
Abstract
Biallelic SHQ1 variant-related neurodevelopmental disorder is extremely rare. To date, only six affected individuals, from four families, have been reported. Here, we report eight individuals, from seven unrelated families, who exhibited neurodevelopmental disorder and/or dystonia, received whole-genome sequencing, and had inherited biallelic SHQ1 variants. The median age at disease onset was 3.5 months old. All eight individuals exhibited normal eye contact, profound hypotonia, paroxysmal dystonia, and brisk deep tendon reflexes at the first visit. Varying degrees of autonomic dysfunction were observed. One individual had cerebellar atrophy at the initial neuroimaging study, however, three individuals showed cerebellar atrophy at follow-up. Seven individuals who underwent cerebral spinal fluid analysis all had a low level of homovanillic acid in neurotransmitter metabolites. Four individuals who received 99mTc-TRODAT-1 scan had moderate to severe decreased uptake of dopamine in the striatum. Four novel SHQ1 variants in 16 alleles were identified: 9 alleles (56%) were c.997C > G (p.L333V); 4 (25%) were c.195T > A (p.Y65X); 2 (13%) were c.812T > A (p.V271E); and 1 (6%) was c.146T > C (p.L49S). The four novel SHQ1 variants transfected into human SH-SY5Y neuronal cells resulted in a retardation in neuronal migration, suggestive of SHQ1 variant correlated with neurodevelopmental disorders. During the follow-up period, five individuals still exhibited hypotonia and paroxysmal dystonia; two showed dystonia; and one had hypotonia only. The complex interactions among movement disorders, dopaminergic pathways, and the neuroanatomic circuit needs further study to clarify the roles of the SHQ1 gene and protein in neurodevelopment.




Similar content being viewed by others
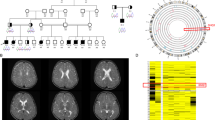


Data availability
All data generated or analyzed during this study are included in this published article.
References
Andermann F, Ohtahara S, Andermann E, Camfield P, Kobayashi K (1994) Infantile hypotonia and paroxysmal dystonia: a variant of alternating hemiplegia of childhood? Mov Disord 9:227–229. https://doi.org/10.1002/mds.870090219
Ayala R, Shu T, Tsai LH (2007) Trekking across the brain: the journey of neuronal migration. Cell 128:29–43. https://doi.org/10.1016/j.cell.2006.12.021
Beinvogl BC, Rosman NP, Baumer FM, Rodan LH, Forster CS, Kwon AH, Berry GT (2016) A 10-month-old with intermittent hypotonia and paralysis. Pediatrics 138:e20151896. https://doi.org/10.1542/peds.2015-1896
Bajaj S, Bagley JA, Sommer C, Vertesy A, Wong SN, Krenn V, Lévi-Strauss J, Knoblich JA (2021) Neurotransmitter signaling regulates distinct phases of multimodal human interneuron migration. EMBO J 40:e108714. https://doi.org/10.15252/embj.2021108714
Bernhermer H (1964) Distribution of homovanillic acid in the human brain. Nature 204:587–588. https://doi.org/10.1038/204587b0
Bizarro J, Meier UT (2017) Inherited SHQ1 mutations impair interaction with NAP57/dyskerin, a major target in dyskeratosis congenita. Mol Genet Genomic Med 5:805–808. https://doi.org/10.1002/mgg3.314
Brignani S, Pasterkamp RJ (2017) Neuronal subset-specific migration and axonal wiring mechanisms in the developing midbrain dopamine system. Front Neuroanat 11:55. https://doi.org/10.3389/fnana.2017.00055
Charlesworth G, Bhatia KP, Wood NW (2013) The genetics of dystonia: new twists in an old tale. Brain 136:2017–2037. https://doi.org/10.1093/brain/awt138
Chi CS, Lee HF, Tsai CR (2012) Tyrosine hydroxylase deficiency in Taiwanese infants. Pediatr Neurol 46:77–82. https://doi.org/10.1016/j.pediatrneurol.2011.11.012
Furukawa Y, Kish S (2017) Tyrosine hydroxylase deficiency. 2008 Feb 8 [Updated 2017 May 11]. In: Adam MP, Everman DB, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A (eds) GeneReviews ® [Internet]. University of Washington, Seattle, Seattle (WA), 1993–2022
