
Abstract
SARS-CoV-2 is one of the main serious challenges of human societies, which emerged in December 2019 from China and quickly extends to all parts of the world. The virus was previously believed to only affect the lungs and respiratory system, but subsequent research has revealed that it affects a variety of organs. For this reason, this disease is known as a multiorgan disease. Current article aimed to highlight latest information and updates about molecular studies regarding pathogenesis of SARS-CoV-2 in kidney, liver, and cardiovascular and respiratory systems, as well as the mechanisms of interaction of these organs with each other to cause clinical manifestations in patients.
Similar content being viewed by others


Introduction
Coronaviruses are a family of RNA viruses causing many acute as well as chronic diseases in both humans and in animals. Of the six known human strains of the corona virus, four cause the common cold. However, two groups of these viruses SARS-CoV and MERS-CoV (Middle East respiratory syndrome coronavirus) are the known roots for deadly respiratory disorders. Like SARS-CoV, SARS-CoV-2 is the deadliest virus in this family (Pedersen and Ho 2020). This new corona virus strain was initially reported early of December 2019 in Wuhan city of Hubei province in China. Since then, the virus quickly disseminated to all parts of the world and has had devastating effects on the global health system, while social and economic activities of the world have also deteriorates (Nishiga et al. 2020).
The virus is transmitted mainly through respiration, respiratory droplets, aerosols, and mucosal membranes. Certain recent studies showed that RNA of this virus has also been detected in human feces (Mokhtari et al. 2020). Patients with this disease show different symptoms, but initial symptoms observed in COVID-19 patients include repetitive cycles of fevers, headache, dry cough, and eventually pneumonia. However, with the spread of this disease in different parts of the world, other symptoms of this disease has also been reported (da Rosa Mesquita et al. 2020).
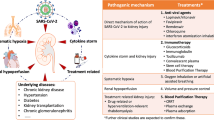
Although, initially, it was thought that major reasons for causalities in COVID-19 patients are primarily because of respiratory disorders and damage to lungs, but advancements in knowledge through extensive recent studies shown that it may affect most of the body organs particularly lungs, liver, heart, and kidneys leading patient’s mortality (Zaim et al. 2020). The aim of this article is to present the pathogenesis of SARS-CoV-2 in four vital organs. Lungs, liver, kidneys, and cardiovascular system. Furthermore, the study cross-link these organs with each other to elucidate clinical manifestations in patients affected with Covid-19.
Respiratory system in pathogenesis of COVID-19
The β-coronaviruses (MERS-CoV, SARS-CoV, and SARS-CoV-2) make their penetration into the human body via respiratory tract epithelium (Astuti 2020, Shereen et al. 2020). This functions as a barrier for any foreign particles or pathogens to provide protection against infections and tissue injuries via secreting mucus and mucociliary clearances while keeping the original airflow in the lungs (Fig. 1). During inhalation, SARS-CoV-2 particles are supposed to infect various types of epithelial cells. The initial SARS-CoV-2 contact starts inside the nasal mucosal cavity when the viral S (spike) proteins binds with that of the angiotensin-converting enzyme-2 (ACE2) receptors, and later, these S proteins cleaved by transmembrane serine protease 2 (TMPRSS2) and the viral particles start replicating normally within these cells (Hoffmann et al. 2020b; Wan et al. 2020; Sungnak et al. 2020). In vitro studies suggested that the ciliated airway epithelium serves as a primary host for SARS-CoV-2 infection if the airway epithelium cells express sufficient ACE2 that allows virus entry (Reyfman et al. 2019). ACE2 is a type I transmembrane metallocarboxypeptidase that promotes conversion of angiotensin 2 into different metabolites that help exert vasodilator effects or either interfere with the renin–angiotensin–aldosterone system. Moreover, manipulating the levels of ACE2 or its activities could alter the chances for the onset of COVID-19.

Pathobiological consequences of alveolar epithelial injury by SARS-CoV-2 infection
After successful penetration and replication inside the nasal mucosal cells, SARS-CoV-2 starts traveling towards conducting airways to set off an immune modulatory and inflammatory response that results in apparent clinical signs and symptoms of COVID-19 (Mason 2020). After their entry inside the epithelial cells, pro-inflammatory C-X-C motif chemokine-10 (CXCL-10) is expressed along, while type I and III interferons expressed and activated as the front line defensive molecules by recognizing SARS-CoV-2 attack (Hancock et al. 2018; Tang et al. 2005; Choi and Shin 2021). Initially, ACE2 expression increased inside small airways of smokers and people with chronic obstructive pulmonary disease (COPD), and may partly explain why individuals with underlying cardiopulmonary disease appear to be more likely to die from severe COVID-19 (Leung et al. 2020; Wu and McGoogan 2020). On the contrary, low expression lack of ACE2 in aged people and the one suffering either from diabetes mellitus or cardiovascular disease together with increased clearance of ACE2 from the cell surface with infection may result in over activity of the ACE-angiotensin 2-angiotensin 1 receptor axis, leading to increased inflammation and thrombosis (Verdecchia et al. 2020).
Although SARS-CoV-2 infection starts at upper airway epithelium, but in certain patients, the virus infects deeply in the alveolar epithelium to impair gaseous exchange and finally respiratory failure occurs (Fig. 1). The SARS-CoV-2 infection initiated by interaction of the viral S protein with ACE2, leading to internalization of the virion into endosomes. Host proteases such as TMPRSS2 or either furin help cleave the S protein giving rise to a fusion protein which facilitate the cytoplasmic entry of the virus (Hoffmann et al. 2020b; Shang et al 2020). Similar to SARS-CoV, SARS-CoV-2 shows alveolar type I (AT1) and AT2 cells which are equipped with increased ACE2 expression, but the prolific infections perhaps occurs primarily in surfactant-producing AT2 cells (Mossel et al. 2008). Few studies also claimed some alternative cellular-entry route for the SARS-CoV-2 such as Fc-receptor-mediated internalization of antibody-bound virions (Yip et al. 2014). The infected epithelial cells produce multiple virions which immediately invades adjacent endothelial cells and macrophages. Pathological examinations of late-stage cases depicted virus protein, while complete absence of prominent interstitial inflammation and vasculitis that suggest a persistent infection of alveolar epithelium occurs in severe disease.
Apoptosis is an ultimate fate of an infected alveolar epithelial cell, but some other types of cell deaths are still largely unknown. A wide expression of virus proteins has been observed in a rodent model of SARS-CoV-2 infection. The terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) assay confirmed that majority of the cells are apoptosed in this rodent model of SARS-CoV-2 (Chan et al. 2020). These virus proteins destabilize cellular physiology, induce apoptosis and release of interferon, and facilitate the production of virion (Lim et al. 2016). The infected epithelial cells fuse to create syncytia, a process mediated by the fusion machinery helping the virus entry, which promotes cell–cell spread of the SARS-CoV-2 and evasion of immune surveillance. The infected epithelial cells detach and leave behind a porous alveolar-capillary barrier. This alveolar epithelium offers majority of the barrier functions of the alveolar-capillary interface; hence, the loss of epithelium gets linked with the plasma exudation or hemorrhage, while the development of hyaline membranes full of factor VIII, cytokeratins and fibrin happens (Gorin and Stewart 1979). The epithelium infection not only results in the production of the virus but also the loss of the barrier function. The infected or strictly injured epithelium generates cytokine burst. In vitro studies of alveolar epithelial cells infected with coronavirus or either influenza virus showed production of a series of proinflammatory molecules, e.g., interleukins (ILs) (IL1β, IL6, IL8, and IL29), CXCL9, CXCL10, CXCL11, and CCL5. The loss of AT2 cells declines the secretion of the surfactants and may facilitate alveolar collapse. In an in vitro study, influenza infection of AT2 cells down regulated the release of surfactant proteins A and D (Wang et al. 2011). Surfactant protein D is a lectin that bind with SARS-CoV S protein but not confirmed in case of SARS-CoV-2 S protein (Leth-Larsen et al. 2007). Alveolar epithelium regulates coagulation and fibrinolysis on the alveolar surface, mainly by producing urokinase and plasminogen activator inhibitor 1 (PAI1) (IDELL 2003). SARS-CoV-2 pathology includes both hemorrhage and fibrin deposition in the alveolar spaces and microvasculature, implying perturbations in coagulation and fibrinolysis. A system biology analysis of experimental SARS-CoV infection demonstrated that urokinase-related pathways predict lung injury (Gralinski et al. 2013). Thus, alveolar epithelium as well as endothelium may promote coagulation disorders in COVID-19.
The biomarkers are of extreme importance especially in this time of COVID-19 pandemic to help diagnosis and developing novel drugs and treatment methods especially the vaccine formation. At clinical levels, biomarkers or biological indicators of the COVID-19 have gained much focus recently which depict the severity of the disease and its possible treatment options (Strimbu and Tavel 2010, Dobler 2019). The expressions of biomarkersespecially IL-8, intracellular adhesion molecule 1 (ICAM-1), and NO that have already been reported in ARDS are linked with increased mortalities and modulation in survival respectively (Jain 2010) and give a general idea for their implementation in COVID-19 prognosis. The genome of SARS-CoV-2 comprised of an RNA molecule which may act as primary biomarker for straightforward diagnosis of COVID-19. Furthermore, SARS-CoV-2 RNA encodes multiple structural proteins. i.e., nucleocapsid (N), envelope (E), membrane (M), and spike (S) protein along with various other proteins that mediate the viral entry into the human cells and the subsequent replication in the host cells (Chen 2020; Schoeman and Fielding 2019) may also be used as alternative biomarkers using appropriate detection techniques such as enzyme linked immunosorbent assay (ELISA). But there are certain hurdles that limit the use of these proteins as the biomarkers for the detection of SARS-CoV-2 in particular organs. These hurdles include the complexity in structure of these proteins (N, E, M, and S) and require huge quantity of the sample from that particular organ. Majority of these virus proteins, however, could serve as potential targets for anti-viral drugs or biomarkers of drug development for COVID-19 treatment.
