
Abstract
Smacoviruses are small circular single-stranded DNA viruses that appear to be prevalent in faeces of a range of animals and have also been found in a few insect species. In this study, we report the first viral genomes from faeces of free-roaming wild felids on two continents. Two smacoviruses were recovered from the faeces of two North American bobcats (Lynx rufus), and one was recovered from an African lion (Panthera leo). All three genomes are genetically different, sharing 59-69% genome-wide sequence identity to other smacoviruses. These are the first full smacovirus genome sequences associated with a large top-end feline predator, and their presence in these samples suggests that feline faeces are a natural niche for the organisms that these viruses infect.
Similar content being viewed by others


The Smacoviridae are a family of circular single-stranded DNA viruses that have been identified in the faecal excrement of many mammals and birds, and a few arthropods [19]. The genomes of smacoviruses range from 2.3 to 3 kb in size and contain at least two bidirectionally transcribed open reading frames (ORFs), which encode a replication-associated protein (Rep) and a capsid protein (CP). These ORFs are separated by two intergenic regions, one of which contains an origin of replication. At present, more than 170 smacoviruses have been documented, with six established genera; Bovismacovirus, Cosmacovirus, Dragsmacovirus, Drosmacovirus, Huchismacovirus and Porprismacovirus. Recently, it was shown that DNA matching a smacovirus was found in the archaeon “Candidatus Methanomassiliicoccus intestinalis”, suggesting that smacoviruses may infect faeces-dwelling archaea [7]. Despite the broad range of animals with which smacoviruses have been associated, none have been documented in a member of the family Felidae. Felids, both domestic and wild, are known to harbour a multitude of viruses, including; feline immunodeficiency virus [13, 20], feline foamy virus [4, 11], feline leukaemia virus [5, 16], feline anellovirus [9, 21], feline gammaherpesvirus [2, 18], feline coronavirus [15], and several more.
In two separate studies, faecal samples were collected from bobcats (Lynx rufus) in California, USA, and African lions (Panthera leo) in the Serengeti National Park in Tanzania. Faecal samples described in this study were collected from three wild cats. One bobcat sample was collected during necropsy, the other from a live trap on the day of capture, and the African lion sample was collected shortly after defecation was observed. Viral DNA was extracted, and circular molecules were enriched from the faecal samples according to a previously described protocol [12]. Enriched DNA was sequenced on an Illumina 2500 platform, and contigs were generated through de novo assembly of paired-end reads using metaSPAdes v. 3.12.0 [1]. Three contigs with similarities to smacoviruses were identified in BLASTx searches and amplified from the three individual samples using abutting primers (Table 1) by PCR. The amplicons were then cloned and sequenced using the Sanger method [12].
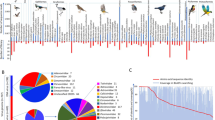
We have tentatively named the three novel smacoviruses reported in this study “Lynx rufus associated smacovirus 1” (LruSmV 1, MK796234), “Lynx rufus associated smacovirus 2” (LruSmV 2, MK796235) and “Panthera leo associated smacovirus” (PlSmV, MK796236). Their genomes are 2,435 nt, 2595 nt and 2,650 nt, respectively, in size (Table 1). All contain two large open reading frames oriented in opposite directions, encoding the Rep and CP proteins. 174 complete smacovirus genome sequences available in the GenBank database were compiled and used for comparison with the three sequences reported here. Full-genome, Rep and CP amino acid sequence datasets were assembled and used to determine pairwise identity values using SDT 1.2 [14]. Genome-wide pairwise comparison showed that the three genomes share 54-57% nucleotide sequence identity. With all other smacoviruses, the closest relative for each is as follows: LruSmV 1 shares 59% sequence identity with chimpanzee associated porprismacovirus 2 (GQ351273) [3], LruSmV 2 shares 61% sequence identity with Chlorocebus cynosuros associated smacovirus (LC386199), and PlSmV shares 67% sequence identity with sheep faeces associated smacovirus 3 (KT862219) [17] (Supplementary Data 1). Rep and CP amino acid comparison with all smacoviruses showed they share 39-63% sequence identity in Rep and 35-60% sequence identity in CP (Fig. 1 and Supplementary data 1).

