-ePorator™, Carpi, MO, Italy) into the epaxial muscles [27]. The profile of electroporation parameters for each DNA vaccine administration was checked in real time, and the data for each animal were stored into the Vet-ePorator™ archive. A vaccine booster was administered on day 28 to animals in groups G1 and G3 following the procedures described above. For sample collection, ferrets were sedated with dexmedetomidine. Whole blood was collected through cranial vena cava (CVC) puncture using a 3-ml sterile syringe and a 23G × 1” needle and transferred to heparin or serum separator tubes on days 0, 28, and 42 post-vaccination. Blood was centrifuged at 1200 × g for 10 min, and the serum was aliquoted and stored at -20°C until further analysis. Body weight and temperature were recorded on days 0, 1, and 2 post-immunization (pi) for all of the animals, and on days 28, 29, and 30 pi for the animals in groups G1 and G3.
All animals were moved to the ABSL-3 facility at the ECRF at Cornell University on day 39 pi. Following an acclimation period of ~ 72 h, on day 42 after prime vaccination, all ferrets were challenged intranasally with 1 ml (0.5 ml per nostril) of a virus suspension containing 5 × 105 PFU of SARS-CoV-2 of the Alpha variant B.1.1.7 lineage (isolate NYC853-21). All animals were maintained in pairs or individually in Horsfall HEPA-filtered cages, which were connected to the ABSL-3's exhaust system. Clinical evaluation was performed daily, including body temperature and body weight measurement, observation of activity level, and signs of respiratory disease. Blood, oropharyngeal swab (OPS), nasal swab (NS), and rectal swab (RS) samples were collected under sedation (dexmedetomidine) on days 0, 1, 3, 5, 7, and 10 post-challenge (pc) as described previously [28]. All animals were humanely euthanized on day 10 pc.
Serological responses
Antibody responses were assessed using a bead-based multiplex assay based on the SARS-CoV-2 spike receptor binding domain (S-RBD). The assay was performed as described previously with a few modifications [29]. Briefly, beads were incubated with ferret serum samples diluted 1:200. Antibodies were detected using biotinylated mouse anti-ferret IgG (H + L) followed by streptavidin-phycoerythrin (Invitrogen, Carlsbad, CA, USA). All incubation steps were performed for 30 min at room temperature, and wells were washed after each incubation step. The assay was developed in a Luminex 200 instrument (Luminex Corp., Austin, TX, USA). Assay results for individual animals were expressed as fold change from day 0 (pre-immunization).
Neutralizing antibody (NA) responses to SARS-CoV-2 were assessed using a virus neutralization (VN) assay performed under BSL-3 conditions at the Cornell AHDC following a previously established protocol [30]. Serum samples obtained on days 0 and 42 post-immunization and on days 5 and 10 post-challenge were tested by VN. Briefly, two-fold serial dilutions (1:8 to 1:1024) of serum samples were incubated with 100–200 TCID50 of SARS-CoV-2. Following incubation of serum and virus, a suspension of Vero E6 cells was added to each well of a 96-well plate and incubated for 48 h at 37°C in a 5% CO2 incubator. The cells were fixed with 3.7% formaldehyde for 30 min at room temperature (RT), permeabilized with 0.2% Triton X-100 in phosphate-buffered saline (PBS) for 10 min at RT, and subjected to an immunofluorescence assay. Neutralizing antibody titers were expressed as the reciprocal of the highest dilution of serum that completely inhibited SARS-CoV-2 infection/replication.
Cellular responses
The RBD-specific T cell response was characterized using an ELISpot assay for IFN-γ according to the manufacturer’s instructions (Mabtech, Nacka Strand, Sweden). An RBD peptide pool composed by 132 out of the 338 peptides covering the whole spike protein was used for T cell stimulation. Briefly, the ELISpot assay was performed by stimulating PBMCs collected on day 42 (after boost and before challenge) overnight at 37°C. Intracellular cytokine staining was performed as described previously [26]. PBMCs were stimulated with the RBD peptide pool (final concentration, 5 µg/ml) and brefeldin A (1 µg/ml; BD Pharmingen, San Diego, CA, USA) at 37°C overnight. DMSO and PMA/IONO (Sigma-Aldrich, Burlington, MA, USA) at 10 g/ml were used as an internal negative and positive control, respectively, for the assay. Spot-forming colonies (SFCs) were counted using an automated ELISPOT reader (A.EL.VIS ELIspot reader, Germany). Results are expressed as SFCs/106 PBMCs.
