
Abstract
Ubiquitination, a highly adaptable post-translational modification, plays a pivotal role in maintaining cellular protein homeostasis, encompassing cancer chemoresistance-associated proteins. Recent findings have indicated a potential correlation between perturbations in the ubiquitination process and the emergence of drug resistance in CRC cancer. Consequently, numerous studies have spurred the advancement of compounds specifically designed to target ubiquitinates, offering promising prospects for cancer therapy. In this review, we highlight the role of ubiquitination enzymes associated with chemoresistance to chemotherapy via the Wnt/β-catenin signaling pathway, epithelial–mesenchymal transition (EMT), and cell cycle perturbation. In addition, we summarize the application and role of small compounds that target ubiquitination enzymes for CRC treatment, along with the significance of targeting ubiquitination enzymes as potential cancer therapies.
Similar content being viewed by others

Introduction
Colorectal cancer (CRC) is the third most diagnosed cancer and the second leading cause of cancer-related deaths worldwide, accounting for over 1.9 million new cases and 935,000 new deaths worldwide (Sung et al. 2021). The heterogeneous and metastatic nature is the hallmark of CRC, with a poor prognosis (Van Cutsem et al. 2016). The prognosis of CRC is related to its stage at the time of diagnosis, with a 5-year survival rate of approximately 90% for stage I, 70% for stage II, 58% for stage III, and less than 15% for stage IV (Johnston 2005). Currently, adjuvant chemotherapy is the primary treatment for CRC patients, and systemic adjuvant chemotherapy based on fluoropyrimidine (5-fluorouracil [5-FU] or capecitabine) has been widely used in all patients with stage II and stage III CRC with high-risk clinicopathologic features (Messersmith 2017). A recent meta-analysis of 25 well-reported studies found that the 5-year relapse-free survival (RFS) estimates for patients with stage II and III CRC treated without adjuvant chemotherapy were 82.7% and 49.0%, respectively, compared to 79.3% and 63.6%, respectively, with adjuvant chemotherapy (Böckelman et al. 2015). However, the prognosis for these patients continues to be unfavorable, primarily due to the inherent resistance of these tumors to chemotherapy. Nonetheless, the overall survival of patients with colorectal cancer (CRC) has shown improvement in recent decades, thanks to the introduction of various chemotherapeutic agents despite their considerable drug resistance (Park et al. 2019). Consequently, it is necessary to summarize the potential mechanisms underlying the development of chemotherapy resistance during CRC treatment, which may have the potential to develop effective treatments to prevent chemotherapy resistance of CRC patients.
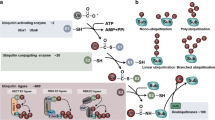
In eukaryotic cells, the ubiquitin proteasome system (UPS) is responsible for protein level control or activity regulation of many proteins in a highly selective manner (Chen and Dou 2010). Numerous studies have demonstrated the vital role of the UPS in CRC resistance to chemotherapy (Wang et al. 2022). Ubiquitin (Ub) is a β-grasp-fold protein with a compact overall structure and tight hydrogen bonds. It contains seven lysine residues involved in the formation of different types of Ub chains that have different roles in regulating and maintaining cellular homeostasis of the cell (Husnjak and Dikic 2012). Ubiquitination is a multifaceted enzymatic process that encompasses a series of regulated steps, involving the activation of ubiquitin through the ubiquitin-activating enzyme E1, the conjugation of ubiquitin through the ubiquitin-conjugating enzyme E2, and the subsequent transfer of the ubiquitin moiety to the target molecule facilitated by the ubiquitin protein ligase E3 (Han et al. 2022) (Fig. 1A). Notably, E3 ligases, as the core component of the UPS, can recognize the most specific substrates and are widely studied as potential targets to intervene in chemotherapy resistance, mainly through the Wnt/β-catenin signaling pathway, EMT, and cell cycle perturbation (Fig. 1B, C).

The flowchart illustrates the contents of the present review. A Ubiquitination process involves Ub, E1, E2, and E3 ubiquitin ligases. ATP activates the E1 enzyme and binds to Ub. The Ub-E1 intermediate then transfers the activated Ub to the E2 enzyme. Finally, Ub is transferred to a specific substrate by E3 ubiquitin ligases, which are categorized as the really interesting new gene (RING)-type E3 ligases, the U-box-type E3 ligases, the HECT-type E3 ligases, or the RBR-type E3 ligases. In RING and U-box E3 ubiquitin ligases, the Ub is transferred directly from the E2 enzyme to the substrate. HECT E3 ligase transfers Ub to the C lobe of HECT, and then Ub is transferred back to the substrate. RBR E3 ubiquitin ligase transfers Ub through the two RING domains. Subsequently, the substrate with the Ub linkage is destined for degradation or to participate in DNA repair, kinase activation, transcriptional regulation, and growth reactions. B, C ubiquitination enzymes are involved in the process of resistance to first-line chemotherapeutic agents, such as 5-FU, CPT-11, L-OHP, and Cetuximab, mainly through their involvement in the Wnt/β-catenin signaling pathway, EMT, cell cycle perturbation, and others. D Various small-molecule drugs that target ubiquitination enzymes are being developed for use in the clinical management of CRC
With the advances in the development of small molecules targeting various ubiquitination enzymes, it may be a promising treatment alone or in combination with other drugs to overcome the severe chemotherapy resistance in CRC (Fig. 1D). This review aims to elucidate the role of ubiquitination enzymes in the development of therapeutic resistance in colorectal cancer (CRC). Additionally, it provides a comprehensive analysis of the molecules examined in clinical trials for CRC treatment, with the intention of offering novel perspectives on strategies to overcome drug resistance.
Types of ubiquitinated enzymes
E1 ubiquitin-activating enzymes and E2 ubiquitin-conjugating enzymes
E1 enzymes (E1s), the first key enzymes in the ubiquitination process, can be shared in the process of ubiquitination, and there are only two kinds of E1 in humans (Varshavsky 2012). The adenosine 5'-triphosphate (ATP)-binding site is currently the sole identified means of inhibiting E1s. E1 triggers the activation of Ub through an ATP-dependent reaction, which necessitates the consumption of energy, resulting in the formation of a thioester-linked E1–Ub conjugate. Subsequently, the activated Ub is transferred to an E2 enzyme (E2s) via a trans-thiolation process involving an E2 Cys residue. E2s consist of 40 members and act as bridges to help the shuttle of Ub from E1s enzymes to E3 enzymes (E3s) or substrates during the ubiquitination process (Zhou et al. 2021a, b). To date, all E2s contain a conserved catalytic core domain of approximately 150 amino acids, termed the Ub-conjugating domain or UBC (Stewart et al. 2016). E2s can be divided into four classes based on having extra N- and/or C-terminal domains with E2-specific functions and enable specific interactions with particular E3s: class I E2s consist of a UBC domain, class II has an additional N-terminal domain, class III E2s have additional C-terminal domains, whereas class IV E2s contain both N- and C-terminal domains (Van Wijk and Timmers 2010). Some E2s have been shown to regulate the Wnt/β-catenin signaling pathway and drug transporter proteins to make CRC cells resistant to chemotherapeutic drugs (Huang et al. 2020).
E3 ubiquitin ligases
E3s display specific recognition of substrates, which plays a critical role in UPS (Toma-Fukai and Shimizu 2021). More than 600 E3s have been identified in humans and classified into four categories based on their distinctive catalytic structural domains and Ub transfer characteristics: really interesting new gene (RING)-type E3 ligases, UFD2 homology (U-box)-type E3 ligases, and homologous to E6AP carboxyl terminus (HECT)-type E3 ligases or the RING-between-RING (RBR)-type E3 ligases(Buetow and Huang 20166).
The most abundant E3 ubiquitin ligases in humans are RING-type E3 ligases, named after its RING domain, which requires the chelation of two zinc ions (Zn2+) for its activity (Zhao et al. 2022; Morreale and Walden 2016). Remarkably, RING-type E3 enzymes can mediate the direct transfer of Ub from the E2 enzyme to the substrate, functioning as monomers, homodimers, heterodimers, or multiple subunits (Morreale and Walden 2016; Berndsen and Wolberger 2014). Among them, the anaphase-promoting complex/cyclosome (APC/C) and cullin–RING E3 ubiquitin ligase (CRL) are both composed of multiple subunits. All CRLs share a common feature with at least four common subunits, including the E2-binding RING protein, scaffold containing the cullin protein, substrate-recognized receptor, and articulation protein between the receptor and scaffold (Harper and Schulman 2021). The well-studied cullin protein family consists of CUL1 to CUL7, which bind to BTB proteins to specifically recognize substrates (Fouad et al. 2019). Moreover, multiple tripartite motif (TRIM) family proteins, which belong to a large class of molecules in the RBCC protein family that contain the RING structural domain, have been shown to play a critical role in chemotherapy resistance in CRC (Liang et al. 2019). U-box ligases, an E3 ubiquitin ligase domain structurally similar to the RING-type E3 ligase domain, link Ub to substrates in a single reaction, but their activity is not dependent on Zn2+ (Morreale and Walden 2016).
As for HECT-type E3s, consisting of approximately 30 have been identified in humans (Rotin and Kumar 2009). The HECT-type E3 ligase possesses a conserved catalytic HECT structural domain with an approximate molecular weight of 40 kDa, which is responsible for specific substrate recognition. Typically, the HECT E3 ligase facilitates substrate ubiquitination through a two-step process. Initially, the HECT domain interacts with the E2 enzyme and transfers Ub to the C lobe via a trans-thioesterification reaction. Subsequently, Ub is subsequently transferred to the substrate (Weber et al. 2019). Based on the N lobe structure, HECT E3s can be further divided into three subfamilies: the NEDD4 subfamily containing multiple tryptophan–tryptophan (WW) motifs, the HECT and RCC1-like structural domain (HERC) subfamily, and HECT E3s that retain different structural domains. Furthermore, HECT-type E3s have also been found to be associated with chemotherapy resistance. Both HECT-type E3 and RBR-type E3 require a two-step reaction to link Ub to the substrate. As a newly identified E3 ligase isoform, more than 10 RBR-type E3s have been identified in humans (Cotton and Lechtenberg 2020). In RBR E3 ligases, there are usually three structural domains: the RING1 domain, which is responsible for binding to Ub-containing E2; the RING2 domain, which transfers Ub to the substrate; and the zinc-binding intermediate-ring domain (IBR) (Qi et al. 2015).
Ubiquitination has been shown to play a pivotal role in the occurrence and progression of many cancers, as well as the advances showing that ubiquitination enzymes are associated with drug resistance in cancer.
Mechanisms of chemotherapy resistance in CRC
Resistance of CRC to chemotherapy involves multiple mechanisms. In the following section, we describe in detail the mechanisms involved in ubiquitination enzymes in the context of chemotherapeutic drug resistance.
Wnt/β-catenin signaling pathway
The Wnt/β-catenin signaling pathway plays a pivotal role in the initiation and progression of CRC. Wnt molecules interact with frizzled (FZD) receptors, belonging to a specific family, to modulate the activity of β-catenin, a crucial downstream protein that exerts significant influence on cellular processes (Shay et al. 2016). When Wnt ligands do not bind to the transmembrane FZD receptors or low-density lipoprotein receptor-related protein 5/6(LRP5/6), the Wnt/β-catenin signaling is in an ‘off’ state (Sawa et al. 2016). The β-catenin could be targeted for ubiquitination and degradation through the adenomatous polyposis coli complex, consisting of glycogen synthase kinase 3β (GSK3β), framework protein Axin, APC and casein kinase 1α (CK1α) (Drew et al. 2016). The APC complex-mediated ubiquitination of β-catenin inhibits its nuclear translocation and activation of its target genes, such as c-Myc (Bernkopf et al. 2019). However, when the Wnt ligand binds to the receptor in normal mature cells, CK1 and GSK3β are recruited to LRP5/6, resulting in the inability of the APC complex to degrade β-catenin and thus promoting target gene transcription (Dominguez-Brauer et al. 2016; Zarkou et al. 2018).
Notably, the Wnt/β-catenin signaling pathway has been reported to be involved in chemotherapeutic resistance of CRC by influencing the presence of highly metastatic cancer stem cells, regulating non-coding RNAs, and modulating the tumor microenvironment (Zhu et al. 2021a, b) (Fig. 2). Considering the regulatory role of ubiquitination on multiple elements of the Wnt/β-catenin signaling axis, it becomes crucial to provide a comprehensive overview of additional E3 ligases. Notably, ring finger 43 (RNF43) and its homolog, zinc and ring finger 3 (ZNRF3), are highly correlated ring finger proteins (RNFs) that function as negative modulators of Wnt/β-catenin signaling. Their mechanism involves facilitating the degradation of Wnt coreceptors FZD and LRP6, thereby impeding intracellular Wnt cascades and promoting tumor suppression (Jiang et al. 2015). However, it is interesting to note that β-TRCP positively regulated Wnt/β-catenin signaling by targeting ZNRF3 (Ci et al. 2018). As mentioned above, axin negatively regulates Wnt/β-catenin signaling by regulating the level of β-catenin, which is a key effector molecule. Knockdown of Smad ubiquitination regulatory factor 2(Smurf2) results in reduced activity of the β-catenin/Tcf reporter gene, specifically ubiquitinylated Lys (505) of Axin, inducing its degradation (Kim and Jho 2010). Moreover, the RING domain E3 ligase SIAH proteins promote the ubiquitination and proteasomal degradation of Axin by interacting with a VxP motif in Axin (Ji et al. 2017).

The role of ubiquitination enzymes in the Wnt/β-catenin signaling pathway of CRC. Under normal cellular conditions (in the absence of WNT), β-catenin in the cytoplasm undergo UPS-mediated degradation by interacting with a destructive complex (DC) consisting of APC, Axin, CK1 and glycogen synthase kinase 3 (GSK3-β). DVL removes FZD6 from the cell membrane via RNF43/ZNRF3 binding. LPR is also deleted. The ubiquitination enzymes β-TrCP and UBES2S promote the degradation of β-catenin. On the other hand, Wnt binding to the LPR and FZD6 prevents β-catenin degradation, the C-terminus of LRP is phosphorylated and the binding of Axin inhibits the interaction of β-catenin with DCs, leading to stable transfer of β-catenin to the nucleus that binds to the TCF/LEF transcription factors to activate transcription of Wnt target genes. Expression of β-catenin is promoted by TRIM29 and TRIM27. RNF14 promotes β-catenin binding to TCF/LEF. β-Catenin degradation has been shown to be inhibited by NEDD4L, Smurf2, CRBN, SIAH and RNF146
E3s RNF146 also mediates ubiquitination and degradation of Axin (Callow et al. 2011). Cereblon (CRBN), the substrate receptor of CRL4CRBN E3 ubiquitin ligase, induces ubiquitination and degradation of CK1-α, leading to an increase in Wnt/β-catenin signaling (Shen et al. 2021). Moreover, the ubiquitination of K6, K27, and K29 in Disheveled (Dvl2) by a neural precursor cell expressed, developmentally down-regulated 4-like (NEDD4L) could regulate β-catenin level (Ding et al. 2013b, a). In addition, β-catenin can be phosphorylated by GSK3β and then recognized by the Skp1–Cdc53/cullin–F-box-protein complex (SCF)/β-TrCP in the absence of Wnt, finally contributing to ubiquitination and degradation of β-catenin (Liu et al. 2018a, b). In addition, the ubiquitin-conjugating enzyme E2S (UBE2S) targets the ubiquitination and degradation of β-catenin (Li et al. 2018). Interestingly, TRIM27, a RING-type E3 ubiquitin ligase identified by differential display (EDD), directly targets GSK-3β for degradation, resulting in the stabilization of β-catenin (Shen et al.2021). TRIM29, a RING-type E3 ubiquitin ligase, indirectly increases β-catenin expression by upregulating CD44 expression, thus inducing activation of the Wnt/β-catenin signaling pathway (Wang et al. 2015). In addition, RNF14 stabilizes its interaction with TCF/LEF by promoting β-catenin recruitment; however, the specific mechanism needs to be elucidated (Wu et al. 2013).