Maximum-likelihood phylogenetic tree based on Rep protein sequences (left), displaying the six described genera. The LruSmV 1 Rep is most similar to that of Papio cynocephalus associated smacovirus (LC386204), sharing 38% amino acid sequence identity. An expanded subtree of the Rep-based phylogeny of the porprismacoviruses is displayed on the right. Names in red letters indicate the feline-faeces-associated Reps of the genomes identified in this study. A pairwise amino acid sequence comparison of the two novel feline smacovirus Rep proteins in the genus Porprismacovirus to those of all other members of the genus is shown next to the phylogenetic tree
Smacovirus Rep amino acid sequences were aligned using MAFFT [10], and the resulting alignment was used to construct a maximum-likelihood phylogenetic tree using PhyML 3.0 [8] with the best-fit amino acid model rtREV+G+I+F selected using ProtTest [6]. Branches with less than 0.8 aLRT branch support were collapsed. The phylogenetic tree was rooted with the Rep sequences of the nanoviruses. Based on the taxonomic guidelines put forward by the Smacoviridae subcommittee for the International Committee on Taxonomy of Viruses [19], the results of phylogenetic analysis and sequence comparisons support the assignment of each of these three smacoviruses to a distinct species.
Phylogenetic analysis showed that the Rep of LruSmV 1 is divergent, forming a singleton clade that is closest to the huchismacoviruses. The Reps of LruSmV 2 and PlSmV group in a clade consisting of the members of the genus Porprismacovirus (Fig. 1). The Rep proteins of LruSmV 2 and PlSmV share a common ancestor with porcine associated porprismacovirus 10 (KT862225) and sheep faeces associated smacovirus 3 (KT862219), respectively (Fig. 1) [17]. It is worth noting that these two LruSmVs were recovered from the faeces of individual bobcats living in the Los Angeles area (California, USA), but they are genetically distinct from each other. If smacoviruses do in fact infect archaea [7], this may indicate that these two distinct viruses are hosted by different archaeal species. Here, we describe the first smacoviruses associated with felid faeces and show that they are highly diverse, representing three distinct species. Continued research in this area is needed to determine the true hosts of these viruses and to elucidate what relationship they have to other felid viruses and those infecting other animals.
References
Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD (2012) SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19:455–477
Beatty JA, Troyer RM, Carver S, Barrs VR, Espinasse F, Conradi O, Stutzman-Rodriguez K, Chan CC, Tasker S, Lappin MR (2014) Felis catus gammaherpesvirus 1; a widely endemic potential pathogen of domestic cats. Virology 460:100–107
Blinkova O, Victoria J, Li Y, Keele BF, Sanz C, Ndjango J-BN, Peeters M, Travis D, Lonsdorf EV, Wilson ML, Pusey AE, Hahn BH, Delwart EL (2010) Novel circular DNA viruses in stool samples of wild-living chimpanzees. J Gen Virol 91:74–86
Cavalcante L, Muniz C, Jia H, Augusto A, Troccoli F, Medeiros S, Dias C, Switzer W, Soares M, Santos A (2018) Clinical and molecular features of feline foamy virus and feline leukemia virus co-infection in naturally-infected cats. Viruses 10:702
Chiu ES, Kraberger S, Cunningham M, Cusack L, Roelke M, VandeWoude S (2019) Multiple introductions of domestic cat feline leukemia virus in endangered florida panthers. Emerg Infect Dis 25:92
Darriba D, Taboada GL, Doallo R, Posada D (2011) ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics 27:1164–1165
Díez-Villaseñor C, Rodriguez-Valera F (2019) CRISPR analysis suggests that small circular single-stranded DNA smacoviruses infect Archaea instead of humans. Nat Commun 10:294