Kidney in pathogenesis of COVID-19
The clinical picture of COVID-19 infection is a disease with systemic involvement of the body, especially dysfunction in vital organs (Ronco et al. 2020). Kidneys are mostly involved in COVID-19, and abnormal proteinuria is reported. Acute kidney injury (AKI) is thought as a key indicator for the severity of disease while a negative prognostic factor for survivorship (Qian et al. 2020). The term of AKI nearby ICU patients hangs on creatinine levels 3.1 mg/dl and urea level 145 mg/dl (Reference creatinine range 0.6–1.2 mg/dl) and (Urea Reference range 15–45 mg/dl) (Ghobadi et al. 2020). The AKI pathogenesis in COVID-19 is generally presumptive; it was mentioned that renal destruction is mainly assaulted by the SARS-CoV-2 via efflux of cytokines along with immune system dysfunctions (Qian et al. 2020). A hormone released from kidneys transfers angiotensinogen proteins into angiotensin-I, which is being converted to angiotensin-II by the ACE present in lung capillaries.(Aleebrahim-Dehkordi et al. 2020; Corvol et al. 2004; Pan et al. 2020; Wang et al. 2020c). ACE2 receptors mainly facilitate the entry of the coronavirus into the cell and hence become target cells susceptible to COVID-19 infection (Zou et al. 2020). It has been confirmed that ACE2 can be effectively bound to the S1 domain of the spike protein on SARS-CoV. So this protein is supposed to be essential receptor of SARS-CoV (Zhou et al. 2020c). This receptor is expressed at several parts of the kidney cells such as several types of epithelial cells of the glomerulus, distal tubular cells, also endothelium of interlobular arteries, and smooth muscle cells of the kidney (Asgharpour et al. 2020). The association between ACE2 and S protein in SARS-CoV depends on numerous variants of the ACE2 gene which may be determinative for outcome, symptoms, and sensitivity of SARS-CoV-2 infection in diverse tissues (Zhou et al. 2020c). The major coronavirus receptor ACE2 is expressed in human kidney cells primarily in the brush border of proximal tubules, afferent arterioles, collecting ducts, and the thick ascending loop of Henle, except in mesangial cells and glomerular endothelial (Santos et al. 2013; Ye et al. 2006). The presence of viral nucleic acid in the urine can also indicate the kidneys might be the target of this virus (Ling et al. 2020). Recently, it was demonstrated that SARS-CoV-2 by infecting kidney tubules can directly induce acute tubular necrosis (Qian et al. 2020). Immunohistochemistry findings demonstrated that renal failure happens before death (Diao et al. 2020). About inducing AKI in COVID-19 patients, these outcomes support that the SARS-CoV-2 virus can directly affect the podocytes and renal tubular epithelium (Su et al. 2020).
Although earlier studies offered a lower incidence (3–9%) of AKI in patient with COVID-19, but currently, it has been recognized as one of the fatal side effects of this infection. As said by previous studies on SARS-CoV and MERS-CoV infections, AKI has a high mortality rate (60–90%) (Aleebrahim-Dehkordi et al. 2020; Gagliardi et al. 2020).
New prognostic glomerular filtration biomarkers are β-trace protein (BTP) and β2-microglobulin (B2M) which comparisons to creatinine and cystatin C are limited by the absence of carefully developed GFR assessing stabilities for the unique biomarkers (Inker et al. 2016). In the general population and high-risk groups, these factors are autonomously associated with outcome of renal function such as end-stage renal disease (ESRD) and mortality (Foster et al. 2016).
Prognosis factors of AKI are creatinine (sCr), urea, and cystatin C. Recognition cystatin C, alone or in association with sCr, could be reflected a strong tool for the prediction of kidney action (Bongiovanni et al. 2015). Also, in the early detection of renal dysfunction, especially to detect mild reductions in GFR, in different types of renal diseases, assessment of cystatin C level may be helpful to estimate GFR (Coll et al. 2000). Nevertheless, it has also been used to estimate level of mortality; higher cystatin C levels are an independent forecaster of death in COVID-19 patients and can be used as an indicator of poor prognosis (Gagliardi et al. 2020). It was shown that COVID-19 patients with elevated urinary β2-microglobulin (β2MG) and α1-microglobulin (α1MG) levels had pointedly lower amounts of hospital discharge equated to those with normal urinary β2MG and α1MG levels (Sun et al. 2020).
With a better understanding of the physiopathology of AKI in COVID- 19 disease, be possible to achieve early diagnosis, optimize treatment plans, and promote life expectancy in these patients.
The critical point in this infection is interaction between lung and kidney damage. Acute respiratory distress syndrome (ARDS) causes acute hypoxia and may contribute to the delopment of acute kidney injury through systemic processes such as altered kidney function, increased renal vascular resistance, venous congestion, and decreased cardiac output, all of which may contribute to renal hypoperfusion and acute tubular necrosis (Fogagnolo et al. 2021). Acute kidney injury, on the other hand, has been demonstrated to promote lung injury by increasing lung capillary permeability, local inflammation, and fluid overload in various conditions (Legrand et al. 2021). According to several pathways that are caused to COVID-19 infections, the main factors are revealed in Table 1 that occur in the acute phase of patients admitted to the intensive care unit so can reach the good concepts of the pathogenesis of COVID-19 disease.
Cardiovascular system in pathogenesis of COVID-19
One of the main involved organs in patients with COVID-19 is cardiovascular system. The evidence and studies suggest that about 20–40% of the patients referring to hospitals report complications such as myocardial injury, acute myocardial infarction, myocarditis, acute coronary syndrome (ACS), venous thromboembolic even, cardiac chest pain, fulminant heart failure, cardiac arrhythmias, and cardiac death. However, chest pain and heartbeat are the most common symptoms in cardiac patients (Fig. 2) (Guzik et al. 2020; Long et al. 2020; Nishiga et al. 2020).

Schematic of the effect of COVID-19 on the cardiovascular system. COVID-19 affects the cardiovascular system through a variety of pathways. (1) COVID-19 affects the cardiovascular system through the production of large amounts of inflammatory cytokines. Large amounts of inflammatory factors released cause acute coronary syndrome, thrombosis, and myocarditis. (2) This virus also has an impact on the cardiovascular system by inducing hypoxia as a result of respiratory system disruptions or acute coronary syndrome. (3) In addition to the indirect effects, COVID-19 causes cardiomyocytes through its direct effect on cardiomyocytes
The hospital evidence suggests that some of the patients without any typical symptoms of COVID-19 report cardiac symptoms as the primary clinical manifestations (Nishiga et al. 2020). Cardiac symptoms are not the specific symptoms of COVID-19; rather, other diseases may be also manifested by the symptoms similar to the ones reported by COVID-19 patients. In 2009, studies on SARS-CoV patients showed that this virus can cause myocardial injury and systolic dysfunction by interacting with ACE2. In 2016, the symptoms of cardiovascular disorders in MERS-CoV patients were evaluated by cardiac MRI (Babapoor-Farrokhran et al. 2020). For example, Stefanini et al. reported that patients with COVID-19 who showed high levels of hs-TnI and BNP had a significant mortality rate than patients without elevated hs-TnI and BNP. The group also showed that concomitant increases in hs-TnI and BNP could be considered as an independent and reliable predictor of all-cause mortality (Stefanini et al. 2020). A meta-analysis included 23 studies and 4631 patients revealed that patients with high levels of TnI were more likely to have severe disease, ICU hospitalization, and mortality. Patients with raised CK, CK-MB, LDH, and IL-6 levels, as well as arrhythmia, are associated to the development of severe disease and the requirement for ICU admission, and patients with elevated LDH and IL-6 levels have a higher mortality rate (Li et al. 2020a) In 2954 COVID-19 patients showed that patients with pre-existing coronary artery disease showed high levels of cardiac biomarkers and high rates of ICU hospitalization and mortality. High levels of hs-TNI, -HBDH, CK-MB, and LDH, regardless of the presence or absence of coronary artery disease, are also prognostic biomarkers in COVID-19 patients (Li et al. 2021).
The molecular pathogenesis of the effect of COVID-19 on cardiovascular system
Studies about the molecular mechanism of COVID-19 in myocardial injury suggest that this virus can probably cause myocardial injury in different ways such as the ACE2 receptor (the direct way), hypoxia induction, systemic inflammation, and production of inflammatory cytokines (indirect way) (Zhu et al. 2020). As we know, ACE2 and serine protease (TMRPSS2) are necessary for intrusion of COVID-19 into the cell. First, S protein is priming by TMPRSS2, and in the following, it binds to ACE2 to enter into the cell. Therefore, simultaneous expression of TMPRSS2 and ACE2 is the main requisite for COVID-19 infection (Tajbakhsh et al. 2020).
ACE2 is a protein with a high expression in many tissues such as testicle, kidney, lung, and heart. Studies on expression of this protein in different cardiovascular cells suggest that ACE2 has a high expression in cardiomyocyte, fibroblast, smooth muscle cells, and pericytes (Unudurthi et al. 2020). According to the recent studies about the molecular mechanism of cardiovascular disorders in COVID-19 patients, there is a high level of ACE2 in pericytes that can be the cause of cardiovascular failure in this virus. On the other hand, expression of ACE2 increases in patients with failing human hearts, and it can be the cause of vulnerability of failing human hearts patients to COVID-19 (Guzik et al. 2020). In addition to the direct damage of COVID-19 to myocardial cells by ACE2, the other mechanism through which COVID-19 leads to myocardial injury is hypoxia and ischemia.
Researchers assume that this ischemia may be caused by the acute respiratory distress syndrome (ARDS) or the direct effects of the virus on macro- or micro-vascular system. It has been reported that ACE2 arteries are expressed on the surface of endothelial cells, and COVID-19 can probably disturb the arterial function and induce ischemia by binding to these receptors. On the other hand, local inflammation in these arteries leads to production of procoagulant factors that can cause thrombosis and ischemia in COVID-19 patients (Zhu et al. 2020). The other mechanism through which COVID-19 induces ischemia and myocardial injury is coagulopathy. One of the main indications of these patients is the increased production of coagulant factors and disseminated intravascular coagulation (DIC) which increases the probability of thrombosis and ischemia in these patients (Tang et al. 2020, LILLICRAP 2020).
The other mechanism through which COVID-19 causes cardiovascular diseases is systemic inflammatory response and immune response dysfunction which leads to production of large amounts of inflammatory cytokines, i.e., cytokine storm (Tajbakhsh et al. 2020). Inflammatory factors are among the factors involved in electrical disorders and cardiac arrhythmia. For example, IL-6 is one of the inflammatory factors that are significantly increased in the systemic inflammation caused by COVID-19 (Grifoni et al. 2020).
This pre-inflammatory factor increases the level of L-type Ca2 + current (ICaL) and decreases the expression of Ca2 + -ATPase (SERCA2) and junction proteins in myocardial cells and so, it causes cardiac arrhythmia (Wang et al. 2020e; Unudurthi et al. 2020).
The other probable mechanism proposed for myocardial injury in COVID-19 patients is the decreased level of ACE2 on the cell surface that results from its binding and internalization with the virus in the process of viral transmission into the cell. It can increase the expression of Ang II and decrease the expression of Ang-(1–7). The evidence and studies suggest that Ang-(1–7) activates the protective signaling pathway from the myocardium by Mas receptor and it inhibits the effects of Ang II (Wang et al. 2020e; Patel et al. 2016). However, several clinical studies have reported that with the increase of Ang II, this factor can induce atherosclerosis, fibrosis, hypertrophy, and consequently cardiovascular disorders by AT1 receptor (Billet et al. 2008; De Mello and Danser 2000; Chakafana et al. 2020).