Virus and cells
Vero-E6/TMPRSS2 cells (JCRB Cell Bank JCRB1819) were cultured in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS), L-glutamine (2 mM), penicillin (100 U/ml), streptomycin (100 µg/ml), and gentamicin (50 µg/ml) and maintained in a 37°C, 5% CO2 incubator. The SARS-CoV-2 Alpha (B.1.1.7 lineage) New York City 853 − 21 (NYC853-21) isolate used in this study was obtained from residual human anterior nares secretions and propagated in Vero-E6/TMPRSS2 cells. Low-passage virus stock (passage 3) was prepared, cleared by centrifugation (2000 × g for 15 min), and stored at -80°C. Genome sequencing was performed to confirm the integrity of the whole genome of the virus stock after amplification in cell culture. The virus titer was determined by plaque assay, calculated by the Spearman and Karber method, and expressed as plaque-forming units per milliliter (PFU/ml).
RNA isolation and real-time reverse transcription PCR
Ribonucleic acid (RNA) was extracted from 200 µL of cleared supernatant of OPS, NS, and RS. RNA extraction was performed using a MagMax Core Extraction Kit (Thermo Fisher, Waltham, MA, USA) and an automated KingFisher Flex Nucleic Acid Extractor (Thermo Fisher, Waltham, MA, USA), following the manufacturer’s recommendations.
Real-time reverse transcription PCR (rRT-PCR) for total (genomic and subgenomic) detection of viral RNA was performed using the EZ-SARS-CoV-2 rRT-PCR assay (Tetracore Inc., Rockville, MD, USA), which targets the viral nucleoprotein (N) gene. An internal inhibition control was included in all reactions. Positive and negative amplification controls were run side by side with test samples. For detection of specific subgenomic RNA, an rRT-PCR assay targeting the virus envelope protein (E) gene was performed using the primers and protocols described previously [31]. A standard curve using tenfold serial dilutions from 100 to 10− 8 of the virus suspension containing 106 TCID50/ml of the SARS-CoV-2 isolate used in the challenge was used for rRT-PCR validation. Based on this standard curve, the relative viral genome copy number in each sample was calculated using GraphPad Prism 9 (GraphPad, La Jolla, CA, USA) and expressed as log genome copy number per ml.
Virus titration
Samples from OPS, NS, and RS that tested positive for SARS-CoV-2 by rRT-PCR were subjected to virus isolation and endpoint titration under biosafety level 3 (BSL-3) conditions at the Animal Health Diagnostic Center (ADHC) Research Suite at Cornell University. For endpoint titrations, the sample supernatants were serially diluted and inoculated onto Vero E6/TMPRSS2 cells prepared 24 h in advance in 96-well plates. At 48 h post-inoculation, the cells were fixed and subjected to an immunofluorescence assay (IFA) as described previously [29]. The viral titer at each time point was calculated using endpoint dilutions and the Spearman and Karber method and expressed as TCID50/ml.
Statistical analysis and data plotting
Statistical analysis was performed using the Mann-Whitney U-test to compare groups. Statistical analysis and data plotting were performed using GraphPad Prism software (version 9.0.1). Error bars indicate the standard error of the mean (SEM). Figures 1A and 3A were created with BioRender.com.
Results
Vaccine safety and tolerance
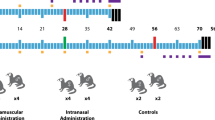
Fifteen ferrets (Mustela putorius furo) were divided into three groups as follows: sham-immunized (sterile water) (G1), single dose (G2), and prime + booster (G3) with 0.25 mg of DNA. The animals were immunized intramuscularly on day 0, and groups G1 and G3 received a booster dose on day 28 (Fig. 1A). The vaccination was immediately followed by intramuscular electroporation. Safety was assessed by monitoring local and systemic adverse reactions for 2 days after each immunization. During the period after the administration of the DNA vaccine or placebo (sterile water), there was no significant difference in body temperature between the vaccinated and sham-immunized groups (Fig. 1B). A slight increase in temperature up to 40 ºC was observed one day after the first vaccination and on the booster day in all groups, including the sham-immunized animals. These results confirm the safety of the linear DNA vaccine delivered intramuscularly concomitantly with co-localized electroporation (Vet-ePorator™) in ferrets.

