
Abstract
Immunoglobulin-derived light-chain (AL) amyloidosis of lungs and bronchi can appear as a systemic and a local form. While systemic AL amyloidosis may need haemato-oncological care, the localised form can be treated restrained. We re-evaluated 207 specimens of lungs and bronchi sent in for amyloid diagnostics. Amyloid was diagnosed by polarization microscopy using Congo red-stained tissue specimens and classified immunohistochemically. Histoanatomical amyloid distribution patterns were documented as well as additional histological findings. For 118 patients with AL amyloidosis, we retrieved clinical data. CT scan results were available from 59 patients. AL amyloidosis was the most common type (183 cases). ALλ was found in 141 and of ALκ in 27 cases. Fifteen cases were AL amyloid not otherwise specified. Twenty cases harboured transthyretin and three serum amyloid A derived amyloid. By correlation of histoanatomy, radiological and clinical data, amyloid was rarely in the initial differential diagnosis. Local AL amyloidosis often presented with a nodular pattern on CT scan and showed a significantly better disease-specific 10-year survival compared with systemic AL amyloidosis (96.0 vs. 51.9%). Localised and systemic pulmonary and bronchial AL amyloidosis are having a completely different prognosis. While CT scan might be indicative, histological and clinical assessment are mandatory to reach a proper diagnosis and guide patient care.
Similar content being viewed by others


Introduction
Amyloid is characterized by the pathological deposition of peptides and proteins in diverse tissues and organs interfering with normal tissue and organ function [1]. More than 35 autologous, physiological proteins have been identified, which can form amyloid [2]. The gold standard of amyloid diagnosis is the histological examination of a Congo red-stained specimen under polarized light and the detection of a characteristic green-yellow-orange birefringence [3,4,5]. Amyloid can affect any organ or tissue type [6]. Different amyloid types can show different clinical pictures depending on organ involvement and deposition pattern. The immunoglobulin light-chain (AL) amyloidosis can occur as a local or systemic variant and is able to involve almost every organ/tissue type [7, 8]. Transthyretin-derived (ATTR) amyloidosis occurs as hereditary form due to a point mutation in the TTR gene or as wild-type variant. Clinical presentation is characterized by sensomotoric polyneuropathy and/or cardiomyopathy [9,10,11]. Amyloid A (AA) amyloidosis mainly presents with renal involvement [1].
Lungs and bronchi can be affected by localised and systemic amyloidosis [7, 12, 13]. The exact diagnosis of the amyloid type and differentiation between a localised and a systemic form is clinically important, as patient prognosis and patient management vary [6, 12, 14]. While localised amyloidosis usually requires restrained symptom-orientated therapy, the treatment of systemic amyloidosis may include, e.g. high-dose chemotherapy, autologous stem cell transplantation, dialysis, and occasionally, organ transplantation [12, 15]. Median survival of systemic AL amyloidosis with cardiac involvement is 15.7 months [16].
Patients with amyloidosis of the lungs and bronchi may present clinically with symptoms of dyspnoea, cough, haemoptysis, pleural effusions, pneumonia and pulmonary arterial hypertension [12, 13, 17, 18]. In chest x-ray or computed tomography scans, findings are e.g. nodules, bronchial wall thickening, cysts, calcifications, septal thickening, atelectasis and ground glass opacities [19,20,21]. As symptoms and imaging findings are not very specific, the differential diagnosis of amyloidosis in lungs and bronchi is broad and includes diseases, such as neoplasms, infectious, interstitial and granulomatous lung diseases [19, 20].
In this retrospective study on amyloidosis in lungs and bronchi, we tested the following hypotheses: (1) lungs and bronchi are affected by diverse types of local and systemic amyloidosis, (2) different types of amyloidosis show specific demographic patient characteristics, (3) systemic and localised AL amyloidosis can get distinguished histopathologically and (4) localised AL amyloidosis is having a better prognosis than systemic AL amyloidosis.
Materials and methods
Patients
From the Amyloid Registry Kiel we retrieved all cases with biopsy and resection specimens of the lung and bronchi. Two hundred and twenty-seven specimens were obtained between January 2006 and January 2017 and referred to the Amyloid Registry for a second opinion, i.e. confirmation of amyloid, and subsequent classification of the amyloid type. Twenty biopsies from 20 patients submitted to the Amyloid Registry Kiel were excluded from this series as the presence of amyloid could not be confirmed.
For all cases of AL amyloidosis, the pathologists, submitting the biopsies to the Amyloid Registry Kiel, were asked for the contact details of the general practitioners and clinicians treating the patients. Then, the general practitioners and clinicians were asked to fill out a survey and submit the last medical report on the patient. The survey was designed to collect information about the clinical presentation (localised or systemic disease), the follow-up of the patients, the chosen form of treatment, the appearance of lymphoma or other haematological diseases and monoclonal gammopathy of undetermined significance (MGUS) in serum immunofixation electrophoresis.