Liver in pathogenesis of COVID-19
Previous reports have revealed that some SARS-CoV-2 patients have gastrointestinal symptoms such as diarrhea, abdominal pain, loss of appetite, vomiting, and nausea (Zhou et al. 2020d; Guan et al. 2020b). In addition, concomitant injury of liver has recently been published in COVID-19 patients (Zhang et al. 2020a). The abnormality of function tests of liver were seen in some patients with COVID-19 upon admission (Xiao et al. 2020). A group of liver function tests was performed, and the results revealed that some COVID-19 patients had liver dysfunction. Alteration of liver cells damages biomarkers, such as alanine aminotransferase (ALT), aspartate aminotransferase (AST), albumin, and bilirubin, is a common laboratory finding in patients with SARS-CoV-2 (Lei et al. 2020a; Bangash et al. 2020; Han et al. 2020; Hu et al. 2020). Furthermore, pathological examination of COVID-19 patients showed that hepatocyte destruction was linked with moderate steatosis, focal necrosis, leukocyte infiltration into the lobular and portal area, and sinusoidal congestion (Chang et al. 2020). Therefore, all of these showed liver involvement in COVID-19 patients. However, the underlying mechanism is not completely discovered, especially in the case of hepatobiliary involvement of the disease (Kovalic et al. 2021). In these patients, as in the case of previous infections seen with SARS-CoV, the binding and uptake through the cholangiocyte receptor of the angiotensin converting enzyme (ACE2) causes liver injury (Chai et al. 2020b). Not only does the increase degree in ALT or AST appear to be related to the severity of SARS-CoV-2 infection, but also suggests that this mechanism is mediated by liver damage through ACE2 up-regulation also among liver cells in a mouse model (Guan et al. 2020a).
Although hepatocytes and bile duct epithelial cells express ACE2 receptor (Chai et al. 2020b), but no significant changed histopathological properties have been tested in such cells from SARS-CoV-2 infected patients (Xu et al. 2020). In addition, glutamyl transpeptidase (GGT) is a biomarker for the diagnosis of cholangiocyte damage. Some COVID-19 patients have GGT enhanced during hospitalization (Chai et al. 2020a). Preliminary reports revealed that ACE2 receptor expression is enriched in cholangiocytes (Hoffmann et al. 2020a), and SARS-CoV-2 enters the target cell via ACE2 receptor (Hoffmann et al. 2020a), which results in serum abnormality. A direct cause of liver damage in SARS-CoV-2-infected patients may be that the virus infects liver cells. Approximately 2–10% of COVID-19 patients developed diarrhea symptoms, and SARS-CoV-2 nucleic acids were detected in their feces and blood specimens (Yeo et al. 2020), which suggests that the virus may invade the liver through blood circulation or digestive tract. On the other hand, hypoalbuminemia and abnormal PT were seen in some patients with COVID-19 (Lei et al. 2020a). Abnormal PT can be due to liver damage or coagulopathy. Hypoalbuminemia is reported via liver damage, kidney injury, heart failure, malnutrition, and protein-losing enteropathy. Hypoalbuminemia and elevated AST levels were frequently seen in severe patients, and correlation analysis revealed a correlation between AST and albumin levels with gradation of severity. Therefore, biomarkers of liver function including albumin and AST should be evaluated in COVID-19 patients (Lei et al. 2020b).
Due to clinical evaluation and biochemical experiments on COVID-19 patients, enhanced levels of liver function abnormality biomarkers have been linked with severe COVID-19 and worse prognosis (Lippi and Plebani, 2020). Severe patients with COVID-19 frequently induce hypercoagulability with both microangiopathy and local thrombus formation, while abnormality in prothrombin time, in disease onset, is relatively uncommon (Connors and Levy 2020; Iba et al. 2020). Evaluation of several COVID-19 studies reveals that laboratory chemistries were collected from patients following their hospitalization. More comprehensive trends in liver chemistries may be considered in the future due to test; the peak value of this biochemical composition results and compares them based on clinical results. For instance, the peak AST and ALT levels noted were 1445 U/L and 7590 U/L, respectively, in severe COVID-19 (Chen et al. 2020; Fan et al. 2020). Literatures revealed that 14–53% of patients with advanced COVID-19 had elevated AST and ALT levels. The alkaline phosphatase (ALP) level increased in 1.8% of COVID-19 patients during hospitalization. There is a higher liver dysfunction incidence in severe patients with COVID-19 (Zhang et al. 2020b). The prothrombin test (PT), D-dimers, fibrinogen, and the platelet count are significant predictors of severity of disease illness as detection alterations herald the onset of disseminated intravascular coagulopathy (DIC) (O'Shea et al. 2020).
In addition, the description of “chronic liver disease” is not yet defined in these reports and requires further study. Many of these laboratory results, such as abnormal liver parameters, reduced albumin, low platelet count, or prolonged PT, can be attributed to acute SARS-CoV-2 infection, but can also be associated with underlying viral hepatitis or cirrhosis that is obviously not observed between these studies. An Italian study reported chronic liver injuries in only 28 of 1591 ICU patients (Grasselli et al. 2020).
Another study from the USA found that of 5700 hospitalized patients, only 11 (0.2%) had HBV or HCV and only 19 (0.4%) had cirrhosis (Richardson et al. 2020). A large readiness study found that individuals with chronic liver disease and COVID-19 were three times more likely to die than patients without liver problems (Singh and Khan 2020). In contrast, two other meta-analyzes have recently failed to show an association between chronic liver disease and the severity or mortality from SARS-CoV-2 infection (Wang et al. 2020d; Lippi et al. 2021).
Other causes of liver damage include underlying liver injury, medication including experimental drugs, and in severe patients, a cytokine storm via tumor necrosis factor α (TNF-α), interleukin [IL] -6 or IL-18), and the ischemic hypoxia-reperfusion that associated with COVID 19 infection with more severity (Hu et al. 2020).
Therefore, SARS-CoV-2 infection–related liver damage should be the result of secondary liver damage due to the systemic inflammatory response, underlying liver diseases, respiratory distress syndrome-induced hypoxia, hypoxic hepatitis, MOF, and administration of hepatotoxic drugs (Feng et al. 2020; Bangash et al. 2020; Han et al. 2020; Hu et al. 2020). The characterization of liver dysfunction among patients with SARS-CoV-2-infected remains paramount with respect to potential therapeutic methods. Regarding to treatment effect on liver function of patients with COVID-19, Xu et al. (Hu et al. 2020) showed the medications applied for SARS-CoV-2 patients probably induced liver injury, such as antibiotics, antiviral agents, etc.
Hydroxychloroquine has been used as a potential option for COVID-19 treatment (Gautret et al. 2020), and although hepatotoxicity is relatively rare, it is metabolized in the liver and has been revealed to cause acute liver damage in some individuals (Hydroxychloroquine 2012). Preliminary reports of the IL-6 inhibitor tosilizumab in COVID-19 patients also suggest that tosilizumab may be linked with unusual hepatotoxicity (Zhang et al. 2020c).
In addition, the use of antiviral therapy has been further investigated. Remdesivir, a new antiviral agent that acts as an RNA-strand terminator, has been shown to be effective in vitro, and in early clinical trials, the results of remdesivir showed liver dysfunction among other side effects (Grein et al. 2020; Beigel et al. 2020; Goldman et al. 2020). Among the baseline data from a study of the first cases of SARS-CoV-2 infected admitted to the hospital, all patients receiving remdesivir experienced transaminase during their clinical course (Kujawski et al. 2020). However, an increase in ALT or AST levels more than five times the normal range is still an exclusion criterion for starting remdesivir in patients with SARS-CoV-2 infected, and this may exclude many cases with liver damage from takingremdesivir (Grein et al. 2020; Beigel et al. 2020; Goldman et al. 2020; Kujawski et al. 2020).
Thus, further characterization and identification of liver damage are very important among SARS-CoV-2-infected patients with severe/critical infections, especially if one of the few treatment options for underlying liver chemical conditions is expected. To overcome this difficulty, in one study, functional liver chemistries were collected within 2 days of hospitalization to prevent drug-induced liver injury (DILI). In addition, liver function abnormalities were observed in patients with chronic liver disease, so these types of cases were excluded. Therefore, the possibility of liver injury in COVID-19 patients may be associated with viral infection of liver cells, inflammatory reactions, and hypoxic hepatitis (Lei et al. 2020b).
As the COVID-19 epidemics continue to affect more people around the world, case studies and the clinical implications of liver damage among this patient population will go a long way.
In addition to coronary artery disease, cerebrovascular disease, leukocytosis, COPD, neutrophilia, elevated creatinine kinase, lymphopenia, elevated LDH, and elevated PT, it appears to be correlation significantly with severe or critical COVI-19 cases. Extensive long-term studies are required to describe the cause and extent of liver damage in patients with COVID-19. The impact of COVID-19 on chronic liver disease needs to be carefully studied and further research is needed in this area (Jothimani et al. 2020).
Finally, the experience from researchers and doctors around the world suggests that liver function should be considered in all SARS-CoV-2-infected patients, but mild to moderate abnormalities may be of secondary importance. However, during illness, one of the most important points to consider is the use of medications that can cause DILI in COVID-19 patients. Also, more careful monitoring of liver enzymes in serum is required, especially in hospitalized patients or patients with liver disease (Papadopoulos et al. 2020). Current knowledge about contributing factors such as liver failure or concomitant drug use remains largely unknown. To continue prospective studies, it is necessary to assess the various factor roles in the pathogenesis of liver damage in infection with SARS-CoV-2.
Cross-link of SARS-CoV-2 with various body organs
The entry of the SARS-CoV-2 inside the host cells is considered important milestone for viral infection and pathogenesis (Li 2016, Perlman and Netland 2009) and remains a key target for immune surveillance in the host cells (Du et al. 2009, 2017). In-depth understanding of underlying mechanism that shows SARS-CoV-2 make its interaction with the lungs epithelial cells is crucial for advancing the knowledge of SARS pathogenesis. Most of coronaviruses initially bind with specialized cell surface receptors for proper attachment, through its surface-anchored spike proteins, enter into the cell by forming an endosome, and finally, the virus and lysosome membranes get fused (Li 2016, Perlman and Netland 2009). The extracellular vehicles (EVs) (30–120 nm) encircled by lipid bilayer are secreted from almost all kind of cells, even the cells of the linings of respiratory tract and are assigned to perform specialized functions by providing immunity to the lung cells, and pathogenesis of multiple disorders of the lungs especially the viral infections (Pocsfalvi et al. 2020). Cells infected with SARS-CoV-2 discharge exosomes, which are linked with infections primarily by transportingvarious constituents of the viruses particularly virus-derived micro RNAs (miRNAs) multiple proteins, also contain ACE2 receptors making the recipient cells highly susceptible and make an easy for viral entry (Hassanpour et al. 2020). Some studies showed the presence of SARS-CoV-2 RNA-containing exosomal bodies shows an alternative route for cellular entry especially in cardiomyocytes which results in quick cardiac dysfunction and no more direct coronavirus infection is needed in this case. Moreover, the examinations of the exosomal bodies filled with coronavirus genes may upregulate inflammatory factors at transcriptional levels in pluripotent stem-cell-derived cardiomyocytes (Kwon et al. 2020). (Fig. 3).