Experimental design and body temperature after vaccination with the linear SARS-CoV-2 DNA vaccine candidate. A A total of fifteen 12- to 16-month-old ferrets (Mustela putorius furo) were divided into three groups with three males and two females per group, corresponding to the vaccine regimen to be administered: control sham-immunized (G1), single dose (G2), and prime + booster (G3). B Body temperature following intramuscular vaccination with the linear SARS-CoV-2 DNA vaccine candidate was measured on the days of vaccine administration and on the subsequent 2 days as indicted in the graphic. Data are presented as the mean ± standard error.
Vaccine immunogenicity
The serological response to vaccination was assessed using a fluorescent bead-based multiplex assay and a virus neutralization (VN) assay. Serum samples collected on day 42 post-immunization (pi) were used to assess S receptor binding domain (RBD)-specific or neutralizing antibody (NA) levels. All animals in group 3 (G3) (prime and booster) seroconverted against SARS-CoV-2, as evidenced by detection of binding antibodies against S-RBD in all five immunized animals and by detection of neutralizing antibodies (NA) in four out of five animals by day 42 pi. Binding anti-RBD antibody titers were higher in G3 than in the control group 1 (G1; p < 0.01) and group 2 (single dose) (G2; p < 0.05) (Fig. 2A). In addition to binding antibodies, four out of five animals in G3 also had higher NA titers on day 42 pi (p < 0.05) (Fig. 2B), with a titer of 8 in three animals and 16 in one animal. Prior to immunization, all animals tested negative for SARS-CoV-2 antibodies. All of the ferrets in group 2 (single dose) were tested on day 28 pi and found to be negative.
T cell responses were examined on day 42, after the booster and before challenge with SARS-CoV-2, and were found to be significantly stronger in the G3 vaccinated group than in the sham-immunized group (G1) (p < 0.05) (Fig. 2C). These results show that 0.25 mg of the linear DNA under a prime-boost vaccination schedule was able to induce both humoral and cellular immune responses in ferrets.

Serological and cellular responses to the linear SARS-CoV-2 DNA vaccine candidate assessed by bead-based multiple, virus neutralization, and ELISpot assays. A Antibody responses following immunization were measured by bead-based multiplex assay to quantify IgG anti-SARS-CoV-2 spike receptor-binding domain (RBD) in serum samples collected on day 42 post-immunization (pi). Results are presented as fold change from day 0 (pre-immunization). B Neutralizing antibody (NA) responses to SARS-CoV-2 in serum samples collected on day 42 pi. Neutralizing antibody titers represent the reciprocal of the highest dilution of serum that completely inhibited infection with 100–200 TCID50 of SARS-CoV-2. C The SARS-CoV-2 spike receptor-binding domain (RBD)-specific T cell response elicited by the linear DNA vaccine was assessed by ELISpot assay for IFN-γ. Proliferation of peripheral blood mononuclear cells (PBMCs) in samples from all of the ferrets was measured after stimulation with RBD pool peptides on day 42 post-immunization (pi). * = p < 0.05; ** = p < 0.01. Data are presented as the mean ± standard error.
Vaccine protection against heterologous challenge
The vaccine efficacy was assessed using a heterologous virus for challenge. On day 42 pi, all control and vaccinated animals were challenged with a SARS-CoV-2 Alpha variant of concern (VOC, isolate NYC853-21). Following the challenge, body temperature and body weight were measured and clinical observations were made on a daily basis (Fig. 3A). No significant differences in body temperature and weight were observed between the vaccinated and control groups (Fig. 3B, C). However, a slight increase in body temperature to 40 ºC on day 2 post-challenge was observed in all groups (Fig. 3B).

Experimental design and clinical observations following intranasal challenge with 5 × 105 PFU of a SARS-CoV-2 Alpha variant of concern (isolate NYC853-21). A Black squares represent the collection/measure time points for each sample type/parameter described. Clinical parameters, including temperature, body weight, activity, and signs of respiratory disease, were monitored daily after challenge. Oropharyngeal (OPS), nasal (NS), and rectal swab (RS) and blood samples were collected at various times points (black squares). Animals were humanely euthanized on day 10 pc. B Body temperature and (C) body weight following intranasal viral challenge were recorded throughout the experimental period. Data are presented as the mean ± standard error. Body weight was normalized to day 0, which represents 100%.