Glousker G, Touzot F, Revy P, Tzfati Y, Savage SA (2015) Unraveling the pathogenesis of Hoyeraal-Hreidarsson syndrome, a complex telomere biology disorder. Brit J Haematol 170:457–471. https://doi.org/10.1111/bjh.13442
Gorodetsky C, Fasano A (2022) Approach of the treatment of pediatric dystonia. Dyst 1:10287. https://doi.org/10.3389/dyst.2022.10287
Gressens P (2000) Mechanisms and disturbances of neuronal migration. Pediatr Res 48:725–729. https://doi.org/10.1203/00006450-200012000-00004
Grozdanov PN, Roy S, Kittur N, Meier UT (2009a) SHQ1 is required prior to NAF1 for assembly of H/ACA small nucleolar and telomerase RNPs. RNA 15:1188–1197. https://doi.org/10.1261/rna.1532109
Grozdanov PN, Fernandez-Fuentes N, Fiser A, Meier UT (2009b) Pathogenic NAP57 mutations decrease ribonucleoprotein assembly in dyskeratosis congenita. Hum Mol Genet 18:4546–4551. https://doi.org/10.1093/hmg/ddp416
Indelicato E, Boesch S, Baumgartner M, Plecko B, Winkelmann J, Zech M (2022) Confirmation of a causal role for SHQ1 variants in early infantile-onset recessive dystonia. Mov Disord. https://doi.org/10.1002/mds.29281
Jernigan TL, Baaré WFC, Stiles J, Madsen KS (2011) Postnatal brain development: structural imaging of dynamic neurodevelopmental processes. Prog Brain Res 189:77–92. https://doi.org/10.1016/B978-0-444-53884-0.00019-1
Kim MK, Lee SJ, Won CK (2020) Dopaminergic neuronal development in the embryonic mesencephalon of mouse. Korean J Vet Res 60:203–207. https://doi.org/10.14405/kjvr.2020.60.4.203
Kiss T, Fayet-Lebaron E, Jády BE (2010) Box H/ACA small ribonucleoproteins. Mol Cell 37:597–606. https://doi.org/10.1016/j.molcel.2010.01.032
Kong CK, Ko CH, Tong SF, Lam CW (2001) Atypical presentation of dopa-responsive dystonia: generalized hypotonia and proximal weakness. Neurology 57:1121–1124. https://doi.org/10.1212/WNL.57.6.1121
Kung MP, Stevenson DA, Plössl K, Meegalla SK, Beckwith A, Essman WD, Mu M, Lucki I, Kung HK (1997) [99mTc] TRODAT-1: a novel technetium-99m complex as a dopamine transporter imaging agent. Eur J Nucl Med 24:372–380. https://doi.org/10.1007/BF00881808
Lee HF, Tsai CR, Chi CS, Chang TM, Lee HJ (2009) Aromatic l-amino acid decarboxylase deficiency in Taiwan. Eur J Pediatr Neurol 13:135–140. https://doi.org/10.1016/j.ejpn.2008.03.008
Lee HF, Chi CS, Tsai CR (2021) Diagnostic yield and treatment impact of whole-genome sequencing in pediatric neurological disorders. Dev Med Child Neurol 63:934–938. https://doi.org/10.1111/dmcn.14722
Lisi EC, Cohn RD (2011) Genetic evaluation of the pediatric patient with hypotonia: perspective from a hypotonia specialty clinic and review the literature. Dev Med Child Neurol 53:586–599. https://doi.org/10.1111/j.1469-8749.2011.03918.x
Luhmann HJ, Fukuda A, Kilb W (2015) Control of cortical neuronal migration by glutamate and GABA. Front Cell Neurosci 9:4. https://doi.org/10.3389/fncel.2015.00004
Machado-Pinilla R, Liger D, Leulliot N, Meier UT (2012) Mechanism of the AAA+ ATPases pontin and reptin in the biogenesis of H/ACA RNPs. RNA 18:1833–1845. https://doi.org/10.1261/rna.034942.112
Okumura A, Maruyama K, Shibata M, Kurahashi H, Ishii A, Numoto S, Hirose S, Kawai T, Iso M, Kataoka S, Okuno Y, Muramatsu H, Kojima S (2018) A patient with a GNAO1 mutation with decreased spontaneous movements, hypotonia, and dystonic features. Brain Dev 40:926–930. https://doi.org/10.1016/j.braindev.2018.06.005
Pearson TS, Gilbert L, Opladen T, Garcia-Cazorla A, Mastrangelo M, Leuzzi V, Tay SKH, Sykut-Cegielska J, Pons R, Mercimek-Andrews S, Kato M, Lücke T, Oppebøen M, Kurian MA, Steel D, Manti F, Meeks KD, Jeltsch K, Flint L (2020) AADC deficiency from infancy to adulthood: symptoms and developmental outcome in an international cohort of 63 patients. J Inherit Metab Dis 43:1121–1130. https://doi.org/10.1002/jimd.12247
Rahimi-Balaei M, Bergen H, Kong J, Marzban H (2018) Neuronal migration during development of the cerebellum. Front Cell Neurosci 12:484. https://doi.org/10.3389/fncel.2018.00484
Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M (2019) CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res 47(D1):D886-894. https://doi.org/10.1093/nar/gky1016
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, Laboratory Quality Assurance Committee ACMG (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for molecular pathology. Genet Med 17:405–424. https://doi.org/10.1038/gim.2015.30
Singh M, Gonzales FA, Cascio D, Heckmann N, Chanfreau G, Feigon J (2009) Structure and functional studies of the CS domain of the essential H/ACA ribonucleoparticle assembly protein SHQ1. J Biol Chem 284:1906–1916. https://doi.org/10.1074/jbc.M807337200
Sleiman S, Marshall AE, Dong X, Mhanni A, Alidou-D’Anjou I, Frosk P, Marin SE, Stark Z, Del Bigio MR, McBride A, Sadedin S, Gallacher L, Care4Rare Canada Consortium, Christodoulou J, Boycott KM, Dragon F, Kernohan KD (2022) Compound heterozygous variants in SHQ1 are associated with a spectrum of neurological features, including early-onset dystonia. Hum Mol 31:614–624. https://doi.org/10.1093/hmg/ddab247
Steinrücke S, Lohmann K, Domingo A, Rolfs A, Bäumer T, Spiegler J, Hartmann C, Münchau A (2016) Novel GNB1 missense mutation in a patient with generalized dystonia, hypotonia, and intellectual disability. Neurol Genet 2:e106. https://doi.org/10.1212/NXG.0000000000000106
Theisen U, Hennig C, Ring T, Schnabel R, Köster RW (2018) Neurotransmitter-medicated activity spatially controls neuronal migration in the zebrafish cerebellum. PLoS Biol 16:e2002226. https://doi.org/10.1371/journal.pbio.2002226
Vaswani AR, Weykopf B, Hagemann C, Fried HU, Brüstle O, Blaess S (2019) Correct setup of the substantia nigra requires Reelin-mediated fast, laterally-directed migration of dopaminergic neurons. Elife 8:e41623. https://doi.org/10.7554/eLife.41623
Walbott H, Machado-Pinilla R, Liger D, Blaud M, Réty S, Grozdanov PN, Godin K, van Tilbeurgh H, Varani G, Meier UT, Leulliot N (2011) The H/ACA RNP assembly factor SHQ1 functions as an RNA mimic. Gene Dev 25:2398–2408. https://doi.org/10.1101/gad.176834.111
Yu YT, Meier UT (2014) RNA-guided isomerization of uridine to pseudouridine-pseudouridylation. RNA Biol 11:1483–1494. https://doi.org/10.4161/15476286.2014.972855
Acknowledgements
The authors gratefully thank Professor Huei-Jane Lee who works at Department of Biochemistry, School of Medicine, Chung Shan Medical University, Taichung, Taiwan, for her generous contribution with interpretation of experimental data.
Funding
This work was supported by TCVGH-1096502B and TCVGH-1106504B for the molecular research and in vitro study.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. C-SC participated in recruitment of patients, acquisition, analysis, and interpretation of data. C-RT participated in the sequence alignment and carried out the molecular genetic studies and the in vitro functional study. H-FL made great contributions to recruitment of patients and interpretation of data, revised the manuscript critically for important intellectual content, and gave final approval of the version. The first draft of the manuscript was written by C-SC and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors have non-financial interests to disclose. The founders had no role in the design of the study; in the collection, analysis, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Ethical approval
This study was performed in line with the principles of the Declaration of Helsinki. Approval was granted by the Ethics Committee of Taichung Veterans General Hospital (Date April 20th 2022/ No TCVGH IRB CE22134B).
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Chi, CS., Tsai, CR. & Lee, HF. Biallelic SHQ1 variants in early infantile hypotonia and paroxysmal dystonia as the leading manifestation. Hum. Genet. 142, 1029–1041 (2023). https://doi.org/10.1007/s00439-023-02533-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-023-02533-5