The extracellular vesicle (EV) formation in SARS-CoV-2 entry into the cell, replication, and exocytosis to approach other cells/organs of the body
In pursuance to give an effective reaction against COVID-19 disease, a deep understanding of the mechanism of infection along with identified and authentic biomarkers facilitates the availability, precision, and efficacy of COVID-19 testing. The respiratory system of the people who have developed clinical illness due to SARS-CoV-2, remained highly vulnerable. The virus may have a potential to affect any organ of the body and even in severe patient’s multiple organs get severely affected (Fig. 4). It is postulated that cellular responses to SARS-CoV are uncertain, and COVID-19 disease is divided in three stages, and corresponds to multiple clinical phases based on infected cells (Wu and McGoogan 2020). Just after the entry into the nasal cavity via inhalation SARS-CoV-2 potentially binds and begins to replicate inside epithelial cells (Fig. 3). SARS-CoV-2 have higher reproduction number (R0) of 2.2–5.7 that gives rise to its exponential spread (Patil et al. 2020).

A cross-talk of SARS-CoV-2 spread to different body organs
ACE2 receptor facilitates SARS-CoV-2 attachment on the cell surface (Wan et al. 2020; Hoffmann et al. 2020c), and lung ciliated cells are the main destination in the contaminated airways (Sims et al. 2005). ACE2 receptors are present in the membranes of vascular endothelial cells, lung cells, cardiomyocytes, brain cells, kidney cells, intestinal cells, hepatocytes, pharynx, and lot more to count (Meredith Wadman et al. 2020). but less in the airway cell membranes (Reyfman et al. 2019), ACE cleaves angiotensin (Ang)-I and converts into Ang-II, that may help in inflammations and vasoconstriction, while ACE2 help process Ang-II further into Ang(I-VII) that produce vasodilation, anti-inflammatory, antioxidant, and anti-apoptotic effects. Moreover, ACE2 in addition to the cellular receptors for SARS-CoV-2 gets down-regulated following binding by SARS-CoV-2 particles, and the Ang-II levels increased, which promote vasoconstriction, inflammation, oxidative stress, and cell apoptosis (Chen and Subbarao 2007).
Initially, systemic influx of inflammatory cytokine may result in cytokine release syndrome (CRS), is the key underlying mechanism for creating severe complications, and increased mortalities in COVID-19. Inflammations started in lung tissues because of SARS-CoV-2 mediated damage to the alveolar epithelial cells, triggering an immense infiltration of the immune cells (Li and Ma 2020). These micro inflammatory responses mediate the systemic discharge of proinflammatory factors (cytokines), that results in the hyperinflammations of numerous body organs, and subsequently, tissues are damaged and the patients dies (Ye et al. 2020). Various pathophysiological mechanisms particularly endotheliosis, dysregulation of RAAS (Renin–Angiotensin–Aldosterone System), thrombosis, lymphocytopenia, and T cell energy are the contributing factors for enhanced morbidities (Amraei and Rahimi 2020; Jesenak et al. 2020).
It is crucial to find the risk linked with the mortalities in COVID-19 patients especially the vulnerable ones. The SARS-CoV-2 is known to develop serious conditions in different body organs such as inflammation, endotheliitis, vasoconstriction, hyper-coagulability, and edema (Jain 2020). Furthermore, lymphocytopenia, increasing levels of D-dimers and FDPs (fibrin degradation products), and disseminated intravascular coagulation (DIC) are observed in COVID-19. Many diseases such as venous thromboembolism, pulmonary embolism (PE), systemic and pulmonary arterial thrombosis or embolism, deep vein thrombosis (DVT), ischemic strokes, and myocardial infarctions (MI) have been studied extensively (Jain 2020). Attempts are being made for the development of novel COVID-19 biomarkers; herein, the cross talks between various body organs, the diagnostic, and prognostics to shield against the COVID-19 are discussed. The COVID-19 patients primarily depict respiratory disease symptoms being treated for pneumonia. It is crucial to focus the cardiovascular system specifically to identify prior symptoms of acute myocardial injuries. ACE2 receptors are present on cardiomyocytes and endothelial cells but not on the cell membranes lining of liver sinusoids, lungs, bile ducts, intestines, or kidney cells (Hamming et al. 2004).
About one quarter of COVID-19 patients showed an unusual prevalence of cardiovascular disease (CVD) remained in ICU (intensive care unit) along with hypertension patients (58%) (Wang et al. 2020a). During myocardial injuries, an increased level in serum cardiac troponin-I (cTn-I) is seen, and same has also been seen in COVID-19 patients which might be the reason of half of deaths (Zhou et al. 2020a).
Several recent reports examined that abnormal liver functions is linked with enhanced disease-mediated deaths. SARS-CoV-2 may cause hepatic injury even in initial medication started, suggesting the primary threshold is un-related to medical management but rather due to either direct effect of the coronavirus or might be consequence of any systemic disease. The hepatologists are continuously facing a great job of looking after COVID-19 patients suffering liver diseases or liver transplant patients who have been SARS-CoV-2 positive, and also patients on routine surveillance without COVID-19 (Sahin et al. 2020).
In one-third of COVID-19 patients, cardiomyopathy is frequent condition in SARS-CoV-2-positive patients in USA (Arentz et al. 2020). It is emphasized that both cardiac dysfunction along with hepatic congestion contributes to hepatic injury in COVID-19 infection and is mainly linked with an elevated levels of amino-transferases and gamma-glutamyl transferase (GGT) (Weisberg and Jacobson, 2011; Van Deursen et al. 2010). Severe ischemic hepatitis is a condition characterized by severe AST-predominant hepatitis (Tapper et al. 2015) and may be observed in critically ill patients with COVID-19. The infrequently observed alkaline phosphatase elevation occurs late in COVID-19 disease progression and could reflect the cholestasis of sepsis, critical illness, or medication effect (Fuchs and Sanyal 2008).
In the heart, SARS-CoV-2 may develop acute coronary syndrome, myocarditis, congestive heart failure, and arrhythmias. Many young’s face stroke and cardiac complications can lead and occur in lack of pulmonary and other complications (Madjid et al. 2020; Akhmerov and Marbán 2020). Many of the patients faced ischemic cardiac injury who have developed coronary artery disease (CAD), latent CAD and without CAD, where former remained vulnerable due to attack of SARS-CoV-2 which alters the pulmonary system to cause acute respiratory distress syndrome (ARDS), diffuse alveolar hemorrhage, and the failure of the lungs (Yang et al. 2020).
The primary cause of the former two is plaque rupture and thrombosis. The last one is due to inadequate oxygen supply and mimics a myocardial infarction (MI). For acute coronary syndrome due to plaque rupture, antiplatelet and anticoagulation therapy may be beneficial. Fibrinolytic therapy and percutaneous coronary intervention may be considered. However, the reported incidence of acute MI has declined in the COVID-19 period (Solomon et al. 2020). The SARS-CoV-2 invades myocytes to generate a cytokine burst to produce a systemic inflammatory response to cause myocarditis without direct viral infiltration. Moreover, it may lead heart failure and arrhythmias even at the acute phase of the viral infection has resolved without any damage to the lung. Almost 50% of non survivors of COVID-19 reported having acute cardiac injury and heart failure.
During early stages of the COVID-19, respiratory failure occurs while cardiac injury is more obvious and severe in the later stages of the disease. Various risk factors associated with severity and deaths, diabetes, obesity, age, and hypertension than does respiratory disease alone. Failure of the heart and elevated brain-type natriuretic peptide (BNP) are also observed, along with increased troponin are linked with mortalities (Akhmerov and Marbán 2020). In older patients with existing CAD or hypertension, heart failure is mainly due to poor demand–supply relationship, while in younger patients, myocarditis is the major reason. The hike in cardiac issues is reported due to inflammatory stress, myocarditis, hypoxemia, metabolic abnormalities, or medications. Cardiovascular issues (dyslipidemia, pulmonary fibrosis, and a vascular necrosis) appear long after virus clearance and even after recovery in survivors, while evolution and persistence of inflammations are silent (Solomon et al. 2020; Akhmerov and Marbán 2020). In such cases, frequently applied medicines include ACE inhibitors and angiotensin II receptor blockers, which have not been explained for increased risks of COVID-19 infection or its associated complications (Vaduganathan et al. 2020).
These observations led to define theories surrounding the interplay between the pathophysiology of COVID-19 and the cardiovascular system (Clerkin et al. 2020; Zheng et al. 2020). Moreover, COVID-19 may exacerbate cardiovascular risk factors and pre-existing CVD or may increase susceptibility for the development of new cardiovascular complications. Alternatively, CVD or myocardial injury may predispose to worse outcomes in COVID-19 patients, reflected in huge literature, whereby established CVD is linked with severe COVID-19, leading to higher morbidity and mortality.
The injuries to the kidney remained secondary to systemic abnormalities in COVID-19. Many of the studies indicate that ACE2 is highly expressed in renal tubular cells, Sertoli cells, Leydig cells, and cells in seminiferous ducts in testis (Fan et al. 2021). Moreover, recombinant SARS-CoV-2 spike protein (RBD) domain and ACE2 of RPTEC/SerC cell-binding assays also confirmed that SARS-CoV-2 can bind to ACE2 on the surface of these cells (Fan et al. 2021). Multiple factors may cause the renal injuries following SARS-CoV-2 infection. Various kidney cells (glomerular cells, tubular epithelium, and podocytes) also have got the ACE2 receptors on cell membranes and reported the existence of the SARS-CoV-2 virus (Puelles et al. 2020). Through highly expressed ACE2 in renal tissue, SARS-CoV-2 infection fundamentally initiates a mechanism of renal injury. Systemic effects such as host immune clearance and immune tolerance disorders, endothelial cell injury, thrombus formation, glucose and lipid metabolism disorder, and hypoxia aggravate this renal injury (Wang et al. 2020b). Due to severe but huge number of COVID-19 patients and the associated complications make it hard to manage patients on dialysis and during kidney transplantation (Alberici et al. 2020). About 15% patients expired in the UK were reported suffering chronic kidney disease. The inflammatory bursts especially the cytokines play a role in severe hypo-perfusion and acute kidney injury (AKI) and are usually secondary to systemic abnormalities such as diabetes, hypertension, chronic kidney disease, hypoxemia, and coagulopathy (Braun et al. 2020). AKI may also be caused by the rhabdomyolysis mainly by hyperventilation or anti-viral medicines especially remdesivir. In addition, over 90% patients suffering mechanical ventilation in New York city had been developed AKI in temporal association with respiratory failures (Braun et al. 2020; Hirsch et al. 2020). The lack of the renal replacement therapy system and related hemodialysis machines and supplies and peritoneal dialysis were used extensively especially for unstable patients. On the other hand, the patients of kidney transplant, initially fever, is present in 50% and diarrhea was reported in every fourth patient, and such patients have a faster progression of COVID-19 and higher death rates (Hirsch et al. 2020; Akalin et al. 2020; Boyarsky et al. 2020). The SARS-CoV-2-induced kidney damage is expected to be multifactorial, i.e., directly it can infect the kidney podocytes and proximal tubular cells and based on an ACE2-pathway may lead to acute tubular necrosis, protein leakage in Bowman’s capsule, collapsing glomerulopathy and mitochondrial impairment (Ahmadian et al. 2020). The SARS-CoV-2-driven dysregulation of the immune responses including cytokine storm, macrophage activation syndrome, and lymphopenia can be other causes of the AKI. Lack of oxygen delivery to kidney may cause an ischemic injury.