Following virus challenge, the dynamics of viral replication and RNA shedding were assessed in all animals. Oropharyngeal and nasal secretions and feces obtained through OPS, NS, and RS were tested for the presence of SARS-CoV-2 RNA by real-time reverse transcription PCR (rRT-PCR). Viral RNA was detected throughout the post-challenge (pc) period, between days 1 and 10 pc in all secretions and in all groups, regardless of the vaccination regimen used (Fig. 4). The highest viral RNA loads were detected between days 1 and 3 pc in oropharyngeal secretions of all groups, which decreased thereafter through day 10 pc. Prior to immunization, all animals were screened and tested negative for SARS-CoV-2 RNA.
In addition to the rRT-PCR for total viral RNA, we also performed RT-PCR for subgenomic RNA (sgRNA). sgRNA was detected in oropharyngeal swabs from day 1 to day 10 pc, peaking on day 3 pc and slowly decreasing until day 7 pc (Fig. 4D). A significant decrease in sgRNA in nasal swabs on day 1 pc was detected in both vaccinated groups (G2 and G3, p < 0.05) when compared to sham-immunized ferrets (G1) (Fig. 4E). By day 10 pc, only the sham-immunized group still had detectable levels of sgRNA in nasal secretions (Fig. 4E). In rectal swabs, the sgRNA was found in very low amounts in all groups (Fig. 4F). A significant decrease was found in the vaccinated groups compared to the sham-immunized group (G1), and the results showed that the ferrets in G3 had the lowest amount of shedding of sgRNA through the bodily secretions tested in our study.

Viral genomic and subgenomic RNA in nasal and oropharyngeal secretions and feces after SARS-CoV-2 challenge. Detection of viral RNA in A oropharyngeal swab (OPS), B nasal swab (NS), and C rectal swab (RS) samples from ferrets challenged with a SARS-CoV-2 Alpha variant of concern. Samples were tested for the presence of SARS-CoV-2 RNA by real-time reverse transcription PCR (rRT-PCR) (genomic and subgenomic viral RNA load). Detection of subgenomic SARS-CoV-2 RNA (sgRNA) in (D) OPS, (E) NS, and (F) RS samples from challenged ferrets. Day 0 represents swab samples collected prior to challenge (day 42 post-immunization). Data are presented as the mean ± standard error. * = p < 0.05; ** = p < 0.01.
In addition to detection of viral RNA, all samples from oropharyngeal and nasal swabs that were positive for SARS-CoV-2 by rRT-PCR were tested for the presence of infectious virus. Group 3, which received the prime-boost vaccination regimen, showed a significant decrease in viral loads in oropharyngeal and nasal secretions on days 3 and 5 pc (Fig. 5A). In addition, while all (5/5) animals from G1 shed infectious virus on day 1 pc, shedding was observed in four (4/5) and only two (2/5) ferrets from group G2 and G3, respectively (Table 1). Notably, after day 1 pc until the end of the study (day 10 pc), no infectious virus was detected in the animals of G3 (Table 1, Fig. 5A). As was observed with viral RNA, infectious virus titers were markedly lower in nasal secretions than in oropharyngeal samples, and all of the vaccinated ferrets had a significantly lower viral load on days 1 and 3 pc than the sham-immunized ferrets (Fig. 5B). No shedding of infectious virus was detected in feces.
All animals seroconverted by day 10 pc (experimental day 52), with NA titers increasing at least four-fold in comparison to the day of challenge (day 0 pc). Interestingly, the highest neutralizing titers were observed in group G3, which was the only group with significantly higher NA titers (p < 0.001) (Fig. 5C). These results suggest that the prime-boost vaccination regimen might have been more efficient in priming the immune system, resulting in a robust secondary response to SARS-CoV-2 challenge infection. Together, these results demonstrate that the linear DNA vaccine administered in a prime-boost vaccination schedule efficiently reduced infectious viral shedding through oropharyngeal and nasal secretions.

Titer of infectious virus in oropharyngeal secretions and feces and titers of neutralizing antibodies following SARS-CoV-2 challenge. A, B Shedding of infectious virus in oropharyngeal and B nasal swab samples from ferrets challenged with a SARS-CoV-2 Alpha variant of concern. C Titers of neutralizing antibodies in the animals after SARS-CoV-2 challenge. Day 0 represents swab samples collected prior to challenge (day 42 post-immunization). Data are presented as the mean ± standard error. * = p < 0.05; ** = p < 0.01; *** = p < 0.005; **** = p < 0.001.