EMT
The process of epithelial–mesenchymal transition (EMT) involves the temporary induction of a quasi-mesenchymal cellular phenotype in epithelial cells, leading to the gradual loss of their characteristic cobblestone appearance in monolayer cultures and acquisition of a spindle-shaped mesenchymal morphology. It is important to note that despite this transition, the mesenchymal cells still possess the capacity to revert back to their original epithelial state (Dongre and Weinberg 2019). Cells undergoing EMT usually exhibit decreased expression of epithelial genes, including occludin, E-cadherin, and ZO-1, and increased expression of mesenchymal genes, such as fibronectin, N-cadherin, and vimentin (Lamouille et al. 2014). Changes in the expression of EMT-associated genes may influence physiological processes, including cell morphology, loss of adhesion, and the acquisition of stem cell-like features (Du and Shim 2016). EMT is a highly dynamic and reversible process during chemotherapy resistance (Vander Heiden and DeBerardinis 2017).
Numerous RING-type E3 ligases, particularly those within the TRIM family, have been implicated in the modulation of epithelial-mesenchymal transition (EMT). For example, TRIM21, an E3 ubiquitin ligase belonging to the TRIM family, exhibits a significant decrease in colitis-associated cancers and exerts a negative regulatory influence on colon carcinogenesis through the modulation of genes associated with tumor cell adhesion, such as E-calmodulin (Zhou et al. 2021a, b). Additionally, TRIM58 actively participates in EMT by regulating the expression of key transcription factors Snail and Slug, as well as cytoskeletal proteins vimentin and E-cadherin (Liu et al. 2018a, b). Moreover, TRIM66 knockdown inhibited EMT by increasing E-cadherin expression and decreasing N-cadherin and waveform protein expression (He et al. 2019). TRIM27 has also been shown to promote EMT in CRC cells (Zhang et al. 2018). Another RING-type E3 ubiquitin ligase, TRIM37, enhances CRC metastasis by inducing EMT process (Hu and Gan 2017). S-phase kinase-associated protein 2 (Skp2) is a key component of the SCF complex belonging to the RING finger type and enhances cellular migration through ubiquitination and destruction of E-cadherin (Wang et al. 2012). TNF receptor-associated factor 6 (TRAF6) promotes the formation of the LC3B–ATG7 complex by interacting with LC3B and catalyzing K63-linked polyubiquitination, which is essential for the subsequent recognition of catenin beta1 (CTNNB1) for selective autophagic degradation to efficiently inhibit EMT efficiently (Wu et al. 2019). Moreover, the E3-ubiquitin ligase F-Box and WD repeat domain containing 7 (FBXW7) directly bind and degrade the EMT-inducing transcription factor zinc finger E-box-binding homeobox 2(ZEB2) in a phosphorylation-dependent manner (Li et al. 2019). Furthermore, Fbox45, TRIM62, retinoblastoma-binding protein 6 (RBBP6), and Hakai have been implicated in EMT (Xu et al. 2015; Chen et al. 2013; Xiao et al. 2019; Díaz-Díaz et al. 2017).
The HECT E3 ligase HERC3 has been found to facilitate the degradation of eukaryotic translation initiation factor 5A2 (EIF5A2) through K27- and K48-linked ubiquitination, utilizing its HECT domain. This degradation process has been shown to regulate the epithelial–mesenchymal transition (EMT) through the EIF5A2/TGF-/Smad2/3 signaling pathway (Zhang et al. 2022). Additionally, another HECT ligase, Itch, has been observed to positively regulate EMT by promoting TGF-β signaling via ubiquitination of Smad7 (Park et al. 2015). Furthermore, the WW domain-containing E3 ubiquitin protein ligase 2 (WWP2) has been identified as targeting Smad2, Smad3, and Smad7 for degradation, thereby exerting control over transforming growth factor β (TGFβ)-dependent transcription and EMT (Soond and Chantry 2011). Furthermore, the involvement of SNAIL in cancer progression is of paramount importance as it facilitates the process of epithelial–mesenchymal transition (EMT), while the degradation of SNAIL through ubiquitination by the HECT structural domain E3 ubiquitin ligase 1 (HECTD1) is responsible for mediating this phenomenon (Wang et al. 2020a, b). Additionally, β-TrCP1 has been demonstrated to participate in the ubiquitination of SNAIL (Zhong et al. 2013). Nevertheless, the precise mechanisms underlying drug resistance in EMT are still not fully understood, necessitating further comprehensive investigations in subsequent research.
Cell cycle perturbation
The regulation of the cell cycle encompasses intricate mechanisms, such as the control of various cyclins, cyclin-dependent kinases, cell cycle checkpoints, and cell cycle signaling pathways. Historically, the eukaryotic cell cycle has been categorized into four distinct phases: G1, S, G2, and mitosis (M phase). During the G1 phase, cells undergo rapid synthesis of RNA and proteins, while also making preparations for DNA synthesis in the subsequent S phase. The S phase holds significant importance in the cell cycle as it involves the replication of DNA. The G2 phase denotes the stage where DNA replication has been completed, occurring prior to the initiation of mitosis. The M phase encompasses prophase, metaphase, anaphase and telophase, which collectively facilitate the precise and equitable division of chromosomes into two daughter cells (Sun et al. 2021). However, most cells do not enter the next cycle, but temporarily exist in a non-dividing state called the G0 phase.
Emerging evidence has shown that perturbation of cell cycle control enables continuous cell division primarily by impairing the ability of cells to exit the cell cycle, thus enhancing drug resistance (Matthews et al. 2022). Several RING-type E3 ligases have been shown to be involved in the regulation of this process (Fig. 3). The FHA domain of the RING-type E3 ligase checkpoint protein with forkhead associated and ring finger domains (CHFR) excludes cyclin B1 from the nucleus, leading to cell cycle arrest at G2/M, indicating a functional link between the anti-proliferative effects and checkpoint function of this tumor suppressor protein (Fukuda et al. 2008).

Cell cycle regulation of ubiquitination enzymes in CRC. The stages of the cell cycle are divided into four major phases: (1) G1 phase. (2) S phase. (3) G2 phase. (4) M phase. CHFR promotes G2/ -stage cell cycle arrest. c-Myc also promotes cell cycle arrest in G2/M phase and S phase. APC/CCDC20 regulates cell cycle progression through the M and S phase. Furthermore, SKP2 promotes cell cycle arrest at the G1/S transition. TRIM72 promotes G1-phase cell cycle arrest. Cyclin F inhibits the activity of transcription motors in the G2 cell cycle. RFPL4A has been shown to induce G1 arrest in CRC cells
In contrast, knockdown of baculoviral inhibitor of apoptosis (IAP) repeat containing 6(BIRC6) inhibited cell proliferation, stalled the cell cycle in the S phase and downregulated the levels of cell cycle proteins A2, B1, D1 and E1, thereby sensitizing CRC cells to chemotherapy (Hu et al. 2015). Notably, the downregulation of c-Myc expression facilitated the inhibition of the cell cycle in CRC when exposed to cytotoxic drugs, leading to growth arrest in the G2/M and S phases (Abaza et al. 2008). APC/C is a multi-subunit CRL that regulates cell cycle progression through the M and S phases. Among these, activation of APC/CCDC20 subsequently mediates proteasomal degradation of cyclin B1 and securin, promoting chromosome segregation and late-phase onset (Singleton and Uhlmann 2017; Zhou et al. 2019). In addition, APC/CCDH1 mediates the ubiquitination and proteasomal degradation of a large number of mitotic and G1 regulators, including cyclin B1, PLK1, CDC20, FOXM1, and SKP2, facilitating irreversible mitotic exit and G1 maintenance (Li and Zhang 2009). The SCF complex, which belongs to the CRL family, is active throughout the cell cycle. Four substrates, SKP2, cyclin F, FBXW7 and β-TrCP, have been well characterized in cell cycle regulation (Ang and Wade Harper 2005).
Phosphorylated p27 is ubiquitinated by activated SCF–Skp2 ubiquitin E3 ligase in the late G1 phase, triggering its proteasomal degradation, which contributes to RB1 full phosphorylation and the G1/S transition (Roilo et al. 2018). In addition, abnormally high TRIM72 expression catalyzes K48-linked ubiquitination and degradation of cell cycle protein D1, leading to cell cycle arrest in the G1 phase (Fang et al. 2023). As cells enter G2 phase, cyclin F ubiquitinates and restricts the activity of E2F, the major and most critical transcription engine of the cell cycle (Clijsters et al. 2019). Furthermore, βTrCP has demonstrated its ability to modulate the stability of cyclin F, thereby facilitating the degradation of ribonucleotide reductase M2 (RRM2) and CP110, thereby exerting control over genome integrity and centrosome homeostasis. Additionally, βTrCP influences CDH1 activity, thereby impacting the cell cycle (D'Angiolella et al. 2012, 2010). FBXW7 regulates the cell cycle by inhibiting the destruction of c-Myc and cell cycle protein E (Dang et al. 2021; Mavrommati et al. 2018). In addition, abnormally high expression of RET finger-like protein 4A (RFPL4A), a RING-type E3 ligase, induces G1-phase retention in CRC cells, decreasing their sensitivity to chemotherapy; however, the specific mechanism remains to be elucidated (Naito et al. 2015). Moreover, X-linked inhibitor of apoptosis protein (XIAP), which has E3 ubiquitin ligase activity, upregulates the phosphorylation of PP2A to inhibit phosphatase activity, thus leading to the phosphorylation and activation of its downstream target c-Jun, which in turn contributes to enhanced cyclin D1 expression and cell cycle transition (Cao et al. 2013).
Others
Multidrug resistance and membrane drug transporter proteins also influence drug resistance in CRC. P-glycoprotein (P-gp)/ABCB1 is an ABC transporter protein that acts as a vital determinant of the multidrug resistance phenotype of cancer cells (Srikant and Gaudet 2019). Knockdown of the RING-type E3 ligase SCFFbx15 decreased the degradation and ubiquitination of P-gp, leading to resistance to chemotherapy (Katayama et al. 2013). Disturbance of apoptosis also contributes to resistance to chemotherapy in CRC. The E3 ubiquitin ligase TRIM25, which belongs to the TRIM family, inhibits CRC cell death by destabilizing caspase-2 and caspase-7, thereby mediating its resistance to chemotherapy (Nasrullah et al. 2023). Moreover, SCF-box and WD repeat domain-containing protein 7 (FBW7) regulate cellular apoptosis by mediating ubiquitination and degradation of myeloid cell leukemia sequence 1(MCL1) (Inuzuka et al. 2011). Mechanistically, TRAF6 ubiquitinates the K63 site of p53 to limit its mitochondrial translocation of p53, contributing to tumor development and drug resistance (Zhang et al. 2016). Homologous to the E6-associated protein carboxyl terminus domain containing 3 (HECTD3) promoted the polyubiquitination of solute carrier family 7 member 11 (SLC7A11) to trigger the degradation of SLC7A11, thereby promoting ferroptosis of CRC (Huang et al. 2023a, b). Interestingly, all the E3s described above are RING-type E3s; however, additional studies are needed to further elucidate other E3s in the chemotherapeutic mechanism of colon cancer (CC). An improved understanding of the mechanisms involved in the ubiquitination-associated resistance to chemotherapy in CRC may reveal novel therapeutic strategies.
Chemotherapy drugs
It is widely acknowledged that CRC exhibits diverse responses to chemotherapy agents, including 5-FU, irinotecan (CPT-11), oxaliplatin (L-OHP), and cetuximab, with drug resistance being a prevalent occurrence. Consequently, this section aims to elucidate the interplay between drug resistance and ubiquitination in CRC.
5-FU
5-FU, an essential component of palliative and adjuvant systemic chemotherapy for CRC, is a synthetic fluorinated pyrimidine analog that replaces hydrogen with fluorine at the C-5 position of uracil (Vodenkova et al. 2020). 5-FU was one of the first chemotherapeutic agents reported to have anticancer activity. In CRC, treatment with 5-FU or other fluoropyrimidines (FPs) has been the backbone of systemic therapy since 1990 (Morawska et al. 2018). 5-FU is absorbed into the cell and activated to 5-FU deoxyribonucleotide for subsequent reactions; it exerts its antitumor effect by inhibiting thymidylate synthase (TS), preventing the conversion of deoxyuridine (dUMP) methylation to deoxythymidine (dTMP), and disrupting DNA replication (Siddiqui et al. 2019). It can also replace more than 50% of the uracil incorporated into RNA, inhibiting RNA synthesis (Shelton et al. 2016). Unfortunately, the use of oral 5-FU alone was abandoned because of its unpredictable gastrointestinal absorption and significant changes in pharmacokinetics (Saif et al. 2009). To maximize anticancer effects and minimize toxic effects, 5-FU is often used in combination with other drugs, such as folinic acid (Leucovorin, LV), bevacizumab, etc. (Thirion et al. 2004). Although new cancer therapies, such as targeted drug therapy, have made significant progress recently, 5-FU remains one of the most influential and commonly used drugs in CRC treatment and is a significant component of combination chemotherapy regimens (Sargent et al. 2009).
E3 ubiquitin ligases have been shown to play a role in the efficacy of 5-FU in patients with CRC. A few of these promote the therapeutic effects of 5-FU. For example, membrane-associated RING-CH-1 (MARCH 1), a RING-type E3 ubiquitin ligase whose expression is targeted by 5-FU, and the consequent downregulation of the PI3K/AKT pathway impact the progression of EMT (Wang et al. 2021). However, most E3 ubiquitin ligases directly contribute to 5-FU resistance. Inhibitors of apoptosis (IAPs), which belong to the RING-type E3 ubiquitin ligase family, have been shown to mediate 5-FU resistance (Oberoi-Khanuja and Rajalingam 2012). The expression level of XIAP increased with the number of 5-FU treatments, ultimately promoting the development of drug resistance (Flanagan et al. 2015). Inhibition of XIAP expression enhances the sensitization of CRC cells to 5-FU (Zhao et al. 2017). High expression of cIAP1 and cIAP2 was significantly associated with poor prognosis in patients with CRC treated with 5-FU, and further studies revealed that downregulation of cIAP2 effectively enhanced 5-FU sensitivity through the apoptotic pathway (Crawford et al. 2021). Ubiquitination and degradation of SMAD Family Member 4(SMAD4) increased upon physical interaction with TRIM47, leading to upregulation of the C–C motif chemokine ligand 15(CCL15) expression and chemotherapy resistance in response to 5-FU therapy (Liang et al.2019). The DNA damage-activated RING-type E3 ubiquitin ligase RAD18 was elevated after 5-FU treatment. Inhibition of its expression significantly attenuates proliferation and promotes apoptosis, thereby enhancing cellular radiosensitivity and 5-FU sensitivity. In contrast, elevated expression levels can induce DNA damage repair and promote drug resistance formation (Yan et al. 2019). UBE2M plays a role in resistance to 5-FU by regulating the expression of β-catenin (Xu et al. 2020).