Conclusion
Global pandemic COVID-19 caused by SARS-CoV-2 is being unraveling to open an array of effects through an extensive research spans on mild respiratory tract illness through brutal ARDS (acute respiratory distress syndrome) distinguished by acute interstitial and alveolar pneumonia, then failure of multiple organs (kidneys, heart, digestive tract, and blood circulatory system), and death. This study revealed the mechanism of pathogenesis in vital organs affected with COVID-19. The main common way is binding the virus to ACE2 receptor, overexpressed in the cells of all mentioned organs, then inducing the inflammatory and coagulant factors and leading disease to severe phase with cytokine storm and DIC, finally increasing risk of mortality. Overall, it is highly recommended that physicians may check the critical biomarkers and carefully perform follow up the molecular signaling pathways to crosslink different organs to make an early diagnostic and good prognostics prior to start irreversible complication picture for infected patients with COVID-19 all over the world.
References
Ahmadian E, Hosseiniyan Khatibi SM, Razi Soofiyani S, Abediazar S, Shoja MM, Ardalan M, and Zununi Vahed S (2020) Covid-19 and kidney injury: Pathophysiology and molecular mechanisms. Rev Med Virol e2176
Akalin E, Azzi Y, Bartash R, Seethamraju H, Parides M, Hemmige V, Ross M, Forest S, Goldstein YD, and Ajaimy M (2020) Covid-19 and kidney transplantation. N Engl J Med
Akhmerov A, Marbán E (2020) COVID-19 and the heart. Circ Res 126:1443–1455
Alberici F, Delbarba E, Manenti C, Econimo L, Valerio F, Pola A, Maffei C, Possenti S, Piva, S. and Latronico, N (2020) Management of patients on dialysis and with kidney transplant during SARS-COV-2 (COVID-19) pandemic in Brescia, Italy. Kidney Int
Aleebrahim-Dehkordi E, Reyhanian A, Saberianpour S, and Hasanpour-Dehkordi A (2020) Acute kidney injury in COVID-19; a review on current knowledge. J Nephropathol 9:e31
Amraei R, Rahimi N (2020) COVID-19, renin-angiotensin system and endothelial dysfunction. Cells 9:1652
Arentz M, Yim E, Klaff L, Lokhandwala S, Riedo FX, Chong M, Lee M (2020) Characteristics and outcomes of 21 critically ill patients with COVID-19 in Washington State. JAMA 323:1612–1614
Asgharpour M, Zare E, Mubarak M, Alirezaei A (2020) COVID-19 and kidney disease: update on epidemiology, clinical manifestations, pathophysiology and management. Journal of the College of Physicians and Surgeons 30:19–25
Astuti I (2020) Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2): an overview of viral structure and host response. Diabetes Metab Syndr 14:407–412
Babapoor-Farrokhran S, Gill D, Walker J, Rasekhi RT, Bozorgnia B, and Amanullah A (2020) Myocardial injury and COVID-19: possible mechanisms. Life Scie 117723
Bangash MN, Patel J, Parekh D (2020) COVID-19 and the liver: little cause for concern. Lancet Gastroenterol Hepatol 5:529–530
Batlle D, Soler MJ, Sparks MA, Hiremath S, South AM, Welling PA, and Swaminathan S (2020) Acute kidney injury in COVID-19: emerging evidence of a distinct pathophysiology. J Am Soc Nephrol.
Beigel JH, Tomashek KM, Dodd LE, Mehta AK, Zingman BS, Kalil AC, Hohmann E, Chu HY, Luetkemeyer A, Kline S, Lopez De Castilla D, Finberg RW, Dierberg K, Tapson V, Hsieh L, Patterson TF, Paredes R, Sweeney DA, Short WR, Touloumi G, Lye DC, Ohmagari N, Oh MD, Ruiz-Palacios GM, Benfield T, Fätkenheuer G, Kortepeter MG, Atmar RL, Creech CB, Lundgren J, Babiker AG, Pett S, Neaton JD, Burgess TH, Bonnett T, Green M, Makowski M, Osinusi A, Nayak S, and Lane HC (2020) Remdesivir for the treatment of Covid-19—final report. N Engl J Med 383:1813–1826
Bellomo R, Kellum JA, Ronco C, Wald R, Martensson J, Maiden M, Bagshaw SM, Glassford NJ, Lankadeva Y, Vaara ST, Schneider A (2017) Acute kidney injury in sepsis. Intensive Care Med 43:816–828
Billet S, Aguilar F, Baudry C, Clauser E (2008) Role of angiotensin II AT1 receptor activation in cardiovascular diseases. Kidney Int 74:1379–1384
Bongiovanni C, Magrini L, Salerno G, Gori CS, Cardelli P, Hur M, Buggi M, and Di Somma S (2015) Serum cystatin C for the diagnosis of acute kidney injury in patients admitted in the emergency department. Disease Markers Article ID 416059, 7 pages.
Boyarsky BJ, Werbel WA, Durand CM, Avery RK, Jackson KR, Kernodle AB, Snyder J, Hirose R, Massie IM, Garonzik-Wang JM (2020) Early national and center-level changes to kidney transplantation in the United States during the COVID-19 epidemic. Am J Transplant 20:3131–3139
Braun F, Lütgehetmann M, Pfefferle S, Wong MN, Carsten A, Lindenmeyer MT, Nörz D, Heinrich F, Meißner K, Wichmann D (2020) SARS-CoV-2 renal tropism associates with acute kidney injury. The Lancet 396:597–598
Chai X, Hu L, Zhang Y, Han W, Lu Z, Ke A, Zhou J, Shi G, Fang N, Fan J, Cai J, Fan J, and Lan F (2020a) Specific ACE2 expression in cholangiocytes may cause liver damage after 2019-nCoV infection. bioRxiv
Chai X, Hu L, Zhang Y, Han W, Lu Z, Ke A, Zhou J, Shi G, Fang N, Fan J, Cai J, Fan J, and Lan, F (2020b) Specific ACE2 expression in cholangiocytes may cause liver damage after 2019-nCoV infection. bioRxiv 2020.02.03.931766
Chakafana G, Mutithu D, Hoevelmann J, Ntusi N, Sliwa K (2020) Interplay of COVID-19 and cardiovascular diseases in Africa: an observational snapshot. Clin Res Cardiol 109:1460–1468
Chan JFW, Zhang AJ, Yuan S, Poon VKM, Chan CCS, Lee ACY, Chan WM, Fan Z, Tsoi HW, Wen L (2020) Simulation of the clinical and pathological manifestations of Coronavirus Disease 2019 (COVID-19) in a golden Syrian hamster model: implications for disease pathogenesis and transmissibility. Clin Infect Dis 71:2428–2446
Chang D, Xu H, Rebaza A, Sharma L, and Dela Cruz CS (2020) Protecting health-care workers from subclinical coronavirus infection. Lancet Respir Med 8:e13
Chen J, Subbarao K (2007) The immunobiology of SARS. Annu Rev Immunol 25:443–472
Chen N, Zhou M, Dong X, Qu J, Gong F, Han Y, Qiu Y, Wang J, Liu Y, Wei Y, Xia JA, Yu T, Zhang X, Zhang L (2020) Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. The Lancet 395:507–513
Chen YLQ, Guo D (2020) Emerging coronaviruses: genome structure, replication, parthenogenesis. J Virol 92:418423
Choi H, Shin EC (2021) Roles of type I and III interferons in COVID-19. Yonsei Med J 62:381
Clerkin KJ, Fried JA, Raikhelkar J, Sayer G, Griffin JM, Masoumi A, Jain SS, Burkhoff D, Kumaraiah D, Rabbani L (2020) COVID-19 and cardiovascular disease. Circulation 141:1648–1655
Coll E, Botey A, Alvarez L, Poch E, Quintó L, Saurina A, Vera M, Piera C, Darnell A (2000) Serum cystatin C as a new marker for noninvasive estimation of glomerular filtration rate and as a marker for early renal impairment. Am J Kidney Dis 36:29–34
Connors JM, Levy JH (2020) COVID-19 and its implications for thrombosis and anticoagulation. Blood 135:2033–2040
Corvol P, Eyries M, and Soubrier F (2004) Peptidyl-dipeptidase A/angiotensin I-converting enzyme. Handbook of proteolytic enzymes 332–346
Da Rosa Mesquita R, Francelino Silva Junior LC, Santos Santana FM, Farias De Oliveira T, Campos Alcântara R, Monteiro Arnozo G, Rodrigues Da Silva Filho E, Galdino Dos Santos AG, Oliveira Da Cunha EJ, Salgueiro De Aquino SH, and Freire De Souza CD (2020) Clinical manifestations of COVID-19 in the general population: systematic review. Wien Klin Wochenschr 1–6
de Mello WC, Danser AH (2000) Angiotensin II and the heart: on the intracrine renin-angiotensin system. Hypertension 35:1183–1188
Diao B, Wang C, Wang R, Feng Z, Tan Y, Wang H, Wang C, Liu L, Liu Y, Liu Y, Wang G, Yuan Z, Ren L, Wu Y, and Chen Y (2020) Human kidney is a target for novel severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) Infection. medRxiv.https://doi.org/10.1101/2020.03.04.20031120
Dobler CC (2019) Biomarkers in respiratory diseases. Eur Respiratory Soc.