Discussion
DNA-based vaccines are promising platforms due to their potential for rapid and scalable production and manufacturing. Moreover, the stability of DNA at room temperature makes this platform of particular interest for a quick response to a new emerging pathogen or to a new variant of a circulating agent, as seen with SARS-CoV-2 [26, 32]. Of note, DNA uptake by the target cells is a critical step for efficient immunization, which can be significantly enhanced by electroporation [21, 27, 33, 34].
Ferrets have been used as animal models to study various respiratory viruses, including influenza virus and SARS-CoV-2 [28, 35,36,37], and to assess pathogenesis, vaccine and drug efficacy and safety, and immune responses [36]. In addition to its extensive use in investigations of influenza A virus, respiratory syncytial virus, and SARS-CoV infections, the ferret model has been considered a reliable tool for analyzing pathogenesis and virus transmission and for assessing therapy and vaccination options for SARS-CoV-2 [28, 36, 38,39,40]. Previously, the safety and efficacy of ChAdOx1 nCoV-19 (AstraZeneca) against SARS-CoV-2 challenge was preclinically assessed in ferrets in parallel to non-human primates [41, 42]. Here, we used the ferret model to test a linear DNA vaccine candidate against SARS-CoV-2 delivered by electroporation into the epaxial muscle of ferrets.
The immune response to SARS-CoV-2 includes a cellular and a humoral component [43]. Although the aim of an ideal vaccine would be to achieve sterilizing immunity, vaccines that induce neutralizing antibodies and T cell responses may be able to restrain infected cells, restrict viral spread, accelerate viral clearance, prevent disease, or improve disease outcome [43]. Here, the linear DNA vaccine in a prime-boost regimen not only induced neutralizing antibodies and T cell immune responses but also reduced the viral load in nasal and oral secretions. In addition, the vaccine was shown to be safe, with no unexpected adverse reactions following intramuscular vaccination. The minor increase in temperature observed here is a common adverse effect after vaccination against COVID-19 that also occurs with the current commercially available vaccines [44, 45]. Thus, no evidence of immune-enhanced disease in vaccinated animals was found in this study. This has not been the case with all other vaccine studies, and immunopathological findings have been observed in some vaccination-challenge studies against SARS-CoV [46,47,48,49,50].
Although no differences were observed in the viral RNA load between the control and vaccinated groups, there was a significant reduction in shedding of infectious virus in oropharyngeal and nasal secretions from days 1 to 5 pc in the group that received the DNA vaccine in a prime-boost regimen (G3) in comparison to the unvaccinated animals. Reduced nasal shedding of SARS-CoV-2 in ferrets was also observed in a preclinical study of a vaccine that later became commercially available for use in humans [41]. The findings of the present study, together with previous investigations of the SARS-CoV-2 linear DNA vaccine delivered by EP, support the conclusion that this vaccine candidate is suitable for clinical development. Accordingly, a combined phase I-II trial has recently started.