Capecitabine is an oral fluoropyrimidine carbamate produced by the modified administration of 5-FU. It inhibits DNA synthesis through a series of enzyme-catalyzed reactions that convert it to 5-FU (Mehta et al. 2018). A large clinical trial demonstrated that capecitabine shows at least comparable efficacy and a good safety profile to push 5-FU in the treatment of metastatic CRC (Yamaguchi et al. 2006). Cancer cells with high nuclear factor erythroid 2-related factor 2 (NRF2) expression levels are less sensitive to chemotherapeutic agents, and multiple RING-type E3 ubiquitin ligase complexes, KEAP1-CUL3-RBX1, β-TrCP-SKP1-CUL1-RBX1, and HRD1, mediate ubiquitination and proteasomal degradation of NRF2, affecting the relationship between NRF2 overexpression and increased 5-FU resistance in CRC (Homma et al. 2009). The HECT structural domain and RCC1-like structural domain 5 (HERC5), a HECT type E3 ubiquitin ligase, were shown to degrade C-terminal-binding protein 1 (CtBP1) by ubiquitination in CRC cells. In contrast, HERC5 expression is downregulated in CRC, promoting the accumulation of CtBP1 and the formation of transcriptional complexes that inhibit apoptotic signaling and promote tumorigenesis. Interestingly, when inhibitors of HERC5 were combined with capecitabine, the inhibitory effects on cell proliferation and tumor growth were much more robust than when using those drugs alone (Zhu et al. 2021b, a).
CPT-11
CPT-11 is the first topoisomerase I inhibitor approved for cancer treatment (Martino et al. 2017). CPT-11,7-ethyl-10-[4-(1-piperidinyl)-1-piperidinyl]-carbonyloxycamptothecin is a pentacyclic alkaloid that undergoes structural changes depending on the physiological pH of the cellular environment (Tsilimigras et al. 2017). CPT-11 forms ternary CPT-11-topoisomerase I-incision DNA complexes in vivo in the form of collisions with replication forks that stall them, leading to double-strand break (DSB) formation and ultimately apoptosis (Stenvang et al. 2013). Interestingly, the dose dependence of CPT-11 increased with increasing cellular topoisomerase concentration, which directly affected the sensitivity of cells to CPT-11 (Burris and Fields 1994). CPT-11 is hydrolyzed in the body by carboxylesterases and converted in the liver to SN-38, which is 100–1000 times more toxic than CPT-11 (Rivory and Robert 1995). In contrast, SN-38 is converted to SN-38 glucosinolate (SN-38G) by UDP-glucuronosyltransferase UGT1A1 in an enzymatic reaction and is inactivated by β-glucuronidase hydrolysis after intestinal excretion (Morton et al. 1999). The correlation between the resistance of CRC tissues to CPT-11 monotherapy and ubiquitination remains relatively limited. Ubiquitin-conjugating enzyme 2C (UBE2C), which is highly expressed in most CRC patients, not only promotes the growth rate of CRC cells, but also enhances resistance to CPT-11 (Cacciola et al. 2016). It has been reported that the Cullin2/ElonginB-CIS complex,which belongs to the CRL, rendered cells more resistant to CPT-11 by promoting the degradation of the pro-apoptotic protein Bim (Ambrosini et al. 2009). Furthermore, upregulation of FBXW7 attenuated the response to CPT-11 by significantly reducing c-Myc expression (Izumi et al. 2017). In addition, downregulation of c-IAP1 and c-IAP2, which leads to the inhibition of NF-κB activation, could enhance the chemosensitivity of CRC cells to CPT-11 (Yu et al. 2014). Interestingly, CPT-11 regulates several ubiquitination enzymes. CPT-11 inhibits the expression of Bcl-x and XIAP and promotes p53 non-dependent apoptosis in CRC cells (Ravi et al. 2004). Moreover, CPT-11 inhibits mouse double minute 2(MDM2), thereby releasing p53 and blocking G2/M phase apoptosis (Lee et al. 2019).
L-OHP
Oxalato(trans-(-)-1,2-cyclohexanediamine)platinum(II), a third-generation platinum drug, was introduced in 2000 for the treatment of CRC and has gradually been explored for its therapeutic effects in other cancers. L-OHP acts intracellularly after its intravenous administration. It usually interacts with DNA-based nucleophilic molecules, forming an intra-strand adduct between two adjacent guanine residues or between guanine and adenine, disrupting DNA replication and transcription (Vander Heiden and DeBerardinis 2017). Interestingly, L-OHP–DNA adducts are not recognized and repaired by the mismatch repair system (MMR), resulting in tumors with a mismatch repair system (MMR) deficiency being more sensitive to L-OHP than the first-generation platinum drug cisplatin(Ahmad 2010). Unfortunately, intrinsic or acquired resistance to L-OHP remains a major impediment to achieving therapeutic benefits in CRC patients. Exploration of resistance mechanisms to L-OHP has become a hot topic in recent years (Martinez-Balibrea et al. 2015). The discovery of the correlation between ubiquitination enzymes and L-OHP resistance has helped in the development of new therapies aimed at overcoming this resistance. RAD6 with E2 ubiquitin-coupled activity and its homolog RAD18 with E3 ubiquitin ligase activity are essential for platinum-based chemotherapy-induced trans-lesion synthesis or post-replication repair. Inhibition of both is a potential new strategy for the treatment of chemotherapy in L-OHP-resistant CRC cells (Sanders et al. 2017).
In TGFβ signaling, the ubiquitination function of Smurf2 regulates the expression of inhibitor of differentiation 1(ID1) in CRC, which in turn causes resistance to L-OHP (Niu et al. 2021). In the p53 signaling pathway, MDM2 interacts with cyclophilin B (CypB), thereby enhancing p53 ubiquitination and degradation. This mechanism is also thought to contribute to the poor prognosis of L-OHP resistance (Choi et al. 2018). Furthermore, c-Myc promotes the transcriptional regulation and expression of WD-repeat protein 43(WDR43), a mechanism that facilitates the binding of WDR43 to ribosomal protein L11 (RPL11) and enhances the ubiquitination of p53 by MDM2, which reduces the stability of p53 proteins and induces chemotherapy resistance in CRC cells (Di et al. 2023). Several studies have demonstrated that the upregulation of XIAP, which mediates apoptosis, in CRC is also responsible for L-OHP-acquired resistance. When XIAP is inhibited, cells regain sensitivity to L-OHP-mediated apoptosis (Hua et al. 2018). In addition, the lack of E3 ubiquitin ligase FBXW7 promotes acquired resistance to L-OHP in CRC cells (Li et al. 2015). Exosome-mediated circ-FBXW7 increases L-OHP-induced apoptosis and inhibits L-OHP efflux, providing a new strategy to address L-OHP resistance in patients (Xu et al. 2021). Interestingly, the target protein cryptochrome2 (CRY2) was recognized by FBXW7. Although CRY2 upregulation caused by FBXW7 downregulation may be a novel prognostic biomarker, CRY2 knockdown increased the sensitivity of CRC to L-OHP (Fang et al. 2015). BRCA1-associated RING domain protein 1(BARD1), a RING-type E3 ubiquitin ligase, forms a positive mutually regulated ternary complex with breast cancer type 1 susceptibility protein (BRCA1) and mammalian metallothionein-2A (MT2A). High expression of MT2A greatly increases CRC cell resistance to L-OHP and increases CRC cell proliferation and viability (Zhao et al. 2020). In addition, TRAF6, a RING-type E3 ubiquitin ligase that binds and promotes enhancer of zeste homolog 2(EZH2) degradation, is inhibited by TRIM25, thereby promoting the stabilization of EZH2 and the characteristic state of CRC stem cells, promoting the resistance of cancer tissues to L-OHP (Zhou et al.2021a, b). Moreover, TRIM25 is involved in the negative regulation of caspase-2 in different CRC cell lines, a phenomenon that protects tumor cells from chemotherapeutic drug-induced apoptosis, and may serve as a novel mechanism of drug resistance in CRC (Nasrullah et al. 2019).
In addition, cisplatin, one of the previous generations of platinum-based drugs, is still used clinically for the treatment of CC by producing DNA lesions, blocking proliferative function, and promoting apoptotic cell death. Increased E3 ubiquitin ligase XIAP induces cisplatin resistance in CRC by activating the PI3K/Akt pathway, which is reversed when XIAP is inhibited (Xiong et al. 2017). Downregulation of the nucleotide excision repair (NER) member ubiquitin ligase cullin (CUL) 4A, which belongs to the RING-type E3 family, effectively improves cisplatin sensitivity but mediates trabectedin resistance (Englinger et al. 2017). TRIM8 overexpression inhibits p53 stability and activity, leading to cisplatin resistance (Mastropasqua et al. 2017). Furthermore, upregulation of Mcl-1 reduces cisplatin sensitivity in HUWE1 knockout mice (Myant et al. 2017).
Cetuximab
Cetuximab is a chimeric human murine derivative IgG1 monoclonal antibody (mAb) directed against the ligand-binding structural domain of the bound epidermal growth factor receptor (Kim 2004). The affinity of cetuximab to the epidermal growth factor receptor (EGFR) is much greater than that of any of the endogenous ligands, which, in turn, inhibits the activation of the receptor tyrosine kinase and associated downstream signaling, thereby exerting an antitumor effect (Humblet 2004). Cetuximab is currently approved for use in combination with chemotherapy in patients with CRC, non-small cell lung cancer (NSCLC), and head and neck cancer. The therapeutic efficacy of cetuximab is often compromised by acquired drug resistance; however, the mechanisms underlying this phenomenon are still unknown. Computer simulations have shown that multiple ubiquitination-related protein gene polymorphisms are involved in EGFR turnover and predict the efficacy of cetuximab, such as the ubiquitin-binding enzyme UBE2M and ubiquitin-conjugating enzyme E2L3 (UBE2L3), which may affect secondary resistance to cetuximab in metastatic CRC (Stintzing et al. 2015). Moreover, previous reports have shown that resistance to cetuximab in CRC is associated with increased expression of c-Myc, which significantly reduces apoptosis (Boos et al. 2022). Interestingly, inhibition of glutamine transporter protein solute carrier 1 family member 5 (SLC1A5) increased EGFR degradation via the ubiquitin–proteasome pathway, thereby significantly enhancing the inhibitory effect of cetuximab on CRC proliferation (Ma et al. 2018). Casitas B spectrum lymphoma-b (Cbl-b), a RING-type E3 ubiquitin ligase, regulates cetuximab sensitivity through the ubiquitin–proteasome system in human gastric cancer cells (Yu et al. 2016).
Multi-agent combination
Indeed, multi-agent combination chemotherapy is the central tenet of all therapies in patients with CRC. The concomitant phenomenon is often the emergence of simultaneous resistance to multi-drug combinations, which poses a significant problem for treatment.
FOLFOX, a combination regimen of folinic acid (FnA; “FOL”), 5-FU, and L-OHP, is a standard care strategy for the development of stage II/III CRC and liver metastases (Allegra et al. 2013). FOLFOX, L-OHP, and 5-FU were able to produce additive or synergistic anticancer activity, with stronger anticancer effects than when used alone. Interestingly, FnA enhanced the sensitivity of cancer cells to 5-FU (Matt et al. 2011). For clinical treatment, FnA and OxP are first administered to patients simultaneously via intravenous infusion, followed by intravenous 5-FU to achieve optimal efficacy (Kuebler et al. 2007). Although FOLFOX treatment has improved survival rates for patients with CRC, it is associated with low efficacy, dose-limiting side effects, poor quality of life, and increased costs (André et al. 2004). In addition, drug resistance to FOLFOX is still an inevitable phenomenon (Chang et al. 2020). It is essential to elucidate the mechanisms of resistance to explore new combination therapies for multi-drug resistance in CRC. Multiple ubiquitination enzymes have been found to be involved in FOLFOX treatment resistance in CRC patients. The ubiquitin-conjugating enzyme E2M (UBE2M) mediates 5-FU and L-OHP resistance in CRC cells through the Wnt/β-catenin signaling pathway (Xu et al. 2020). Furthermore, a novel RING-type E3 ubiquitin-linked enzyme, RNF126, promoted CRC progression and induced FOLFOX treatment resistance by enhancing p53 ubiquitination and degradation (Wang et al. 2020a, b). Another RING-type E3 ligase, TRIM6, which is highly expressed in patients with CRC, was inhibited and subsequently induced cell cycle arrest in the G2/M phase and increased sensitivity to 5-FU and L-OHP (Zheng et al. 2020). Moreover, FBXW7 binds to and degrades the transcription factor ZEB2, thereby inhibiting the resistance of CRC cells to 5-FU and L-OHP chemotherapeutic agents (Li et al.2019). In contrast, FBXW7 inhibition by miR-92a-3p reduces mitochondrial apoptosis and induced resistance (Hu et al. 2019). In addition, CDK2-associated cullin structural domain 1 (CAC1), a member of the CRL family, acts as a cell cycle regulator whose expression positively correlates with P-gp and MRP-1 protein expression and promotes the development of 5-FU and L-OHP resistance (Chen et al. 2019). RANBP2 and C3HC4 containing zinc finger 1 (RBCK1), a RING-type E3 ubiquitin ligase belonging to the IAPs family, and inhibition of their expression levels increased the sensitivity of CRC cells to 5-FU and L-OHP. After performing protein back-complementation, drug resistance in CRC cells was improved (Liu et al. 2019).
Targeting E3 ligases for CRC therapy
The modulation of crucial cellular pathways by ubiquitination enzymes makes them appealing targets for cancer treatment. The inhibition of these enzymes has shown promising effect on the field of cancer therapy (Lee et al. 2016). The research on drug discovery based on E3 ubiquitin ligase is progressing towards a hopeful future, with multiple biotech startups and companies developing small-molecule drugs targeting E3 ubiquitin ligase in various preclinical and clinical stages. The following section provides a description of small-molecule compounds or antibodies that specifically target ubiquitination enzymes, which are currently being investigated in various preclinical cancer models and ongoing clinical trials (Table 1).
Small molecules that specifically target ubiquitination enzymes in CRC
Inhibitors targeting the Wnt/β-catenin signaling pathway remain promising antitumor therapeutic agents. M435-1279, a novel UBE2T inhibitor that inhibits the over-excitation of the Wnt/β-catenin signaling pathway by blocking the UBE2T-mediated degradation of RACK1, has been shown to play a role in gastric cancer and provides insights for the treatment of CC (Anastas and Moon 2013).