Du L, He Y, Zhou Y, Liu S, Zheng B-J, Jiang S (2009) The spike protein of SARS-CoV—a target for vaccine and therapeutic development. Nat Rev Microbiol 7:226–236
Du L, Yang Y, Zhou Y, Lu L, Li F, Jiang S (2017) MERS-CoV spike protein: a key target for antivirals. Expert Opin Ther Targets 21:131–143
Fan C, Lu W, Li K, Ding Y and Wang J (2021) ACE2 expression in kidney and testis may cause kidney and testis infection in COVID-19 patients. Front med 7
Fan Z, Chen L, Li J, Cheng X, Yang J, Tian C, Zhang Y, Huang S, Liu Z, Cheng J (2020) Clinical features of COVID-19-related liver functional abnormality. Clin Gastroenterol Hepatol 18:1561–1566
Feng G, Zheng KI, Yan QQ, Rios RS, Targher G, Byrne CD, Poucke SV, Liu WY, Zheng MH (2020) COVID-19 and liver dysfunction: current insights and emergent therapeutic strategies. J Clin Transl Hepatol 8:18–24
Fogagnolo A, Grasso S, Dres M, Gesualdo L, Murgolo F, Morelli E, Ottaviani I, Marangoni E, Volta CA, and Spadaro S (2021) Focus on renal blood flow in mechanically ventilated patients with SARS-CoV-2: a prospective pilot study. J Clin Monit Comput1–7
Foster MC, Coresh J, Hsu C-Y, Ojo AO, Inker LA (2016) Serum β-trace protein and β-microglobulin as predictors of ESRD, mortality, and cardiovascular disease in adults with CKD in the Chronic Renal Insufficiency Cohort (CRIC) study. Original Investigation Pathogenesis and Treatment of Kidney Disease 68:68–76
Fuchs M, Sanyal AJ (2008) Sepsis and cholestasis. Clin Liver Dis 12:151–172
Gabarre P, Dumas G, Dupont T, Darmon M, Azoulay E, Zafrani L (2020) Acute kidney injury in critically ill patients with COVID-19. Intensive Care Med 46:1339–1348
Gagliardi I, Patella G, Michael A, Serra R, Provenzano M, Andreucci M (2020) COVID-19 and the kidney: from epidemiology to clinical practice J Clin Med 9 https://doi.org/10.3390/jcm9082506
Gautret P, Lagier JC, Parola P, Hoang VT, Meddeb I, Mailhe M, Doudier B, Courjon J, Giordanengo V, Vieira VE, Tissot Dupont H, Honoré S, Colson P, Chabrière E, La Scola B, Rolain JM, Brouqui P and Raoult D (2020) Hydroxychloroquine and azithromycin as a treatment of COVID-19: results of an open-label non-randomized clinical trial. Int J Antimicrob Agents 56:105949
Ghobadi H, Kalan ME, Mohammad-Shahi J, Taleb ZB, Kalan AE, and Fazkzadeh M (2020) COVID-19 and acute kidney injury; a case report.
Goldman JD, Lye DCB, Hui DS, Marks KM, Bruno R, Montejano R, Spinner CD, Galli M, Ahn M-Y, Nahass RG, Chen Y-S, Sengupta D, Hyland RH, Osinusi AO, Cao H, Blair C, Wei X, Gaggar A, Brainard DM, Towner WJ, Muñoz J, Mullane KM, Marty FM, Tashima KT, Diaz G, Subramanian A (2020) Remdesivir for 5 or 10 days in patients with severe Covid-19. N Engl J Med 383:1827–1837
Gorin A, Stewart PA (1979) Differential permeability of endothelial and epithelial barriers to albumin flux. J Appl Physiol 47:1315–1324
Gralinski LE, Bankhead A, Jeng S, Menachery VD, Proll S, Belisle SE, Matzke M, Webb-Robertsonj BJM, Luna ML, and Shukla AK (2013) Mechanisms of severe acute respiratory syndrome coronavirus-induced acute lung injury. MBio 4
Grasselli G, Zangrillo A, Zanella A, Antonelli M, Cabrini L, Castelli A, Cereda D, Coluccello A, Foti G, Fumagalli R, Iotti G, Latronico N, Lorini L, Merler S, Natalini G, Piatti A, Ranieri MV, Scandroglio AM, Storti E, Cecconi M, Pesenti A (2020) Baseline Characteristics and Outcomes of 1591 Patients Infected With SARS-CoV-2 Admitted to ICUs of the Lombardy Region, Italy. JAMA 323:1574–1581
Grein J, Ohmagari N, Shin D, Diaz G, Asperges E, Castagna A, Feldt T, Green G, Green ML, Lescure F-X, Nicastri E, Oda R, Yo K, Quiros-Roldan E, Studemeister A, Redinski J, Ahmed S, Bernett J, Chelliah D, Chen D, Chihara S, Cohen SH, Cunningham J, D’Arminio Monforte A, Ismail S, Kato H, Lapadula G, L’Her E, Maeno T, Majumder S, Massari M, Mora-Rillo M, Mutoh Y, Nguyen D, Verweij E, Zoufaly A, Osinus AO, Dezure A, Zhao Y, Zhong I, Chokkalingam A, Elboudwarej E, Telep L, Timbs L, Henne I, Sellers S, Cao H, Tan SK, Winterbourne L, Desai P, Mera R, Gaggar A, Myers RP, Brainard DM, Childs R, and Flanigan T (2020) Compassionate use of remdesivir for patients with severe Covid-19. N Engl J Med 382:2327–2336
Grifoni E, Valoriani A, Cei F, Lamanna R, Gelli AMG, Ciambotti B, Vannucchi V, Moroni F, Pelagatti L, and Tarquini R (2020) Interleukin-6 as prognosticator in patients with COVID-19: IL-6 and Covid-19. J Infect.
Guan GW, Gao L, Wang JW, Wen XJ, Mao TH, Peng SW, Zhang T, Chen XM, and Lu FM (2020a) [Exploring the mechanism of liver enzyme abnormalities in patients with novel coronavirus-infected pneumonia]. Zhonghua gan zang bing za zhi = Zhonghua ganzangbing zazhi = Chinese journal of hepatology, 28:100–106
Guan WJ, Ni ZY, Hu Y, Liang WH, Ou CQ, He JX, Liu L, Shan H, Lei CL, Hui DSC, Du B, Li LJ, Zeng G, Yuen KY, Chen RC, Tang CL, Wang T, Chen PY, Xiang J, Li SY, Wang JL, Liang ZJ, Peng YX, Wei L, Liu Y, Hu YH, Peng P, Wang JM, Liu JY, Chen Z, Li G, Zheng ZJ, Qiu SQ, Luo J, Ye CJ, Zhu SY, Zhong NS (2020) Clinical characteristics of coronavirus disease 2019 in China. N Engl J Med 382:1708–1720
Guzik TJ, Mohiddin SA, Dimarco A, Patel V, Savvatis K, Marelli-Berg FM, Madhur MS, Tomaszewski M, Maffia P, and D’acuisto F (2020) COVID-19 and the cardiovascular system: implications for risk assessment, diagnosis, and treatment options. Cardiovasc Res
Hamming I, Timens W, Bulthuis M, Lely A, Navis GV, van Goor H (2004) Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. The Journal of Pathology: A Journal of the Pathological Society of Great Britain and Ireland 203:631–637
Han J, Zhu L, Wang Y, Zeng Z, Zhang S (2020) Resumption of daily services in a gastroenterology department in Guangzhou, China, in the wake of COVID-19. Lancet Gastroenterol Hepatol 5:645–646
Hancock AS, Stairiker CJ, Boesteanu AC, Monzón-Casanova E, Lukasiak S, Mueller YM, Stubbs AP, Garcia-Sastre A, Turner M, and Katsikis PD (2018) Transcriptome analysis of infected and bystander type 2 alveolar epithelial cells during influenza A virus infection reveals in vivo Wnt pathway downregulation. Virol J 92
Hassanpour M, Rezaie J, Nouri M, and Panahi Y (2020) The role of extracellular vesicles in COVID-19 virus infection. Infect Genet Evol 104422
Hirsch JS, Ng JH, Ross DW, Sharma P, Shah HH, Barnett RL, Hazzan AD, Fishbane S, Jhaveri KD, and Abate M (202) Acute kidney injury in patients hospitalized with COVID-19. Kidney Int
Hoffmann M, Kleine-Weber H, Krüger N, Müller M, Drosten C and Pöhlmann S (2020a) The novel coronavirus 2019 (2019-nCoV) uses the SARS-coronavirus receptor ACE2 and the cellular protease TMPRSS2 for entry into target cells. bioRxiv 2020.01.31.929042
Hoffmann M, Kleine-Weber H, Schroedes S, Krüger N, Herrle T, Erichsen S, Schiergens TS, Herrler G, Wu NH, and Nitsche A (2020b) SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181:271–280. e8
Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, Schiergens TS, Herrler G, Wu NH, and Nitsche A (2020c) SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell
Hu LL, Wang WJ, Zhu QJ, Yang L (2020) Novel coronavirus pneumonia-related liver injury: etiological analysis and treatment strategy. Zhonghua Gan Zang Bing Za Zhi 28:97–99
Hydroxychloroquine (2012) LiverTox: clinical and research information on drug-induced liver injury. Bethesda MD.
Iba T, Levy JH, Levi M, Connors JM, Thachil J (2020) Coagulopathy of Coronavirus Disease 2019. Crit Care Med 48:1358–1364
Ichimura T, Mori Y, Aschauer P, Das KMP, Padera RF, Wein A, Nasr ML, and Bonventre JV (2020) KIM-1/TIM-1 is a Receptor for SARS-CoV-2 in Lung and Kidney. medRxiv.
IDELL, S. (2003) Coagulation, fibrinolysis, and fibrin deposition in acute lung injury. Crit Care Med 31:S213–S220
Inker LA, Tighiouart H, Coresh J, Foster MC, Anderson AH, Beck GJ, Contreras G, Greene T, Karger A, Kusek JW, Lash J, Lewis J, Schelling JR, Navaneethan SD, Sondheimer J, Shafi T, and Levey AS (2016) GFR Estimation Using β-Trace Protein and β2-Microglobulin in CKD. Am J Kidney Dis. January 67:40–48
Izzedine H, and Jhaveri KD (2020a) Acute kidney injury in patients with COVID-19: an update on the pathophysiology. Nephrol Dial Transplant gfaa184
Izzedine H, and Jhaveri KD (2020b) Acute kidney injury in patients with COVID-19: an update on the pathophysiology. Nephrol Dial Transplant.
Jain KK (2010) The handbook of biomarkers, Springer.