In summary, our study shows that intramuscular vaccination with a SARS-CoV-2 linear DNA vaccine immediately followed by electroporation (Vet-ePorator™) is safe, elicits both humoral and cellular immune responses, and significantly reduces shedding of infectious SARS-CoV-2 through oral and nasal secretions in ferrets.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Wang C, Horby PW, Hayden FG, Gao GF (2020) A novel coronavirus outbreak of global health concern. Lancet 395:470–473. https://doi.org/10.1016/S0140-6736(20)30185-9
Zhu N, Zhang D, Wang W et al (2020) A Novel Coronavirus from Patients with Pneumonia in China, 2019. N Engl J Med. https://doi.org/10.1056/nejmoa2001017
Chan JFW, Yuan S, Kok KH et al (2020) A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: a study of a family cluster. Lancet 395:514–523. https://doi.org/10.1016/S0140-6736(20)30154-9
Chen N, Zhou M, Dong X et al (2020) Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet 395:507–513. https://doi.org/10.1016/S0140-6736(20)30211-7
Zhou P, Yang X, Lou, Wang XG et al (2020) A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. https://doi.org/10.1038/s41586-020-2012-7
Boni MF, Lemey P, Jiang X et al (2020) Evolutionary origins of the SARS-CoV-2 sarbecovirus lineage responsible for the COVID-19 pandemic. Nat Microbiol 5:1408–1417. https://doi.org/10.1038/s41564-020-0771-4
Holmes EC, Goldstein SA, Rasmussen AL et al (2021) The origins of SARS-CoV-2: A critical review. Cell 184:4848–4856. https://doi.org/10.1016/j.cell.2021.08.017
Gorbalenya AE, Baker SC, Baric RS et al (2020) The species severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol
Li F (2016) Structure, Function, and Evolution of Coronavirus Spike Proteins. Annu Rev Virol 3:237–261. https://doi.org/10.1146/annurev-virology-110615-042301
Belouzard S, Chu VC, Whittaker GR (2009) Activation of the SARS coronavirus spike protein via sequential proteolytic cleavage at two distinct sites. Proc Natl Acad Sci U S A 106:5871–5876. https://doi.org/10.1073/pnas.0809524106
Li W, Moore MJ, Natalya et al (2003) Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 426:450–454. https://doi.org/10.1254/fpj.147.120
Li F (2015) Receptor Recognition Mechanisms of Coronaviruses: a Decade of Structural Studies. J Virol 89:1954–1964
Yi C, Sun X, Ye J et al (2020) Key residues of the receptor binding motif in the spike protein of SARS-CoV-2 that interact with ACE2 and neutralizing antibodies. Cell Mol Immunol 17:621–630. https://doi.org/10.1038/s41423-020-0458-z
Polack FP, Thomas SJ, Kitchin N et al (2020) Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine. N Engl J Med 383:2603–2615. https://doi.org/10.1056/nejmoa2034577
Baden LR, El Sahly HM, Essink B et al (2021) Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine. N Engl J Med 384:403–416. https://doi.org/10.1056/nejmoa2035389
Sadoff J, Gray G, Vandebosch A et al (2021) Safety and Efficacy of Single-Dose Ad26.COV2.S Vaccine against Covid-19. N Engl J Med 384:2187–2201. https://doi.org/10.1056/nejmoa2101544
WHO (World Health Organization) (2022) COVID19 vaccine tracker. In: WHO
Khobragade A, Bhate S, Ramaiah V et al (2022) Efficacy, safety, and immunogenicity of the DNA SARS-CoV-2 vaccine (ZyCoV-D): the interim efficacy results of a phase 3, randomised, double-blind, placebo-controlled study in India. Lancet 399:1313–1321. https://doi.org/10.1016/S0140-6736(22)00151-9
Sheng Z, Gao N, Cui X et al (2016) Electroporation enhances protective immune response of a DNA vaccine against Japanese encephalitis in mice and pigs. Vaccine 34:5751–5757. https://doi.org/10.1016/j.vaccine.2016.10.001
Liu MA (2011) DNA vaccines: An historical perspective and view to the future. Immunol Rev 239:62–84. https://doi.org/10.1111/j.1600-065X.2010.00980.x
Sardesai NY, Weiner DB (2011) Electroporation delivery of DNA vaccines: Prospects for success. Curr Opin Immunol 23:421–429. https://doi.org/10.1016/j.coi.2011.03.008
Wang B, Godillot AP, Madaio MP et al (1997) Vaccination against pathogenic cells by DNA inoculation