Specific targeting of MDM2 or mouse double minute X(MDMX) is one approach for the treatment of CRC through the p53 pathway (Wade et al. 2013). HLI98, a novel small molecule targeting the ubiquitin ligase MDM2, specifically inhibits MDM2 to activate the p53 signaling pathway to inhibit cancer occurrence and development in CRC (Vassilev 2007). Gu screened novel inhibitors that antagonize MDM2 protein-XIAP mRNA interactions and inhibit MDM2 stability while blocking XIAP translation levels, producing anti-proliferative and pro-apoptotic effects. It is interesting to note that the inhibitors degraded MDM2, leading not only to inhibition of XIAP expression, but also activated p53 and induced apoptosis in p53 wild-type cancers (Gu et al. 2016). It is worth noting that NSC59984, a small molecule that induces degradation of mutated p53 protein through the MDM2 and ubiquitin–proteasome pathways, has been used in combination with CPT-11 to synergistically induce cell death in CRC cells expressing p53 mutant, indicating a promising therapeutic strategy (Zhang et al. 2015). MMRi71, a novel small-molecule compound with dual targeting of MDM4/FTH1, not only accumulates p53 proteins in wt-p53 bearing cancer cells, but also effectively kills leukemia cells that are p53 deficient. Whose development provides a prototypical structure for potential anticancer therapeutics (Lama et al. 2022).
Good progress has been made in the development of small-molecule compounds to target key subunits of SCF or APC/C complexes in CRC (Milhollen et al. 2011). MLN4924, a novel inhibitor targeting the ubiquitin ligase SCF E3, sensitizes CRC cells to irradiation by inducing cell cycle arrest and increasing apoptosis and DNA damage, which can be used in combination therapy with chemotherapeutic agents on the basis of broken DNA levels in the future (Wan et al. 2016). In contrast, MLN4924 inactivates CRL and leads to the accumulation of CRL substrates, thereby inhibiting the growth of tumor cells in vitro and in vivo. Because of its favorable results, MLN4924 has entered several clinical trials for anticancer therapies (Tan et al. 2011). It is important to note that MLN4924 is currently undergoing multiple Phase 1b trials to determine its safety and feasibility in combination with conventional chemotherapy for CRC treatment of CRC (Zhao et al. 2014).
Strategies for targeted cancer therapy in the clinic also include the development of IAP protein inhibitors. Ceramide and its analog LCL85 are potent sensitizers of Fas-mediated apoptosis and inhibit cancer progression by effectively targeting the protein degradation of cIAP1 and XIAP(Paschall et al. 2014). Tolinapant (ASTX660), a novel IAP antagonist, efficiently and rapidly downregulated cIAP1 expression in a CRC model. Additionally, FOLFOX was shown to promote tolinapant-induced apoptosis in human CRC and murine organ models, providing evidence for the clinical exploration of tolinapant in combination with FOLFOX for the treatment of cIAP1-expressing CRC with poor prognosis and high microsatellite stability (Crawford et al.2021).
Currently, there are other inhibitors of ubiquitination enzymes that have proven to be potential therapeutics for CRC. SMI#9 is a potent novel RAD6-selective small-molecule inhibitor that targets the RAD6 catalytic site to affect translesion synthesis (TLS) and enables CRC to overcome resistance to L-OHP and is required to overcome cisplatin-induced replication fork stalling (Sanders et al.2017). CC0651, an inhibitor that selectively inhibits the E2 enzyme Cdc34, has also been found to effectively inhibit p27Kip1 ubiquitination via E3 ligase SCFSkp2 (Ceccarelli et al. 2011). Further studies revealed that CC0651 and its analogs promoted p27 accumulation and inhibited the proliferation of CRC (Ceccarelli et al.2011). Another inhibitor that effectively inhibits APC-Cdh1 activation, in addition to inducing cell cycle arrest, is pro-TAME (Zeng et al. 2010).
The newest technology developed to connect cell surface E3 ubiquitin ligases to transmembrane proteins for degradation is protein hydrolysis-targeted antibodies (PROTABs). Antibodies targeting zinc finger and ring finger 3 (ZNRF3) protein hydrolysis allow for “on demand” degradation specific to CRC. Currently, one of the drawbacks of targeting E3 ubiquitin ligase in the clinic is the lack of specific targeting agents, leading to unexpected toxicity and side effects in clinical treatment. The experimental finding of targeting E3 ubiquitin ligase is of great benefit for cancer treatment. Several high-throughput screening assays are now available for the rapid filtration of small-molecule compounds from E3 ligases, providing a route to screen for clinically available E3 targeted drugs. An alternative approach is to directly target E3 ligase activity and antagonize the E3 ligase homolog of the target protein.
Small molecules that specifically target ubiquitination processes in CRC
PS-341 is a proteasome inhibitor that blocks chemotherapy-induced NF-κB activation, resulting in IkappaB degradation via the ubiquitin–proteasome pathway. In addition, PS-341 was demonstrated to significantly increase the chemosensitivity of CRC cells to CPT-11,which provides new ideas for clinical treatment (Cusack et al. 2001).Herbal therapy targeting ubiquitination enzymes is also a highlight of drug-resistant CRC therapies. Aidi injections antagonize the activity of the ubiquitin–proteasome (UPS) system and inhibit the breakdown of cytotoxic proteins by binding to the ubiquitin proteasome (Stein et al.2022). Curcumin increased the interaction between calmodulin-1 (Cdh1) and Skp2, leading to ubiquitination and degradation of Skp2, overcoming resistance to 5-FU in CRC, and inducing apoptosis in CRC cells resistant to 5-FU, suggesting that curcumin is a potential chemo-candidate for the treatment of 5-FU resistant CRC (Gan et al. 2023).
Discussion
In conclusion, ubiquitination enzymes, pivotal components in the ubiquitination process, have demonstrated a critical involvement in the development of chemotherapeutic resistance in CRC. The mechanism by which ubiquitination enzymes contribute to chemotherapeutic resistance primarily revolves around their impact on various signaling pathways, including the Wnt/β-catenin signaling pathway, EMT, and disruption of the cell cycle. It is worth noting that currently approved clinical drugs for CRC chemotherapy, namely 5-FU, CPT-11, L-OHP, and cetuximab, are utilized for this purpose. Emerging evidence shows that aberrant ubiquitination is involved in chemotherapeutic resistance in CRC. Moreover, several small-molecule inhibitors are currently under development, but none have demonstrated sufficient efficacy in clinical trials.
At present, our investigations of ubiquitination enzymes are fragmented, and the role of each member in chemotherapeutic resistance and its precise mechanism of regulation are not fully understood. The recognition of the role of ubiquitination enzymes in chemotherapeutic resistance has been growing alongside the increasing number of studies conducted on these enzymes in various cancers. A notable example is the promotion of resistance to cisplatin in non-small cell lung cancer through TRIM17-mediated ubiquitination and degradation of RBM38 (Zhong et al. 2023);RNF6 promotes cisplatin resistance by transcriptionally activating the expression of proliferating cell nuclear antigens and attenuating DNA damage in lung adenocarcinoma (Sun et al. 2022).In addition, UBE2T promotes temozolomide resistance in glioblastoma by modulating the Wnt/β-catenin signaling pathway (Wang et al. 2023). The results of these experiments may provide insights into the mechanisms of chemoresistance of these ubiquitination enzymes in CRC. Thus, further investigation of each ubiquitination enzyme member is warranted to elucidate its relevance to chemotherapeutic resistance in CRC.
Moreover, the additional exploration of chemotherapeutic resistance caused by ubiquitination enzymes could potentially contribute to the discovery of therapeutic targets, the development of innovative inhibitors, and the formulation of novel therapeutic approaches in clinical settings. In this context, risperidone acts as a pharmacological inhibitor of TRAF4, effectively suppressing the self-renewal of glioblastoma (GBM), eradicating tumor pathogenicity, and reversing resistance to temozolomide (Li et al. 2022). TAK-243, a newly developed and highly targeted inhibitor of the E1 enzyme UAE, demonstrated its ability to effectively counteract resistance to bortezomib and carfilzomib in cell line models. Furthermore, it exhibited notable efficacy against primary cells obtained from patients suffering from relapsed/refractory myeloma. Additionally, TAK-243 displayed significant synergistic effects when combined with several anti-myeloma agents, such as doxorubicin, melphalan, and pabilastat (Zhuang et al. 2019). Although it has not been demonstrated that the E3 ubiquitin ligase Hakai is associated with chemoresistance, recent studies have shown that Hakin-1, a Hakai inhibitor, can inhibit EMT in CC, which may provide new insights into the treatment of chemoresistance (Martinez-Iglesias et al. 2020). In addition, several ubiquitination-associated inhibitors have been found to ameliorate side effects associated with chemotherapy. The anti-MDM2 inhibitor DS-5272 ameliorates cisplatin-induced nephrotoxicity by inhibiting NF-κB signaling (Fujikura et al. 2019).
References
Abaza MS, Al-Saffar A, Al-Sawan S, Al-Attiyah R (2008) c-myc antisense oligonucleotides sensitize human colorectal cancer cells to chemotherapeutic drugs. Tumour Biol 295:287–303. https://doi.org/10.1159/000156706
Adès L, Girshova L, Doronin VA, Díez-Campelo M, Valcárcel D, Kambhampati S et al (2022) Pevonedistat plus azacitidine vs azacitidine alone in higher-risk MDS/chronic myelomonocytic leukemia or low-blast-percentage AML. Blood Adv 617:5132–5145. https://doi.org/10.1182/bloodadvances.2022007334
Ahmad S (2010) Platinum–DNA interactions and subsequent cellular processes controlling sensitivity to anticancer platinum complexes. Chem Biodivers 73:543–566. https://doi.org/10.1002/cbdv.200800340
Allegra CJ, Yothers G, O’Connell MJ, Sharif S, Petrelli NJ, Lopa SH et al (2013) Bevacizumab in stage II-III colon cancer: 5-year update of the national surgical adjuvant breast and bowel project C-08 trial. J Clin Oncol 313:359–364. https://doi.org/10.1200/jco.2012.44.4711
Amaravadi RK, Schilder RJ, Martin LP, Levin M, Graham MA, Weng DE et al (2015) A phase I study of the SMAC-mimetic birinapant in adults with refractory solid tumors or lymphoma. Mol Cancer Ther 1411:2569–2575. https://doi.org/10.1158/1535-7163.Mct-15-0475
Ambrosini G, Seelman SL, Schwartz GK (2009) Differentiation-related gene-1 decreases Bim stability by proteasome-mediated degradation. Can Res 6915:6115–6121. https://doi.org/10.1158/0008-5472.Can-08-3024
Anastas JN, Moon RT (2013) WNT signalling pathways as therapeutic targets in cancer. Nat Rev Cancer 131:11–26. https://doi.org/10.1038/nrc3419
André T, Boni C, Mounedji-Boudiaf L, Navarro M, Tabernero J, Hickish T et al (2004) Oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment for colon cancer. N Engl J Med 35023:2343–2351. https://doi.org/10.1056/NEJMoa032709
Andreeff M, Kelly KR, Yee K, Assouline S, Strair R, Popplewell L et al (2016) Results of the phase I trial of RG7112, a small-molecule MDM2 antagonist in leukemia. Clin Cancer Res 224:868–876. https://doi.org/10.1158/1078-0432.Ccr-15-0481
Ang XL, Wade Harper J (2005) SCF-mediated protein degradation and cell cycle control. Oncogene 2417:2860–2870. https://doi.org/10.1038/sj.onc.1208614
Bardia A, Parton M, Kümmel S, Estévez LG, Huang CS, Cortés J et al (2018) Paclitaxel with inhibitor of apoptosis antagonist, LCL161, for localized triple-negative breast cancer, prospectively stratified by gene signature in a biomarker-driven neoadjuvant trial. J Clin Oncol. https://doi.org/10.1200/jco.2017.74.8392
Bauer S, Demetri GD, Halilovic E, Dummer R, Meille C, Tan DSW et al (2021) Pharmacokinetic-pharmacodynamic guided optimisation of dose and schedule of CGM097, an HDM2 inhibitor, in preclinical and clinical studies. Br J Cancer 1255:687–698. https://doi.org/10.1038/s41416-021-01444-4
Benetatos CA, Mitsuuchi Y, Burns JM, Neiman EM, Condon SM, Yu G et al (2014) Birinapant (TL32711), a bivalent SMAC mimetic, targets TRAF2-associated cIAPs, abrogates TNF-induced NF-κB activation, and is active in patient-derived xenograft models. Mol Cancer Ther 134:867–879. https://doi.org/10.1158/1535-7163.Mct-13-0798
Berndsen CE, Wolberger C (2014) New insights into ubiquitin E3 ligase mechanism. Nat Struct Mol Biol 214:301–307. https://doi.org/10.1038/nsmb.2780