Jain U (2020) Effect of COVID-19 on the Organs. Cureus 12
Jesenak M, Brndiarova M, Urbancikova I, Rennerova Z, Vojtkova J, Bobcakova A, Ostro R, Banovcin P (2020) Immune parameters and COVID-19 infection-associations with clinical severity and diseases prognosis. Front Cell Infect Microbiol 10:364
Jothimani D, Venugopal R, Abedin MF, Kaliamoorthy I, Rela M (2020) COVID-19 and the liver. J Hepatol 73:1231–1240
Kovalic A, Huang G, Thuluvath PJ and Satapathy, SK (2021) Elevated liver biochemistries in hospitalized Chinese patients with severe COVID-19: systematic review and meta-analysis. J Hepatol n/a 73:(4):1521-1530. https://doi.org/10.1002/hep.3147210.1002/hep.31472
Kujawski SA, Wong KK, Collins JP, Epstein L, Killerby ME, Midgley CM, Abedi GR, Ahmed NS, Almendares O, Alvarez FN, Anderson KN, Balter S, Barry V, Bartlett K, Beer K, Ben-aderet MA, Benowitz I, Biggs H, Binder AM, Black SR, Bonin B, Brown CM, Bruce H, Bryant-Genevier J, Budd A, Buell D, Bystritsky R, Cates J, Charles EM, Chatham-Stephens K, Chea N, Chiou H, Christiansen D, Chu V, Cody S, Cohen M, Conners E, Curns A, Dasari V, Dawson Pesalvo T, Diaz G, Donahue M, Donovan S, Duca LM, Erickson K, Esona MD, Evans S, Falk J, Feldstein LR, Fenstersheib M, Fischer M, Fisher R, Foo C, Fricchione MJ, Friedman O, Fry AM, Galang, RR, Garcia MM, Gerber SI, Gerrard G, Ghinai I, Gounder P, Grein J, Grigg C, Gunzenhauser JD, Gutkin GI, Haddix M, Hall AJ, Han G, Harcourt J, Harriman K, Haupt T, Haynes A, Holshue M, Hoover C, Hunter JC, Jacobs MW, Jarashow C, Jhung MA, Joshi K, Kamali T, Kamili S, Kim L, Kim M, King J, Kirking HL, Kita-Yarbro A, Klos R, Kobayashi M, Kocharian A, Komatsu KK, Koppaka R, Layden JE, Li, Y, Lindquist S, Indstrom S, Link-Gelles R, Lively J, Livingston M, et al. (2020) First 12 patients with coronavirus disease 2019 (COVID-19) in the United States. medRxiv 2020:03.09.20032896
Kwon Y, Nukala SB, Srivastava S, Miyamoto H, Ismail NI, Ong SB, Lee WH and Ong SG (2020) Exosomes facilitate transmission of SARS-CoV-2 genome into human induced pluripotent stem cell-derived cardiomyocytes. BioRxiv.
Legrand M, Bell S, Forni L, Joannidis M, Koyner JL, Liu K, and Cantaluppi V (2021) Pathophysiology of COVID-19-associated acute kidney injury. Nat Rev Nephrol.
Lei P, Zhang L, Han P, Zheng C, Tong Q, Shang H, Yang F, Hu Y, Li X, Song Y (2020a) Liver injury in patients with COVID-19: clinical profiles, CT findings, the correlation of the severity with liver injury. Hep Intl 14:733–742
Lei P, Zhang L, Han P, Zheng C, Tong Q, Shang H, Yang F, Hu Y, Li X, Song Y (2020b) Liver injury in patients with COVID-19: clinical profiles, CT findings, the correlation of the severity with liver injury. Hepatol Int 14:733–742
Leth-Larsen R, Zhong F, Chow VT, Holmskov U, Lu J (2007) The SARS coronavirus spike glycoprotein is selectively recognized by lung surfactant protein D and activates macrophages. Immunobiology 212:201–211
Leung JM, Yang CX, Tam A, Shaipanich T, Hackett TL, Singhera GK, Dorscheid DR and Sin DD (2020) ACE-2 expression in the small airway epithelia of smokers and COPD patients: implications for COVID-19. Eur Respir J 55
LI, F. (2016) Structure, function, and evolution of coronavirus spike proteins. Annual Review of Virology 3:237–261
Li P, Wu W, Zhang T, Wang Z, Li J, Zhu M, Liang Y, You W, Li K, Ding R (2021) Implications of cardiac markers in risk-stratification and management for COVID-19 patients. Crit Care 25:1–14
Li X, Ma X (2020) Acute respiratory failure in COVID-19: is it “typical” ARDS? Crit Care 24:1–5
Li X, Pan X, Li Y, An N, Xing Y, Yang F, Tian L, Sun J, Gao Y, Shang H (2020a) Cardiac injury associated with severe disease or ICU admission and death in hospitalized patients with COVID-19: a meta-analysis and systematic review. Crit Care 24:1–16
Li Z, Wu M, Yao J, Guo J, Liao X, Song S, Li J, Duan G, Zhou Y, and Wu X (2020b) Caution on kidney dysfunctions of COVID-19 patients.
LILLICRAP, D. (2020) Disseminated intravascular coagulation in patients with 2019-nCoV pneumonia. J Thromb Haemost 18:786
Lim YX, Ng YL, Tam JP, Liu DX (2016) Human coronaviruses: a review of virus–host interactions. Diseases 4:26
Ling Y, Xu SB, Lin YX, Tian D, Zhu ZQ, Dai FH, Wu F, Song ZG, Huang W, Chen J, Hu BJ, Wang S, Mao EQ, Zhu L, Zhang WH, and Lu HZ (2020) Persistence and clearance of viral RNA in 2019 novel coronavirus disease rehabilitation patients. Chin.Med J 133
Lippi G, de Oliveira MHS, Henry BM (2021) Chronic liver disease is not associated with severity or mortality in Coronavirus disease 2019 (COVID-19): a pooled analysis. Eur J Gastroenterol Hepatol 33:114–115
Lippi G, Plebani M (2020) The critical role of laboratory medicine during coronavirus disease 2019 (COVID-19) and other viral outbreaks. Clin Chem Lab Med 58:1063–1069
Long B, Brady WJ, Koyfman A, and Gottlieb M (2020) Cardiovascular complications in COVID-19. The Am J Emerg Med.
Madjid M, Safavi-Naeini P, Solomon SD and Vardeny O (2020) Potential effects of coronaviruses on the cardiovascular system: a review. JAMA Cardiol.
Mason RJ (2020) Pathogenesis of COVID-19 from a cell biology perspective. Eur Respiratory Soc.
Meredith Wadman J, Jocelyn K, and Catherine M (2020) How does coronavirus kill? Clinicians trace a ferocious rampage through the body, from brain to toes. Science Mag.
Mokhtari T, Hassani F, Ghaffari N, Ebrahimi B, Yarahmadi A, and Hassanzadeh G (2020) COVID-19 and multiorgan failure: a narrative review on potential mechanisms. J Mol Histol 1–16
Mossel EC, Wang J, Jeffers S, Edeen KE, Wang S, Cosgrove GP, Funk CJ, Manzer R, Miura TA, Pearson LD (2008) SARS-CoV replicates in primary human alveolar type II cell cultures but not in type I-like cells. Virology 372:127–135
Nishiga M, Wang DW, Han Y, Lewis DB, Wu JC (2020) COVID-19 and cardiovascular disease: from basic mechanisms to clinical perspectives. Nat Rev Cardiol 17:543–558
O’Shea PM, Lee GR, Griffin TP, Tormey V, Hayat A, Costelloe SJ, Griffin DG, Srinivasan S, O’Kane M, Burke CM, Faul J, Thompson CJ, Curley G, Tormey WP (2020) COVID-19 in adults: test menu for hospital blood science laboratories. Ir J Med Sci 189:1147–1152
Pan XW, Xu D, Zhang H, Zhou W, Wang LH, Cui XG (2020) Identification of a potential mechanism of acute kidney injury during the COVID-19 outbreak: a study based on single-cell transcriptome analysis. Intensive Care Med 46:1114–1116
Papadopoulos N, Vasileiadi S, Deutsch M (2020) COVID-19 and liver injury: where do we stand? Ann Gastroenterol 33:459–464
Patel VB, Zhong J-C, Grant MB, Oudit GY (2016) Role of the ACE2/angiotensin 1–7 axis of the renin–angiotensin system in heart failure. Circ Res 118:1313–1326
Patil M, Singh S, Henderson J and Krishnamurthy P (2020) Mechanisms of COVID‐19‐induced cardiovascular disease: Is sepsis or exosome the missing link? J Cell Physiol.
Pedersen SF, and Ho YC, (2020) SARS-CoV-2: a storm is raging. J Clin Invest 130
Perlman S, Netland J (2009) Coronaviruses post-SARS: update on replication and pathogenesis. Nat Rev Microbiol 7:439–450
Pocsfalvi G, Mammadova R, Juarez APR, Bokka R, Trepiccione F and Capasso G (2020) COVID-19 and extracellular vesicles: an intriguing interplay. Kidney Blood Press Res 1–10
Puelles VG, Lütgehetmann M, Lindenmeyer MT, Sperhake JP, Wong MN, Allweiss L, Chilla S, Heinemann A, Wanner N and Liu S (2020) Multiorgan and renal tropism of SARS-CoV-2. N Engl J Med.
Qian JY, Wang B, Liu BC (2020) Acute kidney injury in the 2019 novel coronavirus disease. Kidney Dis. https://doi.org/10.1159/000509086
Reyfman PA, Walter JM, Joshi N, Anekalla KR, McQuattie-Pimentel AC, Chiu S, Fernandez R, Akbarpour M, Chen C-I, Ren Z (2019) Single-cell transcriptomic analysis of human lung provides insights into the pathobiology of pulmonary fibrosis. Am J Respir Crit Care Med 199:1517–1536
Richardson S, Hirsch JS, Narasimhan M, Crawford JM, McGinn T, Davidson KW and Consortium ATNCR (2020) Presenting characteristics, comorbidities, and outcomes among 5700 patients hospitalized with COVID-19 in the New York City area. JAMA 323:2052–2059
Ronco C, and Reis T (2020) Kidney involvement in COVID-19 and rationale for extracorporeal therapies. Nat Rev Nephrol 1–3
Ronco C, Reis T, Husain-Syed F (2020) Management of acute kidney injury in patients with COVID-19. Lancet Respir Med 8:738–742
Sahin TT, Akbulut S, Yilmaz S (2020) COVID-19 pandemic: Its impact on liver disease and liver transplantation. World J Gastroenterol 26:2987
Santos RA, Ferreira AJ, Verano-Braga T, Bader M (2013) Angiotensin-converting enzyme 2, angiotensin-(1–7) and Mas: new players of the renin-angiotensin system. J Endocrinol 216:R1-17
Schoeman D, Fielding BC (2019) Coronavirus envelope protein: current knowledge. Virology Journal 16:1–22
Shang J, Wan Y, Luo C, Ye G, Geng Q, Auerbach A, Li F (2020) Cell entry mechanisms of SARS-CoV-2. Proc Natl Acad Sci 117:11727–11734
Shereen MA, Khan S, Kazmi A, Bashir N, Siddique R (2020) COVID-19 infection: origin, transmission, and characteristics of human coronaviruses. J Adv Res 24:91–98
Sims AC, Baric RS, Yount B, Burkett SE, Collins PL, Pickles RJ (2005) Severe acute respiratory syndrome coronavirus infection of human ciliated airway epithelia: role of ciliated cells in viral spread in the conducting airways of the lungs. J Virol 79:15511–15524
Singh S, and Khan A (2020) Clinical characteristics and outcomes of coronavirus disease 2019 among patients with preexisting liver disease in the United States: a multicenter research network study. Gastroenterology, 159, 768–771 e3
Solomon MD, Mcnulty EJ, Rana JS, Leong TK, Lee C, Sung SH, Ambrosy AP, Sidney S, Go AS (2020) The Covid-19 pandemic and the incidence of acute myocardial infarction. N Engl J Med.