Wolfgang W, Leitner H, Ying and NPR (2007) DNA and RNA vaccines—progress and prospects. Vaccine 18:765–777
Saade F, Petrovsky N (2012) Technologies for enhanced efficacy of DNA vaccines. Expert Rev Vaccines 11:189–209. https://doi.org/10.1586/erv.11.188
Tzeng T-T, Chai KM, Shen K-Y et al (2022) A DNA vaccine candidate delivered by an electroacupuncture machine provides protective immunity against SARS-CoV-2 infection. npj Vaccines 7:1–12. https://doi.org/10.1038/s41541-022-00482-0
Conforti A, Marra E, Palombo F et al (2022) COVID-eVax, an electroporated DNA vaccine candidate encoding the SARS-CoV-2 RBD, elicits protective responses in animal models. Mol Ther 30:311–326. https://doi.org/10.1016/j.ymthe.2021.09.011
Impellizeri J, Aurisicchio L, Forde P, Soden DM (2016) Electroporation in veterinary oncology. Vet J 217:18–25. https://doi.org/10.1016/j.tvjl.2016.05.015
Martins M, Fernandes MHV, Joshi LR, Diel DG (2022) Age-related susceptibility of ferrets to SARS-CoV-2 infection. J Virol. https://doi.org/10.1128/JVI.01455-21
Palmer MV, Martins M, Falkenberg S et al (2021) Susceptibility of white-tailed deer (Odocoileus virginianus) to SARS-CoV-2. J Virol. https://doi.org/10.1128/jvi.00083-21
Martins M, do Nascimento GM, Nooruzzaman M, Yuan F, Chen C, Caserta LC, Miller AD, Whittaker GR, Fang YDD (2022) The Omicron Variant BA.1.1 Presents a Lower Pathogenicity than B.1 D614G and Delta Variants in a Feline Model of SARS-CoV-2 Infection. J Virol. https://doi.org/10.1128/jvi.00961-22
Dagotto G, Mercado NB, Martinez DR et al (2021) Comparison of subgenomic and total RNA in SARS-CoV-2-challenged rhesus macaques. J Virol. https://doi.org/10.1128/jvi.02370-20
Conforti A, Marra E, Roscilli G et al (2020) Are Genetic Vaccines the Right Weapon against COVID-19? Mol Ther 28:1555–1556. https://doi.org/10.1016/j.ymthe.2020.06.007
Xu Z, Patel A, Tursi NJ et al (2020) Harnessing Recent Advances in Synthetic DNA and Electroporation Technologies for Rapid Vaccine Development Against COVID-19 and Other Emerging Infectious Diseases. Front Med Technol. https://doi.org/10.3389/fmedt.2020.571030
Lin F, Shen X, McCoy JR et al (2011) A novel prototype device for electroporation-enhanced DNA vaccine delivery simultaneously to both skin and muscle. Vaccine 29:6771–6780. https://doi.org/10.1016/j.vaccine.2010.12.057
Kim Y, Il, Kim SG, Kim SM et al (2020) Infection and Rapid Transmission of SARS-CoV-2 in Ferrets. Cell Host Microbe. https://doi.org/10.1016/j.chom.2020.03.023
Enkirch T, von Messling V (2015) Ferret models of viral pathogenesis. Virology 479–480:259–270. https://doi.org/10.1016/j.virol.2015.03.017
Rioux M, Francis ME, Swan CL et al (2021) The intersection of age and influenza severity: Utility of ferrets for dissecting the age-dependent immune responses and relevance to age-specific vaccine development. Viruses. https://doi.org/10.3390/v13040678
van den Brand JMA, Leijten haagmans L, Riel V, Martina (2008) Pathology of Experimental SARS Coronavirus Infection in. Vet Pathol 562:551–562
van den Brand JMA, Haagmans BL, van Riel D et al (2014) The pathology and pathogenesis of experimental severe acute respiratory syndrome and influenza in animal models. J Comp Pathol 151:83–112. https://doi.org/10.1016/j.jcpa.2014.01.004
Ciurkiewicz M, Armando F, Schreiner T et al (2022) Ferrets are valuable models for SARS-CoV-2 research. Vet Pathol 59:661–672. https://doi.org/10.1177/03009858211071012
Marsh GA, McAuley AJ, Au GG et al (2021) ChAdOx1 nCoV-19 (AZD1222) vaccine candidate significantly reduces SARS-CoV-2 shedding in ferrets. npj Vaccines 6:1–8. https://doi.org/10.1038/s41541-021-00315-6
van Doremalen N, Lambe T, Spencer A et al (2020) ChAdOx1 nCoV-19 vaccine prevents SARS-CoV-2 pneumonia in rhesus macaques. Nature 586:578–582. https://doi.org/10.1038/s41586-020-2608-y
Wheatley AK, Juno JA, Wang JJ et al (2021) Evolution of immune responses to SARS-CoV-2 in mild-moderate COVID-19. Nat Commun 12:1–11. https://doi.org/10.1038/s41467-021-21444-5