Bernkopf DB, Brückner M, Hadjihannas MV, Behrens J (2019) An aggregon in conductin/axin2 regulates Wnt/β-catenin signaling and holds potential for cancer therapy. Nat Commun 101:4251. https://doi.org/10.1038/s41467-019-12203-8
Böckelman C, Engelmann BE, Kaprio T, Hansen TF, Glimelius B (2015) Risk of recurrence in patients with colon cancer stage II and III: a systematic review and meta-analysis of recent literature. Acta Oncologica (stockholm, Sweden) 541:5–16. https://doi.org/10.3109/0284186x.2014.975839
Boos SL, Loevenich LP, Vosberg S, Engleitner T, Öllinger R, Kumbrink J et al (2022) Disease modeling on tumor organoids implicates AURKA as a therapeutic target in liver metastatic colorectal cancer. Cell Mol Gastroenterol Hepatol 132:517–540. https://doi.org/10.1016/j.jcmgh.2021.10.008
Brenke JK, Popowicz GM, Schorpp K, Rothenaigner I, Roesner M, Meininger I et al (2018) Targeting TRAF6 E3 ligase activity with a small-molecule inhibitor combats autoimmunity. J Biol Chem 29334:13191–13203. https://doi.org/10.1074/jbc.RA118.002649
Brusa I, Sondo E, Pesce E, Tomati V, Gioia D, Falchi F et al (2023) Innovative strategy toward Mutant CFTR rescue in cystic fibrosis: design and synthesis of thiadiazole inhibitors of the E3 ligase RNF5. J Med Chem 6614:9797–9822. https://doi.org/10.1021/acs.jmedchem.3c00608
Buetow L, Huang DT (2016) Structural insights into the catalysis and regulation of E3 ubiquitin ligases. Nat Rev Mol Cell Biol 1710:626–642. https://doi.org/10.1038/nrm.2016.91
Burris HA 3rd, Fields SM (1994) Topoisomerase I inhibitors. An overview of the camptothecin analogs. Hematol Oncol Clin North Am 82:333–355
Cacciola NA, Calabrese C, Malapelle U, Pellino G, De Stefano A, Sepe R et al (2016) UbcH10 expression can predict prognosis and sensitivity to the antineoplastic treatment for colorectal cancer patients. Mol Carcinog 555:793–807. https://doi.org/10.1002/mc.22322
Cai Q, Sun H, Peng Y, Lu J, Nikolovska-Coleska Z, McEachern D et al (2011) A potent and orally active antagonist (SM-406/AT-406) of multiple inhibitor of apoptosis proteins (IAPs) in clinical development for cancer treatment. J Med Chem 548:2714–2726. https://doi.org/10.1021/jm101505d
Callow MG, Tran H, Phu L, Lau T, Lee J, Sandoval WN et al (2011) Ubiquitin ligase RNF146 regulates tankyrase and Axin to promote Wnt signaling. PLoS ONE 67:e22595. https://doi.org/10.1371/journal.pone.0022595
Cao Z, Zhang R, Li J, Huang H, Zhang D, Zhang J et al (2013) X-linked inhibitor of apoptosis protein (XIAP) regulation of cyclin D1 protein expression and cancer cell anchorage-independent growth via its E3 ligase-mediated protein phosphatase 2A/c-Jun axis. J Biol Chem 28828:20238–20247. https://doi.org/10.1074/jbc.M112.448365
Ceccarelli DF, Tang X, Pelletier B, Orlicky S, Xie W, Plantevin V et al (2011) An allosteric inhibitor of the human Cdc34 ubiquitin-conjugating enzyme. Cell 1457:1075–1087. https://doi.org/10.1016/j.cell.2011.05.039
Chan CH, Morrow JK, Li CF, Gao Y, Jin G, Moten A et al (2013) Pharmacological inactivation of Skp2 SCF ubiquitin ligase restricts cancer stem cell traits and cancer progression. Cell 1543:556–568. https://doi.org/10.1016/j.cell.2013.06.048
Chang CW, Lee HC, Li LH, Chiang Chiau JS, Wang TE, Chuang WH et al (2020) Fecal microbiota transplantation prevents intestinal injury, upregulation of toll-like receptors, and 5-Fluorouracil/Oxaliplatin-induced toxicity in colorectal cancer. Int J Mol Sci. https://doi.org/10.3390/ijms21020386
Chen D, Dou QP (2010) The ubiquitin-proteasome system as a prospective molecular target for cancer treatment and prevention. Curr Protein Pept Sci 116:459–470. https://doi.org/10.2174/138920310791824057
Chen Q, Xie W, Kuhn DJ, Voorhees PM, Lopez-Girona A, Mendy D et al (2008) Targeting the p27 E3 ligase SCF(Skp2) results in p27- and Skp2-mediated cell-cycle arrest and activation of autophagy. Blood 1119:4690–4699. https://doi.org/10.1182/blood-2007-09-112904
Chen N, Balasenthil S, Reuther J, Frayna A, Wang Y, Chandler DS et al (2013) DEAR1 is a chromosome 1p35 tumor suppressor and master regulator of TGF-β-driven epithelial-mesenchymal transition. Cancer Discov 310:1172–1189. https://doi.org/10.1158/2159-8290.Cd-12-0499
Chen N, Kong Y, Wu Y, Gao Q, Fu J, Sun X et al (2019) CAC1 knockdown reverses drug resistance through the downregulation of P-gp and MRP-1 expression in colorectal cancer. PLoS ONE 149:e0222035. https://doi.org/10.1371/journal.pone.0222035
Chen YF, Liu RZ, Ying WW, Yang YN, Xiang SF, Shao XJ et al (2023) Arctigenin impairs UBC12 enzyme activity and cullin neddylation to attenuate cancer cells. Acta Pharmacol Sin 443:661–669. https://doi.org/10.1038/s41401-022-00992-6
Choi TG, Nguyen MN, Kim J, Jo YH, Jang M, Nguyen NNY et al (2018) Cyclophilin B induces chemoresistance by degrading wild-type p53 via interaction with MDM2 in colorectal cancer. J Pathol 2461:115–126. https://doi.org/10.1002/path.5107
Ci Y, Li X, Chen M, Zhong J, North BJ, Inuzuka H et al (2018) SCF(β-TRCP) E3 ubiquitin ligase targets the tumor suppressor ZNRF3 for ubiquitination and degradation. Protein Cell 910:879–889. https://doi.org/10.1007/s13238-018-0510-2
Clijsters L, Hoencamp C, Calis JJA, Marzio A, Handgraaf SM, Cuitino MC et al (2019) Cyclin F controls cell-cycle transcriptional outputs by directing the degradation of the three activator E2Fs. Mol Cell 746:1264-1277.e1267. https://doi.org/10.1016/j.molcel.2019.04.010
Cotton TR, Lechtenberg BC (2020) Chain reactions: molecular mechanisms of RBR ubiquitin ligases. Biochem Soc Trans 484:1737–1750. https://doi.org/10.1042/bst20200237
Crawford N, Stott KJ, Sessler T, McCann C, McDaid W, Lees A et al (2021) Clinical Positioning of the IAP Antagonist Tolinapant (ASTX660) in Colorectal Cancer. Mol Cancer Ther 209:1627–1639. https://doi.org/10.1158/1535-7163.Mct-20-1050
Cusack JC Jr, Liu R, Houston M, Abendroth K, Elliott PJ, Adams J et al (2001) Enhanced chemosensitivity to CPT-11 with proteasome inhibitor PS-341: implications for systemic nuclear factor-kappaB inhibition. Can Res 619:3535–3540
Dang F, Nie L, Wei W (2021) Ubiquitin signaling in cell cycle control and tumorigenesis. Cell Death Differ 282:427–438. https://doi.org/10.1038/s41418-020-00648-0
D’Angiolella V, Donato V, Vijayakumar S, Saraf A, Florens L, Washburn MP et al (2010) SCF(Cyclin F) controls centrosome homeostasis and mitotic fidelity through CP110 degradation. Nature 4667302:138–142. https://doi.org/10.1038/nature09140
D’Angiolella V, Donato V, Forrester FM, Jeong YT, Pellacani C, Kudo Y et al (2012) Cyclin F-mediated degradation of ribonucleotide reductase M2 controls genome integrity and DNA repair. Cell 1495:1023–1034. https://doi.org/10.1016/j.cell.2012.03.043
de Jonge M, de Weger VA, Dickson MA, Langenberg M, Le Cesne A, Wagner AJ et al (2017) A phase I study of SAR405838, a novel human double minute 2 (HDM2) antagonist, in patients with solid tumours. Eur J Cancer 76:144–151
Dhillon N, Aggarwal BB, Newman RA, Wolff RA, Kunnumakkara AB, Abbruzzese JL et al (2008) Phase II trial of curcumin in patients with advanced pancreatic cancer. Clin Cancer Res 1414:4491–4499. https://doi.org/10.1158/1078-0432.Ccr-08-0024
Di Y, Jing X, Hu K, Wen X, Ye L, Zhang X et al (2023) The c-MYC-WDR43 signalling axis promotes chemoresistance and tumour growth in colorectal cancer by inhibiting p53 activity. Drug Resist Updates 66:100909. https://doi.org/10.1016/j.drup.2022.100909
Díaz-Díaz A, Casas-Pais A, Calamia V, Castosa R, Martinez-Iglesias O, Roca-Lema D et al (2017) Proteomic analysis of the E3 ubiquitin-ligase hakai highlights a role in plasticity of the cytoskeleton dynamics and in the proteasome system. J Proteome Res 168:2773–2788. https://doi.org/10.1021/acs.jproteome.7b00046
Ding Y, Zhang Y, Xu C, Tao QH, Chen YG (2013a) HECT domain-containing E3 ubiquitin ligase NEDD4L negatively regulates Wnt signaling by targeting dishevelled for proteasomal degradation. J Biol Chem 28812:8289–8298. https://doi.org/10.1074/jbc.M112.433185
Ding Q, Zhang Z, Liu JJ, Jiang N, Zhang J, Ross TM et al (2013b) Discovery of RG7388, a potent and selective p53-MDM2 inhibitor in clinical development. J Med Chem 5614:5979–5983. https://doi.org/10.1021/jm400487c
Dominguez-Brauer C, Hao Z, Elia AJ, Fortin JM, Nechanitzky R, Brauer PM et al (2016) Mule regulates the intestinal stem cell niche via the Wnt pathway and targets EphB3 for proteasomal and lysosomal degradation. Cell Stem Cell 192:205–216. https://doi.org/10.1016/j.stem.2016.04.002
Dongre A, Weinberg RA (2019) New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat Rev Mol Cell Biol 202:69–84. https://doi.org/10.1038/s41580-018-0080-4
Drew DA, Cao Y, Chan AT (2016) Aspirin and colorectal cancer: the promise of precision chemoprevention. Nat Rev Cancer 163:173–186. https://doi.org/10.1038/nrc.2016.4
Du B, Shim JS (2016) Targeting epithelial-mesenchymal transition (EMT) to overcome drug resistance in cancer. Molecules. https://doi.org/10.3390/molecules21070965
Englinger B, Mair M, Miklos W, Pirker C, Mohr T, van Schoonhoven S et al (2017) Loss of CUL4A expression is underlying cisplatin hypersensitivity in colorectal carcinoma cells with acquired trabectedin resistance. Br J Cancer 1164:489–500. https://doi.org/10.1038/bjc.2016.449
Fang L, Yang Z, Zhou J, Tung JY, Hsiao CD, Wang L et al (2015) Circadian clock gene CRY2 degradation is involved in chemoresistance of colorectal cancer. Mol Cancer Ther 146:1476–1487. https://doi.org/10.1158/1535-7163.Mct-15-0030
Fang M, Wu HK, Pei Y, Zhang Y, Gao X, He Y et al (2023) E3 ligase MG53 suppresses tumor growth by degrading cyclin D1. Signal Transduct Target Ther 81:263. https://doi.org/10.1038/s41392-023-01458-9
Flanagan L, Kehoe J, Fay J, Bacon O, Lindner AU, Kay EW et al (2015) High levels of X-linked Inhibitor-of-Apoptosis Protein (XIAP) are indicative of radio chemotherapy resistance in rectal cancer. Rad Oncol (london, England) 10:131. https://doi.org/10.1186/s13014-015-0437-1
Flygare JA, Beresini M, Budha N, Chan H, Chan IT, Cheeti S et al (2012) Discovery of a potent small-molecule antagonist of inhibitor of apoptosis (IAP) proteins and clinical candidate for the treatment of cancer (GDC-0152). J Med Chem 559:4101–4113. https://doi.org/10.1021/jm300060k
Fouad S, Wells OS, Hill MA, D’Angiolella V (2019) Cullin ring ubiquitin ligases (CRLs) in cancer: responses to ionizing radiation (IR) treatment. Front Physiol 10:1144. https://doi.org/10.3389/fphys.2019.01144
Fujikura T, Yasuda H, Iwakura T, Tsuji T, Anders HJ (2019) MDM2 inhibitor ameliorates cisplatin-induced nephropathy via NFκΒ signal inhibition. Pharmacol Res Perspect 71:e00450. https://doi.org/10.1002/prp2.450
Fukuda T, Kondo Y, Nakagama H (2008) The anti-proliferative effects of the CHFR depend on the forkhead associated domain, but not E3 ligase activity mediated by ring finger domain. PLoS ONE 33:e1776. https://doi.org/10.1371/journal.pone.0001776
Gan Y, Zhou L, Wang R, Zhang Y, Li X, Han S et al (2023) Curcumol reduces aerobic glycolysis and overcomes chemoresistance by inducing Cdh1-mediated Skp2 ubiquitination. Am J Chin Med. https://doi.org/10.1142/s0192415x23500349
Gluck WL, Gounder MM, Frank R, Eskens F, Blay JY, Cassier PA et al (2020) Phase 1 study of the MDM2 inhibitor AMG 232 in patients with advanced P53 wild-type solid tumors or multiple myeloma. Invest New Drugs 383:831–843. https://doi.org/10.1007/s10637-019-00840-1
Gu L, Zhang H, Liu T, Zhou S, Du Y, Xiong J et al (2016) Discovery of dual inhibitors of MDM2 and XIAP for cancer treatment. Cancer Cell 304:623–636. https://doi.org/10.1016/j.ccell.2016.08.015
Han S, Wang R, Zhang Y, Li X, Gan Y, Gao F et al (2022) The role of ubiquitination and deubiquitination in tumor invasion and metastasis. Int J Biol Sci 186:2292–2303. https://doi.org/10.7150/ijbs.69411
Harper JW, Schulman BA (2021) Cullin-RING ubiquitin ligase regulatory circuits: a quarter century beyond the F-Box hypothesis. Annu Rev Biochem 90:403–429. https://doi.org/10.1146/annurev-biochem-090120-013613
He T, Cui J, Wu Y, Sun X, Chen N (2019) Knockdown of TRIM66 inhibits cell proliferation, migration and invasion in colorectal cancer through JAK2/STAT3 pathway. Life Sci 235:116799. https://doi.org/10.1016/j.lfs.2019.116799
Homma S, Ishii Y, Morishima Y, Yamadori T, Matsuno Y, Haraguchi N et al (2009) Nrf2 enhances cell proliferation and resistance to anticancer drugs in human lung cancer. Clin Cancer Res 1510:3423–3432. https://doi.org/10.1158/1078-0432.Ccr-08-2822
Hu CE, Gan J (2017) TRIM37 promotes epithelial-mesenchymal transition in colorectal cancer. Mol Med Rep 153:1057–1062. https://doi.org/10.3892/mmr.2017.6125
Hu T, Weng S, Tang W, Xue R, Chen S, Cai G et al (2015) Overexpression of BIRC6 Is a predictor of prognosis for colorectal cancer. PLoS ONE 105:e0125281. https://doi.org/10.1371/journal.pone.0125281
Hu JL, Wang W, Lan XL, Zeng ZC, Liang YS, Yan YR et al (2019) CAFs secreted exosomes promote metastasis and chemotherapy resistance by enhancing cell stemness and epithelial-mesenchymal transition in colorectal cancer. Mol Cancer 181:91. https://doi.org/10.1186/s12943-019-1019-x