Stefanini GG, Chiarito M, Ferrante G, Cannata F, Azzolini E, Viggiani G, de Marco A, Briani M, Bocciolone M, Bragato R (2020) Early detection of elevated cardiac biomarkers to optimise risk stratification in patients with COVID-19. Heart 106:1512–1518
Strimbu K, Tavel JA (2010) What are biomarkers? Curr Opin HIV AIDS 5:463
Su H, Yang M, Wan C, Yi L-X, Tang F, Zhu H-Y, Yi F, Yang H-C, Fogo AB, Nie X, Zhang C (2020) Renal histopathological analysis of 26 postmorten findings of patients with COVID-19 in China. Kidney Int. 2020. doi: 10.1016/j.kint.2020.04.003. Kidney Int 98:219–227
Sun D-Q, Wang T-Y, Zheng KI, Targher G, Byrne CD, Chen Y-P, Zheng M-H (2020) Subclinical acute kidney injury in COVID-19 patients: a retrospective cohort study. Nephron 144:347–350
Sungnak W, Huang N, Bécavin C, Berg M, Queen R, Litvinukova M, Talavera-López C, Maatz H, Reichart D, Sampaziotis F (2020) SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat Med 26:681–687
Tajbakhsh A, Gheibi Hayat SM, Taghizadeh H, Akbari A, Inabadi M, Savardashtaki A, Johnston TP, and Sahebkar A (2020) COVID-19 and cardiac injury: clinical manifestations, biomarkers, mechanisms, diagnosis, treatment, and follow up. Expert Rev Anti Infect 1–13
Tang N, Li D, Wang X, Sun Z (2020) Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J Thromb Haemost 18:844–847
Tang NLS, Chan PKS, Wong CK, To KF, Wu AKL, Sung YM, Hui DSC, Sung JJY, Lam CWK (2005) Early enhanced expression of interferon-inducible protein-10 (CXCL-10) and other chemokines predicts adverse outcome in severe acute respiratory syndrome. Clin Chem 51:2333–2340
Tapper EB, Sengupta N, Bonder A (2015) The incidence and outcomes of ischemic hepatitis: a systematic review with meta-analysis. Am J Med 128:1314–1321
Unudurthi SD, Luthra P, Bose RJ, Mccarthy J and Kontaridis MI (2020) Cardiac inflammation in COVID-19: Lessons from heart failure. Life Sci 118482
Vaduganathan M, Vardeny O, Michel T, McMurray JJ, Pfeffer MA, Solomon SD (2020) Renin–angiotensin–aldosterone system inhibitors in patients with Covid-19. N Engl J Med 382:1653–1659
van Deursen V, Damman K, Hillege H, van Beek A, van Veldhuisen D, Voors A (2010) Abnormal liver function in relation to hemodynamic profile in heart failure patients. J Cardiac Fail 16:84–90
Verdecchia P, Cavallini C, Spanevello A and Angeli F (2020) The pivotal link between ACE2 deficiency and SARS-CoV-2 infection. Eur J Intern Med.
Wan Y, Shang J, Graham R, Baric RS and Li F (2020) Receptor recognition by the novel coronavirus from Wuhan: an analysis based on decade-long structural studies of SARS coronavirus. Virol J 94
Wang D, Hu B, Hu C, Zhu F, Liu X, Zhang J, Wang B, Xiang H, Cheng Z, Xiong Y (2020a) Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus–infected pneumonia in Wuhan, China. JAMA 323:1061–1069
Wang J, Nikrad MP, Phang T, Gao B, Alford T, Ito Y, Edeen K, Travanty EA, Kosmider B, Hartshorn K (2011) Innate immune response to influenza A virus in differentiated human alveolar type II cells. Am J Respir Cell Mol Biol 45:582–591
Wang M, Xiong H, Chen H, Li Q, and Ruan XZ (2020b) Renal injury by SARS-CoV-2 infection: a systematic review. Am J Kidney Dis 1–11
Wang T, Hu M, Chen X, Fu Y, Lei C, Dong H, Zhou Y, Jia H, Chen X and Yan J (2020c) Caution on kidney dysfunctions of 2019-nCoV patients. MedRxiv.
Wang X, Fang X, Cai Z, Wu X, Gao X, Min J, Wang F (2020d) Comorbid chronic diseases and acute organ injuries are strongly correlated with disease severity and mortality among COVID-19 patients: a systemic review and meta-analysis. Research 2020:2402961
Wang Y, Wang Z, Tse G, Zhang L, Wan EY, Guo Y, Lip GY, Li G, Lu Z, Liu T (2020e) Cardiac arrhythmias in patients with COVID-19. Journal of Arrhythmia 36:827–836
Weisberg IS, Jacobson IM (2011) Cardiovascular diseases and the liver. Clin Liver Dis 15:1–20
Wu Z, McGoogan JM (2020) Characteristics of and important lessons from the coronavirus disease 2019 (COVID-19) outbreak in China: summary of a report of 72 314 cases from the Chinese Center for Disease Control and Prevention. JAMA 323:1239–1242
Xiao D, Chen Z, Wu S, Huang K, Xu J, Yang L, Xu Y, Zhang X, Bai C, Kang J, Ran P, Shen H, Wen F, Yao W, Sun T, Shan G, Yang T, Lin Y, Zhu J, Wang R, Shi Z, Zhao J, Ye X, Song Y, Wang Q, Hou G, Zhou Y, Li W, Ding L, Wang H, Chen Y, Guo Y, Xiao F, Lu Y, Peng X, Zhang B, Wang Z, Zhang H, Bu X, Zhang X, An L, Zhang S, Cao Z, Zhan Q, Yang Y, Liang L, Liu Z, Zhang X, Cheng A, Cao B, Dai H, Chung KF, He J, Wang C (2020) Prevalence and risk factors of small airway dysfunction, and association with smoking, in China: findings from a national cross-sectional study. Lancet Respir Med 8:1081–1093
Xu Z, Shi L, Wang Y, Zhang J, Huang L, Zhang C, Liu S, Zhao P, Liu H, Zhu L, Tai Y, Bai C, Gao T, Song J, Xia P, Dong J, Zhao J, Wang FS (2020) Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir Med 8:420–422
Yang Y, Peng F, Wang R, Guan K, Jiang T, Xu G, Sun J and Chang C (2020) The deadly coronaviruses: the 2003 SARS pandemic and the 2020 novel coronavirus epidemic in China. J Autoimmun 109:102434
Ye M, Wysocki J, William J, Soler MJ, Cokic I, Batlle D (2006) Glomerular localization and expression of angiotensinconverting enzyme 2 and angiotensin-converting enzyme: implications for albuminuria in diabetes. J Am Soc Nephrol 17:3067–3075
Ye Q, Wang B, and Mao J (2020) Cytokine storm in COVID-19 and treatment. J Infect.
Yeo C, Kaushal S, Yeo D (2020) Enteric involvement of coronaviruses: is faecal-oral transmission of SARS-CoV-2 possible? Lancet Gastroenterol Hepatol 5:335–337
Yip MS, Leung NHL, Cheung CY, Li PH, Lee HHY, Daëron M, Peiris JSM, Bruzzone R, Jaume M (2014) Antibody-dependent infection of human macrophages by severe acute respiratory syndrome coronavirus. Virology Journal 11:1–11
Zaim S, Chong JH, Sankaranarayanan V, and Harky A (2020) COVID-19 and multi-organ response. Curr Probl Cardiol 100618
Zhang C, Shi L, Wang F-S (2020a) Liver injury in COVID-19: management and challenges. The Lancet Gastroenterology & Hepatology 5:428–430
Zhang C, Shi L, Wang FS (2020b) Liver injury in COVID-19: management and challenges. Lancet Gastroenterol Hepatol 5:428–430
Zhang S, Li L, Shen A, Chen Y, Qi Z (2020c) Rational use of tocilizumab in the treatment of novel coronavirus pneumonia. Clin Drug Investig 40:511–518
Zhang W, Zhao Y, Zhang F, Wang Q, Li T, Liu Z, Wang J, Qin Y, Zhang X, Yan X, Zeng X and Zhang S (2020d) The use of anti-inflammatory drugs in the treatment of people with severe coronavirus disease 2019 (COVID-19): the perspectives of clinical immunologists from China. J Clin Immunol 214:108393–108393
Zhang Y, Xiao M, Zhang S, Xia P, Cao W, Jiang W, Chen H, Ding X, Zhao H and Zhang H (2020e) Coagulopathy and antiphospholipid antibodies in patients with Covid-19. New Eng J Med 382:e38
Zheng YY, Ma YT, Zhang JY, Xie X (2020) COVID-19 and the cardiovascular system. Nat Rev Cardiol 17:259–260
Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z, Xiang J, Wang Y, Song B, Gu X (2020a) Clinical course and risk factors for mortality of adult in patients with COVID-19 in Wuhan, China: a retrospective cohort study. The Lancet 395:1054–1062
Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z, Xiang J, Wang Y, Song B, Gu X, Guan L, Wei Y, Li H, Wu X, Xu J, Tu S, Zhang Y, Chen H, Cao B (2020b) Clinical course and risk factors for mortality of adult in patients with COVID-19 in Wuhan, China: a retrospective cohort study. The Lancet 395:1054–1062
Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, Si HR, Zhu Y, Li B, Huang CL, Chen HD, Chen J, Luo Y, Guo H, Jiang RD, Liu MQ, Chen Y, Shen XR, Wang X, Zheng XS, Zhao K, Chen QJ, Deng F, Liu LL, Yan B, Zhan FX, Wang YY, Xiao GF, Shi ZL (2020) A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579:270–273
Zhou Z, Zhao N, Shu Y, Han S, Chen B, Shu X (2020d) Effect of gastrointestinal symptoms in patients with COVID-19. Gastroenterology 158:2294–2297
Zhu H, Rhee J-W, Cheng P, Waliany S, Chang A, Witteles RM, Maecker H, Davis MM, Nguyen PK, Wu SM (2020) Cardiovascular complications in patients with COVID-19: consequences of viral toxicities and host immune response. Curr Cardiol Rep 22:1–9
Zou X, Chen K, Zou J, Han P, Hao J, Han Z (2020) The single cell RNA-seq data analysis on the receptor ACE2 expression reveals the potential risk of different human organs vulnerable to Wuhan 2019-nCoV infection. Front Med. https://doi.org/10.1007/s11684-020-0754-0
Acknowledgements
The authors thank the Department of Biochemistry, Faculty of Medicine, Urmia Medical Sciences University (UMSU), for all support supplied. This is a review article and has not received any fund so there is no require ethical issues.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethical approval
None.
Informed consent
None.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Rasmi, Y., Babaei, G., Nisar, M.F. et al. Revealed pathophysiological mechanisms of crosslinking interaction of affected vital organs in COVID-19. Comp Clin Pathol 30, 1005–1021 (2021). https://doi.org/10.1007/s00580-021-03269-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00580-021-03269-2