Al Khames Aga QA, Alkhaffaf WH, Hatem TH et al (2021) Safety of COVID-19 vaccines. J Med Virol 93:6588–6594. https://doi.org/10.1002/jmv.27214
Kaur RJ, Dutta S, Bhardwaj P et al (2021) Adverse Events Reported From COVID-19 Vaccine Trials: A Systematic Review. Indian J Clin Biochem 36:427–439. https://doi.org/10.1007/s12291-021-00968-z
Weingartl H, Czub M, Czub S et al (2004) Immunization with Modified Vaccinia Virus Ankara-Based Recombinant Vaccine against Severe Acute Respiratory Syndrome Is Associated with Enhanced Hepatitis in Ferrets. J Virol 78:12672–12676. https://doi.org/10.1128/jvi.78.22.12672-12676.2004
Yasui F, Kai C, Kitabatake M et al (2008) Prior Immunization with Severe Acute Respiratory Syndrome (SARS)-Associated Coronavirus (SARS-CoV) Nucleocapsid Protein Causes Severe Pneumonia in Mice Infected with SARS-CoV. J Immunol 181:6337–6348. https://doi.org/10.4049/jimmunol.181.9.6337
Tseng C, Te, Sbrana E, Iwata-Yoshikawa N et al (2012) Immunization with SARS coronavirus vaccines leads to pulmonary immunopathology on challenge with the SARS virus. PLoS ONE. https://doi.org/10.1371/journal.pone.0035421
Bolles M, Deming D, Long K et al (2011) A Double-Inactivated Severe Acute Respiratory Syndrome Coronavirus Vaccine Provides Incomplete Protection in Mice and Induces Increased Eosinophilic Proinflammatory Pulmonary Response upon Challenge. J Virol 85:12201–12215. https://doi.org/10.1128/jvi.06048-11
Liu L, Wei Q, Lin Q et al (2019) Anti-spike IgG causes severe acute lung injury by skewing macrophage responses during acute SARS-CoV infection. JCI insight 4:1–19. https://doi.org/10.1172/jci.insight.123158
Acknowledgments
We thank the Center for Animal Resources and Education (CARE) staff and Cornell Biosafety team for their support, and the Animal Health Diagnostic Center at Cornell University for the use of extraction and real-time PCR equipment. We also would like to thank Maureen H. V. Fernandes for her help with euthanasia, and Leonardo C. Caserta for whole-genome sequencing of the challenge virus.
Funding
This work was funded by Applied DNA Sciences.
Author information
Authors and Affiliations
Contributions
Conceptualization, Brian Viscount, James Hayward, Luigi Aurisicchio, and Diego Diel; data curation, Gabriela do Nascimento, Antonella Conforti, and Bettina Wagner; formal analysis, Mathias Martins and Antonella Conforti; funding acquisition, Diego Diel; investigation, Mathias Martins, Gabriela do Nascimento, Antonella Conforti, Jessica Noll, Elisa Sanchez, Bettina Wagner, Lucia Lione, Erika Salvatori, Eleonora Pinto, Mirco Compagnone, Brian Viscount, James Hayward, Clay Shorrock, and Luigi Aurisicchio; methodology, Mathias Martins, Antonella Conforti, Jessica Noll, Joseph Impellizeri, Elisa Sanchez, Bettina Wagner, Lucia Lione, Erika Salvatori, Eleonora Pinto, Mirco Compagnone, Brian Viscount, Luigi Aurisicchio, and Diego Diel; project administration, Brian Viscount and James Hayward; resources, Luigi Aurisicchio and Diego Diel; supervision, Diego Diel; validation, Mathias Martins and Diego Diel; visualization, Mathias Martins; writing – original draft, Mathias Martins, Gabriela do Nascimento and Diego Diel; writing – review & editing, Mathias Martins, Antonella Conforti, Jessica Noll, Joseph Impellizeri, Bettina Wagner, Luigi Aurisicchio, and Diego Diel.
Corresponding author
Ethics declarations
Conflict of interest
A.C. is an Evvivax employee. L.L., E.S., and L.A. are Takis employees. Takis and Rottapharm Biotech are jointly developing COVID-eVax. B.V., J.H., and C.S. are employees of Applied DNA Sciences and LineaRx, which retain the IP rights for the linear DNA platform.
Additional information
Handling Editor: Tim Skern.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Martins, M., do Nascimento, G.M., Conforti, A. et al. A linear SARS-CoV-2 DNA vaccine candidate reduces virus shedding in ferrets. Arch Virol 168, 124 (2023). https://doi.org/10.1007/s00705-023-05746-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00705-023-05746-1