Hua Y, Zhu Y, Zhang J, Zhu Z, Ning Z, Chen H et al (2018) miR-122 targets X-linked inhibitor of apoptosis protein to sensitize oxaliplatin-resistant colorectal cancer cells to oxaliplatin-mediated cytotoxicity. Cellular Physiol Biochem 515:2148–2159. https://doi.org/10.1159/000495832
Huang HL, Weng HY, Wang LQ, Yu CH, Huang QJ, Zhao PP et al (2012) Triggering Fbw7-mediated proteasomal degradation of c-Myc by oridonin induces cell growth inhibition and apoptosis. Mol Cancer Ther 115:1155–1165. https://doi.org/10.1158/1535-7163.Mct-12-0066
Huang WL, Luo CW, Chou CL, Yang CC, Chen TJ, Li CF et al (2020) High expression of UBE2B as a poor prognosis factor in patients with rectal cancer following chemoradiotherapy. Anticancer Res 4011:6305–6317
Huang F, Huang Z, Wei Q, Liu G, Pu J (2023a) E3 ubiquitin ligase HECTD3 is a tumor suppressor and mediates the polyubiquitination of SLC7A11 to promote ferroptosis in colon cancer. Exp Cell Res 4301:113697. https://doi.org/10.1016/j.yexcr.2023.113697
Huang D, Wu PE, Chen ZJ, Pang YC, Xu ZW, Tan J et al (2023b) Ethanol Extract of citrus grandis “Tomentosa” exerts anticancer effects by targeting Skp2/p27 pathway in non-small cell lung cancer. Mol Nutr Food Res. https://doi.org/10.1002/mnfr.202300061
Humblet Y (2004) Cetuximab: an IgG(1) monoclonal antibody for the treatment of epidermal growth factor receptor-expressing tumours. Expert Opin Pharmacother 57:1621–1633. https://doi.org/10.1517/14656566.5.7.1621
Hurwitz HI, Smith DC, Pitot HC, Brill JM, Chugh R, Rouits E et al (2015) Safety, pharmacokinetics, and pharmacodynamic properties of oral DEBIO1143 (AT-406) in patients with advanced cancer: results of a first-in-man study. Cancer Chemother Pharmacol 754:851–859. https://doi.org/10.1007/s00280-015-2709-8
Husnjak K, Dikic I (2012) Ubiquitin-binding proteins: decoders of ubiquitin-mediated cellular functions. Annu Rev Biochem 81:291–322. https://doi.org/10.1146/annurev-biochem-051810-094654
Infante JR, Dees EC, Olszanski AJ, Dhuria SV, Sen S, Cameron S et al (2014) Phase I dose-escalation study of LCL161, an oral inhibitor of apoptosis proteins inhibitor, in patients with advanced solid tumors. J Clin Oncol 3228:3103–3110. https://doi.org/10.1200/jco.2013.52.3993
Inuzuka H, Shaik S, Onoyama I, Gao D, Tseng A, Maser RS et al (2011) SCF(FBW7) regulates cellular apoptosis by targeting MCL1 for ubiquitylation and destruction. Nature 4717336:104–109. https://doi.org/10.1038/nature09732
Issaeva N, Bozko P, Enge M, Protopopova M, Verhoef LG, Masucci M et al (2004) Small molecule RITA binds to p53, blocks p53-HDM-2 interaction and activates p53 function in tumors. Nat Med 1012:1321–1328. https://doi.org/10.1038/nm1146
Izumi D, Ishimoto T, Miyake K, Eto T, Arima K, Kiyozumi Y et al (2017) Colorectal cancer stem cells acquire chemoresistance through the upregulation of F-Box/WD repeat-containing protein 7 and the consequent degradation of c-Myc. Stem Cells 359:2027–2036. https://doi.org/10.1002/stem.2668
Ji L, Jiang B, Jiang X, Charlat O, Chen A, Mickanin C et al (2017) The SIAH E3 ubiquitin ligases promote Wnt/β-catenin signaling through mediating Wnt-induced Axin degradation. Genes Dev 319:904–915. https://doi.org/10.1101/gad.300053.117
Jiang X, Charlat O, Zamponi R, Yang Y, Cong F (2015) Dishevelled promotes Wnt receptor degradation through recruitment of ZNRF3/RNF43 E3 ubiquitin ligases. Mol Cell 583:522–533. https://doi.org/10.1016/j.molcel.2015.03.015
Johansson H, Isabella Tsai YC, Fantom K, Chung CW, Kümper S, Martino L et al (2019) Fragment-based covalent ligand screening enables rapid discovery of inhibitors for the RBR E3 ubiquitin ligase HOIP. J Am Chem Soc 1416:2703–2712. https://doi.org/10.1021/jacs.8b13193
Johnston PG (2005) Stage II colorectal cancer: to treat or not to treat. Oncologist 105:332–334. https://doi.org/10.1634/theoncologist.10-5-332
Katayama K, Noguchi K, Sugimoto Y (2013) FBXO15 regulates P-glycoprotein/ABCB1 expression through the ubiquitin–proteasome pathway in cancer cells. Cancer Sci 1046:694–702. https://doi.org/10.1111/cas.12145
Kim ES (2004) Cetuximab as a single agent or in combination with chemotherapy in lung cancer. Clin Lung Cancer 6(Suppl 2):S80-84. https://doi.org/10.3816/clc.2004.s.019
Kim S, Jho EH (2010) The protein stability of Axin, a negative regulator of Wnt signaling, is regulated by Smad ubiquitination regulatory factor 2 (Smurf2). J Biol Chem 28547:36420–36426. https://doi.org/10.1074/jbc.M110.137471
Kuebler JP, Wieand HS, O’Connell MJ, Smith RE, Colangelo LH, Yothers G et al (2007) Oxaliplatin combined with weekly bolus fluorouracil and leucovorin as surgical adjuvant chemotherapy for stage II and III colon cancer: results from NSABP C-07. J Clin Oncol 2516:2198–2204. https://doi.org/10.1200/jco.2006.08.2974
Lama R, Galster SL, Xu C, Davison LW, Chemler SR, Wang X (2022) Dual targeting of MDM4 and FTH1 by MMRi71 for induced protein degradation and p53-independent apoptosis in leukemia cells. Molecules. https://doi.org/10.3390/molecules27227665
Lamouille S, Xu J, Derynck R (2014) Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol 153:178–196. https://doi.org/10.1038/nrm3758
Lee HJ, Li CF, Ruan D, Powers S, Thompson PA, Frohman MA et al (2016) The DNA damage transducer RNF8 facilitates cancer chemoresistance and progression through twist activation. Mol Cell 636:1021–1033. https://doi.org/10.1016/j.molcel.2016.08.009
Lee B, Min JA, Nashed A, Lee SO, Yoo JC, Chi SW et al (2019) A novel mechanism of irinotecan targeting MDM2 and Bcl-xL. Biochem Biophys Res Commun 5142:518–523. https://doi.org/10.1016/j.bbrc.2019.04.009
Li M, Zhang P (2009) The function of APC/CCdh1 in cell cycle and beyond. Cell Div 4:2. https://doi.org/10.1186/1747-1028-4-2
Li N, Lorenzi F, Kalakouti E, Normatova M, Babaei-Jadidi R, Tomlinson I et al (2015) FBXW7-mutated colorectal cancer cells exhibit aberrant expression of phosphorylated-p53 at Serine-15. Oncotarget 611:9240–9256. https://doi.org/10.18632/oncotarget.3284
Li Z, Wang Y, Li Y, Yin W, Mo L, Qian X et al (2018) Ube2s stabilizes β-Catenin through K11-linked polyubiquitination to promote mesendoderm specification and colorectal cancer development. Cell Death Dis 95:456. https://doi.org/10.1038/s41419-018-0451-y
Li N, Babaei-Jadidi R, Lorenzi F, Spencer-Dene B, Clarke P, Domingo E et al (2019) An FBXW7-ZEB2 axis links EMT and tumour microenvironment to promote colorectal cancer stem cells and chemoresistance. Oncogenesis 83:13. https://doi.org/10.1038/s41389-019-0125-3
Li Y, Wang T, Wan Q, Wang Q, Chen Z, Gao Y et al (2022) TRAF4 maintains deubiquitination of Caveolin-1 to drive glioblastoma stemness and temozolomide resistance. Can Res 8219:3573–3587. https://doi.org/10.1158/0008-5472.Can-21-3882
Li ASM, Kimani S, Wilson B, Noureldin M, González-Álvarez H, Mamai A et al (2023) Discovery of nanomolar DCAF1 small molecule ligands. J Med Chem 667:5041–5060. https://doi.org/10.1021/acs.jmedchem.2c02132
Liang Q, Tang C, Tang M, Zhang Q, Gao Y, Ge Z (2019) TRIM47 is up-regulated in colorectal cancer, promoting ubiquitination and degradation of SMAD4. J Exp Clin Cancer Res 381:159. https://doi.org/10.1186/s13046-019-1143-x
Liao Y, Yuan C, Huang M, Si W, Li D, Wu W et al (2023) AZD7762 induces CRBN dependent BAG3 degradation through ubiquitin-proteasome pathway. Anticancer Drugs. https://doi.org/10.1097/cad.0000000000001532
Liu L, Wong CC, Gong B, Yu J (2018a) Functional significance and therapeutic implication of ring-type E3 ligases in colorectal cancer. Oncogene 372:148–159. https://doi.org/10.1038/onc.2017.313
Liu M, Zhang X, Cai J, Li Y, Luo Q, Wu H et al (2018b) Downregulation of TRIM58 expression is associated with a poor patient outcome and enhances colorectal cancer cell invasion. Oncol Rep 403:1251–1260. https://doi.org/10.3892/or.2018.6525
Liu ML, Zang F, Zhang SJ (2019) RBCK1 contributes to chemoresistance and stemness in colorectal cancer (CRC). Biomed Pharmacotherapy 118:109250. https://doi.org/10.1016/j.biopha.2019.109250
Ma H, Wu Z, Peng J, Li Y, Huang H, Liao Y et al (2018) Inhibition of SLC1A5 sensitizes colorectal cancer to cetuximab. Int J Cancer 14212:2578–2588. https://doi.org/10.1002/ijc.31274
Mahammedi H, Planchat E, Pouget M, Durando X, Curé H, Guy L et al (2016) The new combination docetaxel, prednisone and curcumin in patients with castration-resistant prostate cancer: a pilot phase II study. Oncology 902:69–78. https://doi.org/10.1159/000441148
Malek E, Abdel-Malek MA, Jagannathan S, Vad N, Karns R, Jegga AG et al (2017) Pharmacogenomics and chemical library screens reveal a novel SCF(SKP2) inhibitor that overcomes Bortezomib resistance in multiple myeloma. Leukemia 313:645–653. https://doi.org/10.1038/leu.2016.258
Martinez-Balibrea E, Martínez-Cardús A, Ginés A, Ruiz de Porras V, Moutinho C, Layos L et al (2015) Tumor-related molecular mechanisms of oxaliplatin resistance. Mol Cancer Ther 148:1767–1776. https://doi.org/10.1158/1535-7163.Mct-14-0636
Martinez-Iglesias O, Casas-Pais A, Castosa R, Díaz-Díaz A, Roca-Lema D, Concha Á et al (2020) Hakin-1, a new specific small-molecule inhibitor for the E3 ubiquitin-ligase Hakai, inhibits carcinoma growth and progression. Cancers. https://doi.org/10.3390/cancers12051340
Martino E, Della Volpe S, Terribile E, Benetti E, Sakaj M, Centamore A et al (2017) The long story of camptothecin: From traditional medicine to drugs. Bioorg Med Chem Lett 274:701–707. https://doi.org/10.1016/j.bmcl.2016.12.085
Maser T, Zagorski J, Kelly S, Ostrander A, Goodyke A, Nagulapally A et al (2020) The MDM2 inhibitor CGM097 combined with the BET inhibitor OTX015 induces cell death and inhibits tumor growth in models of neuroblastoma. Cancer Med 921:8144–8158. https://doi.org/10.1002/cam4.3407
Mastropasqua F, Marzano F, Valletti A, Aiello I, Di Tullio G, Morgano A et al (2017) TRIM8 restores p53 tumour suppressor function by blunting N-MYC activity in chemo-resistant tumours. Mol Cancer 161:67. https://doi.org/10.1186/s12943-017-0634-7
Matt P, van Zwieten-Boot B, Calvo Rojas G, Ter Hofstede H, Garcia-Carbonero R, Camarero J et al (2011) The European Medicines Agency review of Tegafur/Gimeracil/Oteracil (Teysuno™) for the treatment of advanced gastric cancer when given in combination with cisplatin: summary of the Scientific Assessment of the Committee for medicinal products for human use (CHMP). Oncologist 1610:1451–1457. https://doi.org/10.1634/theoncologist.2011-0224
Matthews HK, Bertoli C, de Bruin RAM (2022) Cell cycle control in cancer. Nat Rev Mol Cell Biol 231:74–88. https://doi.org/10.1038/s41580-021-00404-3
Mavrommati I, Faedda R, Galasso G, Li J, Burdova K, Fischer R et al (2018) β-TrCP- and casein Kinase II-mediated degradation of cyclin F controls timely mitotic progression. Cell Rep 2413:3404–3412. https://doi.org/10.1016/j.celrep.2018.08.076
Mehta LS, Watson KE, Barac A, Beckie TM, Bittner V, Cruz-Flores S et al (2018) Cardiovascular disease and breast cancer: where these entities intersect: a scientific statement from the american heart association. Circulation 1378:e30–e66. https://doi.org/10.1161/cir.0000000000000556
Messersmith WA (2017) Systemic management of colorectal cancer. J Natl Comprehens Cancer Netw 155:699–702. https://doi.org/10.6004/jnccn.2017.0077
Milhollen MA, Narayanan U, Soucy TA, Veiby PO, Smith PG, Amidon B (2011) Inhibition of NEDD8-activating enzyme induces rereplication and apoptosis in human tumor cells consistent with deregulating CDT1 turnover. Can Res 718:3042–3051. https://doi.org/10.1158/0008-5472.Can-10-2122
Morawska K, Goirand F, Marceau L, Devaux M, Cueff A, Bertaut A et al (2018) 5-FU therapeutic drug monitoring as a valuable option to reduce toxicity in patients with gastrointestinal cancer. Oncotarget 914:11559–11571. https://doi.org/10.18632/oncotarget.24338
Morreale FE, Walden H (2016) Types of ubiquitin ligases. Cell 1651:248-248.e241. https://doi.org/10.1016/j.cell.2016.03.003
Morton CL, Wadkins RM, Danks MK, Potter PM (1999) The anticancer prodrug CPT-11 is a potent inhibitor of acetylcholinesterase but is rapidly catalyzed to SN-38 by butyrylcholinesterase. Can Res 597:1458–1463
Mukherjee S, Saha G, Roy NS, Naiya G, Ghosh MK, Roy S (2023) A small HDM2 antagonist peptide and a USP7 inhibitor synergistically inhibit the p53-HDM2-USP7 circuit. Chem Biol Drug Des 1021:126–136. https://doi.org/10.1111/cbdd.14255
Myant KB, Cammareri P, Hodder MC, Wills J, Von Kriegsheim A, Győrffy B et al (2017) HUWE1 is a critical colonic tumour suppressor gene that prevents MYC signalling, DNA damage accumulation and tumour initiation. EMBO Mol Med 92:181–197. https://doi.org/10.15252/emmm.201606684