Guindon S, Dufayard J-F, Lefort V, Anisimova M, Hordijk W, Gascuel O (2010) New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59:307–321
Jarošová V, Hrazdilová K, Filipejová Z, Schánilec P, Celer V (2015) Whole genome sequencing and phylogenetic analysis of feline anelloviruses. Infect Genet Evol 32:130–134
Katoh K, Misawa K, Ki Kuma, Miyata T (2002) MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 30:3059–3066
Kehl T, Bleiholder A, Roßmann F, Rupp S, Lei J, Lee J, Boyce W, Vickers W, Crooks K, VandeWoude S, Löchelt M (2013) Complete genome sequences of two novel puma concolor foamy viruses from california. Genome Announcements 1:e00201–e00212
Kraberger S, Waits K, Ivan J, Newkirk E, VandeWoude S, Varsani A (2018) Identification of circular single-stranded DNA viruses in faecal samples of Canada lynx (Lynx canadensis), moose (Alces alces) and snowshoe hare (Lepus americanus) inhabiting the Colorado San Juan Mountains. Infect Genet Evol 64:1–8
Lee J, Malmberg JL, Wood BA, Hladky S, Troyer R, Roelke M, Cunningham M, McBride R, Vickers W, Boyce W, Boydston E, Serieys L, Riley S, Crooks K, VandeWoude S (2017) Feline immunodeficiency virus cross-species transmission: implications for emergence of new lentiviral infections. J Virol. https://doi.org/10.1128/JVI.02134-16
Muhire BM, Varsani A, Martin DP (2014) SDT: a virus classification tool based on pairwise sequence alignment and identity calculation. PLoS One 9:e108277
Myrrha LW, Silva FMF, Peternelli EFdO, Junior AS, Resende M, Almeida MRd (2011) The paradox of feline coronavirus pathogenesis: a review. Adv Virol. https://doi.org/10.1155/2011/109849
Powers JA, Chiu ES, Kraberger SJ, Roelke-Parker M, Lowery I, Erbeck K, Troyer R, Carver S, VandeWoude S (2018) Feline leukemia virus disease outcomes in a domestic cat breeding colony: relationship to endogenous FeLV and other chronic viral infections. J Virol. https://doi.org/10.1128/JVI.00649-18
Steel O, Kraberger S, Sikorski A, Young LM, Catchpole RJ, Stevens AJ, Ladley JJ, Coray DS, Stainton D, Dayaram A, Julian L, van Bysterveldt K, Varsani A (2016) Circular replication-associated protein encoding DNA viruses identified in the faecal matter of various animals in New Zealand. Infect Genet Evol 43:151–164
Troyer RM, Beatty JA, Stutzman-Rodriguez KR, Carver S, Lozano CC, Lee JS, Lappin MR, Riley SP, Serieys LE, Logan KA (2014) Novel gammaherpesviruses in North American domestic cats, bobcats and pumas: identification, prevalence and risk factors. J Virol 88(8):3914–3924
Varsani A, Krupovic M (2018) Smacoviridae: a new family of animal-associated single-stranded DNA viruses. Arch Virol 163(7):2005–2015
Winkler IG, Löchelt M, Flower RLP (1999) Epidemiology of feline foamy virus and feline immunodeficiency virus infections in domestic and feral cats: a seroepidemiological study. J Clin Microbiol 37:2848–2851
Zhang W, Wang H, Wang Y, Liu Z, Li J, Guo L, Yang S, Shen Q, Zhao X, Cui L, Hua X (2016) Identification and genomic characterization of a novel species of feline anellovirus. Virol J 13:146
Acknowledgements
Sample collection of bobcat faecal material was supported by the National Park Service and Santa Monica Mountains Fund, and Laurel Serieys was supported by a NSF graduate student fellowship.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Handling Editor: William G. Dundon.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Accession numbers: MK796234–MK796236.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Kraberger, S., Serieys, L., Fountain-Jones, N. et al. Novel smacoviruses identified in the faeces of two wild felids: North American bobcat and African lion. Arch Virol 164, 2395–2399 (2019). https://doi.org/10.1007/s00705-019-04329-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-019-04329-3