Naito A, Yamamoto H, Kagawa Y, Naito Y, Okuzaki D, Otani K et al (2015) RFPL4A increases the G1 population and decreases sensitivity to chemotherapy in human colorectal cancer cells. J Biol Chem 29010:6326–6337. https://doi.org/10.1074/jbc.M114.614859
Nakajima H, Fujiwara H, Furuichi Y, Tanaka K, Shimbara N (2008) A novel small-molecule inhibitor of NF-kappaB signaling. Biochem Biophys Res Commun 3684:1007–1013. https://doi.org/10.1016/j.bbrc.2008.01.166
Nasrullah U, Haeussler K, Biyanee A, Wittig I, Pfeilschifter J, Eberhardt W (2019) Identification of TRIM25 as a negative regulator of caspase-2 expression reveals a novel target for sensitizing colon carcinoma cells to intrinsic apoptosis. Cells. https://doi.org/10.3390/cells8121622
Nasrullah U, Stanke K, Recknagel V, Bozkurt S, Wurzel P, Gauer S et al (2023) The E3 ligase TRIM25 impairs apoptotic cell death in colon carcinoma cells via destabilization of Caspase-7 mRNA: a possible role of hnRNPH1. Cells. https://doi.org/10.3390/cells12010201
Niu B, Liu J, Lv B, Lin J, Li X, Wu C et al (2021) Interplay between transforming growth factor-β and Nur77 in dual regulations of inhibitor of differentiation 1 for colonic tumorigenesis. Nat Commun 121:2809. https://doi.org/10.1038/s41467-021-23048-5
Oberoi-Khanuja TK, Rajalingam K (2012) IAPs as E3 ligases of Rac1: shaping the move. Small GTPases 32:131–136. https://doi.org/10.4161/sgtp.19988
Oikawa D, Sato Y, Ohtake F, Komakura K, Hanada K, Sugawara K et al (2020) Molecular bases for HOIPINs-mediated inhibition of LUBAC and innate immune responses. Commun Biol 31:163. https://doi.org/10.1038/s42003-020-0882-8
Park SH, Jung EH, Kim GY, Kim BC, Lim JH, Woo CH (2015) Itch E3 ubiquitin ligase positively regulates TGF-β signaling to EMT via Smad7 ubiquitination. Mol Cells 381:20–25. https://doi.org/10.14348/molcells.2015.2120
Park SH, Jo MJ, Kim BR, Jeong YA, Na YJ, Kim JL et al (2019) Sonic hedgehog pathway activation is associated with cetuximab resistance and EPHB3 receptor induction in colorectal cancer. Theranostics 98:2235–2251. https://doi.org/10.7150/thno.30678
Paschall AV, Zimmerman MA, Torres CM, Yang D, Chen MR, Li X et al (2014) Ceramide targets xIAP and cIAP1 to sensitize metastatic colon and breast cancer cells to apoptosis induction to suppress tumor progression. BMC Cancer 14:24. https://doi.org/10.1186/1471-2407-14-24
Pei J, Xiao Y, Liu X, Hu W, Sobh A, Yuan Y et al (2023) Piperlongumine conjugates induce targeted protein degradation. Cell Chem Biol 302:203-213.e217. https://doi.org/10.1016/j.chembiol.2023.01.004
Pelletier B, Duhamel S, Tambutet G, Jarvis S, Cléroux P, David M et al (2023) Discovery of benzodiazepine-based inhibitors of the E2 enzyme UBCH10 from a cell-based p21 degradation screen. ACS Chem Biol 185:1039–1046. https://doi.org/10.1021/acschembio.2c00909
Proietti S, Cucina A, Dobrowolny G, D’Anselmi F, Dinicola S, Masiello MG et al (2014) Melatonin down-regulates MDM2 gene expression and enhances p53 acetylation in MCF-7 cells. J Pineal Res 571:120–129. https://doi.org/10.1111/jpi.12150
Psatha K, Kollipara L, Drakos E, Deligianni E, Brintakis K, Patsouris E et al (2023) Interruption of p53-MDM2 interaction by Nutlin-3a in human lymphoma cell models initiates a cell-dependent global effect on transcriptome and proteome level. Cancers. https://doi.org/10.3390/cancers15153903
Qi J, Fan L, Hussain A (2015) Implications of ubiquitin ligases in castration-resistant prostate cancer. Curr Opin Oncol 273:172–176. https://doi.org/10.1097/cco.0000000000000178
Ravandi F, Gojo I, Patnaik MM, Minden MD, Kantarjian H, Johnson-Levonas AO et al (2016) A phase I trial of the human double minute 2 inhibitor (MK-8242) in patients with refractory/recurrent acute myelogenous leukemia (AML). Leuk Res 48:92–100. https://doi.org/10.1016/j.leukres.2016.07.004
Ravi R, Jain AJ, Schulick RD, Pham V, Prouser TS, Allen H et al (2004) Elimination of hepatic metastases of colon cancer cells via p53-independent cross-talk between irinotecan and Apo2 ligand/TRAIL. Can Res 6424:9105–9114. https://doi.org/10.1158/0008-5472.Can-04-2488
Ray-Coquard I, Blay JY, Italiano A, Le Cesne A, Penel N, Zhi J et al (2012) Effect of the MDM2 antagonist RG7112 on the P53 pathway in patients with MDM2-amplified, well-differentiated or dedifferentiated liposarcoma: an exploratory proof-of-mechanism study. Lancet Oncol 1311:1133–1140. https://doi.org/10.1016/s1470-2045(12)70474-6
Rew Y, Sun D (2014) Discovery of a small molecule MDM2 inhibitor (AMG 232) for treating cancer. J Med Chem 5715:6332–6341. https://doi.org/10.1021/jm500627s
Ribeiro F, Alves PKN, Bechara LRG, Ferreira JCB, Labeit S, Moriscot AS (2023) Small-molecule inhibition of MuRF1 prevents early disuse-induced diaphragmatic dysfunction and atrophy. Int J Mol Sci. https://doi.org/10.3390/ijms24043637
Rivory LP, Robert J (1995) Molecular, cellular, and clinical aspects of the pharmacology of 20(S)camptothecin and its derivatives. Pharmacol Ther 682:269–296. https://doi.org/10.1016/0163-7258(95)02009-8
Roilo M, Kullmann MK, Hengst L (2018) Cold-inducible RNA-binding protein (CIRP) induces translation of the cell-cycle inhibitor p27Kip1. Nucleic Acids Res 466:3198–3210. https://doi.org/10.1093/nar/gkx1317
Rotin D, Kumar S (2009) Physiological functions of the HECT family of ubiquitin ligases. Nat Rev Mol Cell Biol 106:398–409. https://doi.org/10.1038/nrm2690
Sackton KL, Dimova N, Zeng X, Tian W, Zhang M, Sackton TB et al (2014) Synergistic blockade of mitotic exit by two chemical inhibitors of the APC/C. Nature 5147524:646–649. https://doi.org/10.1038/nature13660
Saif MW, Syrigos KN, Katirtzoglou NA (2009) S-1: a promising new oral fluoropyrimidine derivative. Expert Opin Investig Drugs 183:335–348. https://doi.org/10.1517/13543780902729412
Sakamoto H, Egashira S, Saito N, Kirisako T, Miller S, Sasaki Y et al (2015) Gliotoxin suppresses NF-κB activation by selectively inhibiting linear ubiquitin chain assembly complex (LUBAC). ACS Chem Biol 103:675–681. https://doi.org/10.1021/cb500653y
Sanders MA, Haynes B, Nangia-Makker P, Polin LA, Shekhar MP (2017) Pharmacological targeting of RAD6 enzyme-mediated translesion synthesis overcomes resistance to platinum-based drugs. J Biol Chem 29225:10347–10363. https://doi.org/10.1074/jbc.M117.792192
Sargent D, Sobrero A, Grothey A, O’Connell MJ, Buyse M, Andre T et al (2009) Evidence for cure by adjuvant therapy in colon cancer: observations based on individual patient data from 20,898 patients on 18 randomized trials. J Clin Oncol 276:872–877. https://doi.org/10.1200/jco.2008.19.5362
Sawa M, Masuda M, Yamada T (2016) Targeting the Wnt signaling pathway in colorectal cancer. Expert Opin Ther Targets 204:419–429. https://doi.org/10.1517/14728222.2016.1098619
Sekeres MA, Watts J, Radinoff A, Sangerman MA, Cerrano M, Lopez PF et al (2021) Correction to: Randomized phase 2 trial of pevonedistat plus azacitidine versus azacitidine for higher-risk MDS/CMML or low-blast AML. Leukemia 3512:3637. https://doi.org/10.1038/s41375-021-01473-1
Sekiguchi N, Kasahara S, Miyamoto T, Kiguchi T, Ohno H, Takagi T et al (2023) Phase I dose-escalation study of milademetan in patients with relapsed or refractory acute myeloid leukemia. Int J Hematol 1171:68–77. https://doi.org/10.1007/s12185-022-03464-z
Sharma S, Kumar P (2023) Decoding the role of MDM2 as a potential ubiquitin E3 ligase and identifying the therapeutic efficiency of alkaloids against MDM2 in combating glioblastoma. ACS Omega 85:5072–5087. https://doi.org/10.1021/acsomega.2c07904
Shay JW, Homma N, Zhou R, Naseer MI, Chaudhary AG, Al-Qahtani M, et al., (2016) Abstracts from the 3rd International Genomic Medicine Conference (3rd IGMC 2015): Jeddah, Kingdom of Saudi Arabia. 30 November - 3 December 2015, BMC genomics 6:487, doi: https://doi.org/10.1186/s12864-016-2858-0
Shelton J, Lu X, Hollenbaugh JA, Cho JH, Amblard F, Schinazi RF (2016) Metabolism, biochemical actions, and chemical synthesis of anticancer nucleosides, nucleotides, and base analogs. Chem Rev 11623:14379–14455. https://doi.org/10.1021/acs.chemrev.6b00209
Shen C, Nayak A, Neitzel LR, Adams AA, Silver-Isenstadt M, Sawyer LM et al (2021) The E3 ubiquitin ligase component, Cereblon, is an evolutionarily conserved regulator of Wnt signaling. Nat Commun 121:5263. https://doi.org/10.1038/s41467-021-25634-z
Shukla S, Ying W, Gray F, Yao Y, Simes ML, Zhao Q et al (2021) Small-molecule inhibitors targeting Polycomb repressive complex 1 RING domain. Nat Chem Biol 177:784–793. https://doi.org/10.1038/s41589-021-00815-5
Siddiqui A, Gollavilli PN, Schwab A, Vazakidou ME, Ersan PG, Ramakrishnan M et al (2019) Thymidylate synthase maintains the de-differentiated state of triple negative breast cancers. Cell Death Differ 2611:2223–2236. https://doi.org/10.1038/s41418-019-0289-6
Singleton MR, Uhlmann F (2017) Separase-securin complex: a cunning way to control chromosome segregation. Nat Struct Mol Biol 244:337–339. https://doi.org/10.1038/nsmb.3393
Sondo E, Falchi F, Caci E, Ferrera L, Giacomini E, Pesce E et al (2018) Pharmacological inhibition of the ubiquitin ligase RNF5 rescues F508del-CFTR in cystic fibrosis airway epithelia. Cell Chem Biol 257:891-905.e898. https://doi.org/10.1016/j.chembiol.2018.04.010
Soond SM, Chantry A (2011) Selective targeting of activating and inhibitory Smads by distinct WWP2 ubiquitin ligase isoforms differentially modulates TGFβ signalling and EMT. Oncogene 3021:2451–2462. https://doi.org/10.1038/onc.2010.617
Srikant S, Gaudet R (2019) Mechanics and pharmacology of substrate selection and transport by eukaryotic ABC exporters. Nat Struct Mol Biol 269:792–801. https://doi.org/10.1038/s41594-019-0280-4
Stein EM, DeAngelo DJ, Chromik J, Chatterjee M, Bauer S, Lin C-C et al (2022) Results from a first-in-human phase I study of siremadlin (HDM201) in patients with advanced wild-type TP53 solid tumors and acute leukemia. Clin Cancer Res 285:870–881. https://doi.org/10.1158/1078-0432.Ccr-21-1295
Stenvang J, Kümler I, Nygård SB, Smith DH, Nielsen D, Brünner N et al (2013) Biomarker-guided repurposing of chemotherapeutic drugs for cancer therapy: a novel strategy in drug development. Front Oncol 3:313. https://doi.org/10.3389/fonc.2013.00313
Stewart MD, Ritterhoff T, Klevit RE, Brzovic PS (2016) E2 enzymes: more than just middle men. Cell Res 264:423–440. https://doi.org/10.1038/cr.2016.35
Stintzing S, Zhang W, Heinemann V, Neureiter D, Kemmerling R, Kirchner T et al (2015) Polymorphisms in genes involved in EGFR turnover are predictive for cetuximab efficacy in colorectal cancer. Mol Cancer Ther 1410:2374–2381. https://doi.org/10.1158/1535-7163.Mct-15-0121
Su W, Han HH, Wang Y, Zhang B, Zhou B, Cheng Y et al (2019) The polycomb repressor complex 1 drives double-negative prostate cancer metastasis by coordinating stemness and immune suppression. Cancer Cell 362:139-155.e110. https://doi.org/10.1016/j.ccell.2019.06.009
Sun XS, Tao Y, Le Tourneau C, Pointreau Y, Sire C, Kaminsky MC et al (2020) Debio 1143 and high-dose cisplatin chemoradiotherapy in high-risk locoregionally advanced squamous cell carcinoma of the head and neck: a double-blind, multicentre, randomised, phase 2 study. Lancet Oncol 219:1173–1187. https://doi.org/10.1016/s1470-2045(20)30327-2
Sun Y, Liu Y, Ma X, Hu H (2021) The influence of cell cycle regulation on chemotherapy. Int J Mol Sci. https://doi.org/10.3390/ijms22136923
Sun Y, Sun H, Qi Y, Pan M, An N, Leng X et al (2022) Ring finger protein 6 enhances chemo-resistance by transcriptionally activating proliferating cell nuclear antigen expression and attenuating DNA damage in lung adenocarcinoma. Cancer Lett 534:215609. https://doi.org/10.1016/j.canlet.2022.215609
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. (2021) Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries, CA 713:209–249
Swords RT, Erba HP, DeAngelo DJ, Bixby DL, Altman JK, Maris M et al (2015) Pevonedistat (MLN4924), a First-in-Class NEDD8-activating enzyme inhibitor, in patients with acute myeloid leukaemia and myelodysplastic syndromes: a phase 1 study. Br J Haematol 1694:534–543. https://doi.org/10.1111/bjh.13323
Takahashi S, Fujiwara Y, Nakano K, Shimizu T, Tomomatsu J, Koyama T et al (2021) Safety and pharmacokinetics of milademetan, a MDM2 inhibitor, in Japanese patients with solid tumors: A phase I study. Cancer Sci 1126:2361–2370. https://doi.org/10.1111/cas.14875
Tan M, Li Y, Yang R, Xi N, Sun Y (2011) Inactivation of SAG E3 ubiquitin ligase blocks embryonic stem cell differentiation and sensitizes leukemia cells to retinoid acid. PLoS ONE 611:e27726. https://doi.org/10.1371/journal.pone.0027726
Thirion P, Michiels S, Pignon JP, Buyse M, Braud AC, Carlson RW et al (2004) Modulation of fluorouracil by leucovorin in patients with advanced colorectal cancer: an updated meta-analysis. J Clin Oncol 2218:3766–3775. https://doi.org/10.1200/jco.2004.03.104
Toma-Fukai S, Shimizu T (2021) Structural diversity of ubiquitin E3 ligase. Molecules. https://doi.org/10.3390/molecules26216682
Tovar C, Graves B, Packman K, Filipovic Z, Higgins B, Xia M et al (2013) MDM2 small-molecule antagonist RG7112 activates p53 signaling and regresses human tumors in preclinical cancer models. Can Res 738:2587–2597. https://doi.org/10.1158/0008-5472.Can-12-2807
Tsilimigras MC, Fodor A, Jobin C (2017) Carcinogenesis and therapeutics: the microbiota perspective. Nat Microbiol 2:17008. https://doi.org/10.1038/nmicrobiol.2017.8
Van Cutsem E, Cervantes A, Adam R, Sobrero A, Van Krieken JH, Aderka D et al (2016) ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann Oncol 278:1386–1422. https://doi.org/10.1093/annonc/mdw235
van Wijk SJ, Timmers HT (2010) The family of ubiquitin-conjugating enzymes (E2s): deciding between life and death of proteins. FASEB J 244:981–993. https://doi.org/10.1096/fj.09-136259
Vander Heiden MG, DeBerardinis RJ (2017) Understanding the intersections between metabolism and cancer biology. Cell 1684:657–669. https://doi.org/10.1016/j.cell.2016.12.039
Varshavsky A (2012) The ubiquitin system, an immense realm. Annu Rev Biochem 81:167–176. https://doi.org/10.1146/annurev-biochem-051910-094049
Vassilev LT (2007) MDM2 inhibitors for cancer therapy. Trends Mol Med 131:23–31. https://doi.org/10.1016/j.molmed.2006.11.002
Vodenkova S, Buchler T, Cervena K, Veskrnova V, Vodicka P, Vymetalkova V (2020) 5-fluorouracil and other fluoropyrimidines in colorectal cancer: past, present and future. Pharmacol Ther 206:107447. https://doi.org/10.1016/j.pharmthera.2019.107447
Wade M, Li YC, Wahl GM (2013) MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat Rev Cancer 132:83–96. https://doi.org/10.1038/nrc3430
Wan J, Zhu J, Li G, Zhang Z (2016) Radiosensitization of human colorectal cancer cells by MLN4924: an inhibitor of NEDD8-activating enzyme. Technol Cancer Res Treat 154:527–534. https://doi.org/10.1177/1533034615588197
Wang Z, Inuzuka H, Zhong J, Liu P, Sarkar FH, Sun Y et al (2012) Identification of acetylation-dependent regulatory mechanisms that govern the oncogenic functions of Skp2. Oncotarget 311:1294–1300. https://doi.org/10.18632/oncotarget.740
Wang L, Yang H, Abel EV, Ney GM, Palmbos PL, Bednar F et al (2015) ATDC induces an invasive switch in KRAS-induced pancreatic tumorigenesis. Genes Dev 292:171–183. https://doi.org/10.1101/gad.253591.114
Wang X, De Geyter C, Jia Z, Peng Y, Zhang H (2020a) HECTD1 regulates the expression of SNAIL: Implications for epithelial-mesenchymal transition. Int J Oncol 565:1186–1198. https://doi.org/10.3892/ijo.2020.5002
Wang S, Wang T, Wang L, Zhong L, Li K (2020b) Overexpression of RNF126 promotes the development of colorectal cancer via enhancing p53 ubiquitination and degradation. Onco Targets Ther 13:10917–10929. https://doi.org/10.2147/ott.S271855
Wang N, Yang L, Dai J, Wu Y, Zhang R, Jia X et al (2021) 5-FU inhibits migration and invasion of CRC cells through PI3K/AKT pathway regulated by MARCH1. Cell Biol Int 452:368–381. https://doi.org/10.1002/cbin.11493
Wang H, Yang W, Qin Q, Yang X, Yang Y, Liu H et al (2022) E3 ubiquitin ligase MAGI3 degrades c-Myc and acts as a predictor for chemotherapy response in colorectal cancer. Mol Cancer 211:151. https://doi.org/10.1186/s12943-022-01622-9
Wang Y, Gao G, Wei X, Zhang Y, Yu J (2023) UBE2T promotes temozolomide resistance of glioblastoma through regulating the Wnt/β-Catenin signaling pathway. Drug Des Dev Ther 17:1357–1369. https://doi.org/10.2147/dddt.S405450
Weber J, Polo S, Maspero E (2019) HECT E3 ligases: a tale with multiple facets. Front Physiol 10:370. https://doi.org/10.3389/fphys.2019.00370
Wu L, Grigoryan AV, Li Y, Hao B, Pagano M, Cardozo TJ (2012) Specific small molecule inhibitors of Skp2-mediated p27 degradation. Chem Biol 1912:1515–1524. https://doi.org/10.1016/j.chembiol.2012.09.015
Wu B, Piloto S, Zeng W, Hoverter NP, Schilling TF, Waterman ML (2013) Ring Finger Protein 14 is a new regulator of TCF/β-catenin-mediated transcription and colon cancer cell survival. EMBO Rep 144:347–355. https://doi.org/10.1038/embor.2013.19
Wu H, Lu XX, Wang JR, Yang TY, Li XM, He XS et al (2019) TRAF6 inhibits colorectal cancer metastasis through regulating selective autophagic CTNNB1/β-catenin degradation and is targeted for GSK3B/GSK3β-mediated phosphorylation and degradation. Autophagy 159:1506–1522. https://doi.org/10.1080/15548627.2019.1586250
Xiao C, Wu G, Zhou Z, Zhang X, Wang Y, Song G et al (2019) RBBP6, a RING finger-domain E3 ubiquitin ligase, induces epithelial-mesenchymal transition and promotes metastasis of colorectal cancer. Cell Death Dis 1011:833. https://doi.org/10.1038/s41419-019-2070-7
Xiong Z, Fu Z, Shi J, Jiang X, Wan H (2017) HtrA1 down-regulation induces cisplatin resistance in colon cancer by increasing XIAP and activating PI3K/Akt pathway. Ann Clin Lab Sci 473:264–270
Xu M, Zhu C, Zhao X, Chen C, Zhang H, Yuan H et al (2015) Atypical ubiquitin E3 ligase complex Skp1-Pam-Fbxo45 controls the core epithelial-to-mesenchymal transition-inducing transcription factors. Oncotarget 62:979–994. https://doi.org/10.18632/oncotarget.2825
Xu J, Lv G, Xu B, Jiang B (2020) Overexpression of UBE2M through Wnt/β-Catenin signaling is associated with poor prognosis and chemotherapy resistance in colorectal cancer. Trans Cancer Res 99:5614–5625. https://doi.org/10.21037/tcr-20-2641
Xu Y, Qiu A, Peng F, Tan X, Wang J, Gong X (2021) Exosomal transfer of circular RNA FBXW7 ameliorates the chemoresistance to oxaliplatin in colorectal cancer by sponging miR-18b-5p. Neoplasma 681:108–118. https://doi.org/10.4149/neo_2020_200417N414
Yamaguchi K, Shimamura T, Tada M (2006) [Capecitabine].Gan to kagaku ryoho. Cancer Chemotherapy 337:891–895
Yan X, Chen J, Meng Y, He C, Zou S, Li P et al (2019) RAD18 may function as a predictor of response to preoperative concurrent chemoradiotherapy in patients with locally advanced rectal cancer through caspase-9-caspase-3-dependent apoptotic pathway. Cancer Med 86:3094–3104. https://doi.org/10.1002/cam4.2203
Yan W, Zhong Y, Hu X, Xu T, Zhang Y, Kales S et al (2023) Auranofin targets UBA1 and enhances UBA1 activity by facilitating ubiquitin trans-thioesterification to E2 ubiquitin-conjugating enzymes. Nat Commun 141:4798. https://doi.org/10.1038/s41467-023-40537-x
Yang C, Wu J, He H, Liu H (2020) Small molecule NSC1892 targets the CUL4A/4B-DDB1 interactions and causes impairment of CRL4(DCAF4) E3 ligases to inhibit colorectal cancer cell growth. Int J Biol Sci 166:1059–1070. https://doi.org/10.7150/ijbs.40235
Yu M, Tong X, Qi B, Qu H, Dong S, Yu B et al (2014) Berberine enhances chemosensitivity to irinotecan in colon cancer via inhibition of NF-κB. Mol Med Rep 91:249–254. https://doi.org/10.3892/mmr.2013.1762
Yu P, Fan Y, Qu X, Zhang J, Song N, Liu J et al (2016) Cbl-b regulates the sensitivity of cetuximab through ubiquitin-proteasome system in human gastric cancer cells. J BUON 214:867–873
Yu Z, Jiang X, Qin L, Deng H, Wang J, Ren W et al (2021) A novel UBE2T inhibitor suppresses Wnt/β-catenin signaling hyperactivation and gastric cancer progression by blocking RACK1 ubiquitination. Oncogene 405:1027–1042. https://doi.org/10.1038/s41388-020-01572-w
Zarkou V, Galaras A, Giakountis A, Hatzis P (2018) Crosstalk mechanisms between the WNT signaling pathway and long non-coding RNAs. Non-Coding RNA Res 32:42–53. https://doi.org/10.1016/j.ncrna.2018.04.001
Zeng X, Sigoillot F, Gaur S, Choi S, Pfaff KL, Oh DC et al (2010) Pharmacologic inhibition of the anaphase-promoting complex induces a spindle checkpoint-dependent mitotic arrest in the absence of spindle damage. Cancer Cell 184:382–395. https://doi.org/10.1016/j.ccr.2010.08.010
Zhang S, Zhou L, Hong B, van den Heuvel AP, Prabhu VV, Warfel NA et al (2015) Small-molecule NSC59984 restores p53 pathway signaling and antitumor effects against colorectal cancer via p73 activation and degradation of mutant p53. Can Res 7518:3842–3852. https://doi.org/10.1158/0008-5472.Can-13-1079
Zhang X, Li CF, Zhang L, Wu CY, Han L, Jin G et al (2016) TRAF6 restricts p53 mitochondrial translocation, apoptosis, and tumor suppression. Mol Cell 644:803–814. https://doi.org/10.1016/j.molcel.2016.10.002
Zhang Y, Feng Y, Ji D, Wang Q, Qian W, Wang S et al (2018) TRIM27 functions as an oncogene by activating epithelial-mesenchymal transition and p-AKT in colorectal cancer. Int J Oncol 532:620–632. https://doi.org/10.3892/ijo.2018.4408
Zhang Z, He G, Lv Y, Liu Y, Niu Z, Feng Q et al (2022) HERC3 regulates epithelial-mesenchymal transition by directly ubiquitination degradation EIF5A2 and inhibits metastasis of colorectal cancer. Cell Death Dis 131:74. https://doi.org/10.1038/s41419-022-04511-7
Zhao CY, Szekely L, Bao W, Selivanova G (2010) Rescue of p53 function by small-molecule RITA in cervical carcinoma by blocking E6-mediated degradation. Can Res 708:3372–3381. https://doi.org/10.1158/0008-5472.Can-09-2787
Zhao Y, Morgan MA, Sun Y (2014) Targeting Neddylation pathways to inactivate cullin-RING ligases for anticancer therapy. Antioxid Redox Signal 2117:2383–2400. https://doi.org/10.1089/ars.2013.5795
Zhao C, Zhao Q, Zhang C, Wang G, Yao Y, Huang X et al (2017) miR-15b-5p resensitizes colon cancer cells to 5-fluorouracil by promoting apoptosis via the NF-κB/XIAP axis. Sci Rep 71:4194. https://doi.org/10.1038/s41598-017-04172-z
Zhao Z, Zhang G, Li W (2020) MT2A promotes oxaliplatin resistance in colorectal cancer cells. Cell Biochem Biophys 784:475–482. https://doi.org/10.1007/s12013-020-00930-5
Zhao Y, Li J, Chen J, Ye M, Jin X (2022) Functional roles of E3 ubiquitin ligases in prostate cancer. J Mol Med (berl) 1008:1125–1144. https://doi.org/10.1007/s00109-022-02229-9
Zheng S, Zhou C, Wang Y, Li H, Sun Y, Shen Z (2020) TRIM6 promotes colorectal cancer cells proliferation and response to thiostrepton by TIS21/FoxM1. J Exp Clin Cancer Res 391:23. https://doi.org/10.1186/s13046-019-1504-5
Zhong J, Ogura K, Wang Z, Inuzuka H (2013) Degradation of the transcription factor Twist, an oncoprotein that promotes cancer metastasis. Discov Med 1580:7–15
Zhong T, Zhang J, Liu X, Li H (2023) TRIM17-mediated ubiquitination and degradation of RBM38 promotes cisplatin resistance in non-small cell lung cancer. Cell Oncol (dordr). https://doi.org/10.1007/s13402-023-00825-6
Zhou Q, Pham KTM, Hu H, Kurasawa Y, Li Z (2019) A kinetochore-based ATM/ATR-independent DNA damage checkpoint maintains genomic integrity in trypanosomes. Nucleic Acids Res 4715:7973–7988. https://doi.org/10.1093/nar/gkz476
Zhou L, Yu X, Li M, Gong G, Liu W, Li T et al (2020) Cdh1-mediated Skp2 degradation by dioscin reprogrammes aerobic glycolysis and inhibits colorectal cancer cells growth. EBioMedicine 51:102570. https://doi.org/10.1016/j.ebiom.2019.11.031
Zhou S, Peng J, Xiao L, Zhou C, Fang Y, Ou Q et al (2021a) TRIM25 regulates oxaliplatin resistance in colorectal cancer by promoting EZH2 stability. Cell Death Dis 125:463. https://doi.org/10.1038/s41419-021-03734-4
Zhou G, Wu H, Lin J, Lin R, Feng B, Liu Z (2021b) TRIM21 Is decreased in colitis-associated cancer and negatively regulates epithelial carcinogenesis. Inflamm Bowel Dis 274:458–468. https://doi.org/10.1093/ibd/izaa229
Zhu GX, Gao D, Shao ZZ, Chen L, Ding WJ, Yu QF (2021a) Wnt/β-catenin signaling: Causes and treatment targets of drug resistance in colorectal cancer (Review). Mol Med Rep. https://doi.org/10.3892/mmr.2020.11744
Zhu L, Wu J, Liu H (2021b) Downregulation of HERC5 E3 ligase attenuates the ubiquitination of CtBP1 to inhibit apoptosis in colorectal cancer cells. Carcinogenesis 428:1119–1130. https://doi.org/10.1093/carcin/bgab053
Zhuang J, Shirazi F, Singh RK, Kuiatse I, Wang H, Lee HC et al (2019) Ubiquitin-activating enzyme inhibition induces an unfolded protein response and overcomes drug resistance in myeloma. Blood 13314:1572–1584. https://doi.org/10.1182/blood-2018-06-859686
Funding
This work was supported by The Natural Science Foundation of Ningbo (Grant No. 2021J017 and 2021J065), The Public Welfare Science and Technology Program Project of Ningbo (Grant No. 2021S116), The National Natural Science Foundation of China (Grant No. 32270821), Zhejiang Provincial Natural Science Foundation of China (No. LY24C050001), the Youth Science and technology leader of Ningbo (No.2023QL052), and The K.C. Wong Magna Fund of Ningbo University.
Author information
Authors and Affiliations
Contributions
Conceptualization, Z.R.; investigation, Z.R.; writing—original draft preparation, Z.R. and K.F.Z.; writing—review and editing, X.F.J. and J.C.; supervision, X.F.J. and J.C.; and visualization and legends, X.F.J. and J.C. All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rong, Z., Zheng, K., Chen, J. et al. The cross talk of ubiquitination and chemotherapy tolerance in colorectal cancer. J Cancer Res Clin Oncol 150, 154 (2024). https://doi.org/10.1007/s00432-024-05659-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00432-024-05659-9