Histology
All tissue specimens were fixed in formalin and embedded in paraffin (FFPE). At the Amyloid Registry, serial sections were cut from each paraffin block and stained with haematoxylin and eosin, Congo red and immunohistochemistry (see below). The presence of amyloid was confirmed when the amyloid-typical green-yellow-orange birefringence was found in cross-polarized light in Congo red-stained sections. The histoanatomical distribution of amyloid for every specimen with regard to alveolar septal, interstitial, nodular, tumour-like and vascular deposition as well as additional histological findings like inflammatory infiltrate, giant cells and ossifications was documented. Alveolar septal amyloid was characterized by deposits lining up along the alveolar septa. Vascular amyloid was defined as deposits which could be easily assigned to pulmonary arteries and veins. Nodular amyloid deposits were small (usually around 100-μm diameter) mass-forming lesions without diffuse affection of the surrounding tissue and without alignment along the alveolar septa. Tumour-like deposits were large (usually measuring more than 1 cm) mass-forming lesions. Interstitial deposits not otherwise specified (nos) were amyloid deposits, which could not be allocated to alveolar septa and did not form small or large masses of amyloid.
Immunohistochemistry
The immunohistochemistry was carried out with commercially available monoclonal antibodies directed against AA amyloid (1:2000) and polyclonal antibodies directed against amyloid P-component (1:2000), fibrinogen (1:1000), κ-light-chain (1:100,000), λ-light-chain (1:1:50,000), lysozyme (1:2000), prealbumin (1:3000; all DAKO, Hamburg Germany) and non-commercially available polyclonal antibodies directed against apolipoprotein A1 (anti-apo A1; dilution 1:1000), λ-light-chain-derived amyloid proteins (AL1 antibody, 1:250), anti-λ-light-chain peptides (AL3, 1:250; AL7, 1:200), transthyretin (TTR3, 1:2000) and kappa-light-chain peptides (AK3, 1:1000). Immunostaining was performed on FFPE sections with the BenchMark®XT immunostainer using the ultraView™ Universal Alkaline Phosphatase Red (in older cases, brown) Detection Kit (both from Ventana Medical Systems, Inc. Tucson, AZ) or with the Bond Max Leica immunostainer using the Bond Polymer Refine Red Detection Kit (Leica Microsystems, Wetzlar, Germany). Antigen retrieval was carried out with ER2-Bond Epitope Retrieval Solution 2 (amyloid P-component, λ-light chain, κ-light chain, TTR3 and prealbumin); ER1-Bond Epitope Retrieval Solution 1 (apo A1 and fibrinogen) or Enzyme 1 (AL7; all Leica Microsystems, Germany) according to the manufacturer’s instructions. Immunohistochemical classification was carried out and had been validated as described in detail elsewhere [22,23,24,25,26,27].
In brief, identification of the amyloid was considered to be positive when there was a strong and homogenous immunostaining of the entire amyloid deposits. Uneven and weak staining of some deposits was not assumed to be proof of the amyloid protein. If the staining was clearly positive with more than one antibody against different amyloid precursor proteins, the case was categorized as mixed amyloidosis. AL amyloid nos was characterized by positive staining with antibodies directed against λ- and κ-light-chain and negative immunostaining for the other amyloid proteins tested.
Immunostaining with antibodies directed against fibrinogen and lysozyme were done routinely until 2011. Subsequently, immunostaining was done with these antibodies only when AFib or ALys amyloidosis was within the differential diagnosis. The anti-prealbumin antibody was replaced in 2010 by the anti-TTR peptide antibody (TTR3). The anti-λ-light-chain antibody AL7 was introduced in 2007, while the antibody directed against AL3 was used until 2011. The anti-κ-light-chain antibody AK3 was routinely used since 2011. On slide positive and negative controls, using a tissue microarray with AA-, ALλ- and ATTR amyloid as well as non-neoplastic liver tissue were used on each staining round.
Statistics
Statistical analyses were carried out with IBM SPSS Statistics Version 24 (International Business Machines, Armonk, NY, USA). Cases with AA- or mixed type amyloidosis were excluded from diagnosis specific analyses due to the small patient numbers (AA = three cases, mixed type amyloid = one case). Significances of correspondence between variables in cross tables were determined using Fisher’s exact test. Significances of differences between age distributions of the amyloidosis types were tested using Mann-Whitney U test. Significances of differences between survival times of different forms of amyloidosis were tested using the Log-rank test (Mantel-Cox test). All p values are given unadjusted. A p ≤ 0.05 was considered statistically significant. Effects of multiple testing were accounted for by group-wise application of the Simes (Benjamini-Hochberg) procedure for control of false discovery rate [28].
Results
Two hundred seven biopsy and resection specimens from 205 patients were available with histologically proven amyloid. As two patients had biopsies submitted twice but in separate referrals, these biopsies were analysed each as a single case with regard to the histological phenotype. The 207 cases included 97 lung resection specimens, 66 lung tissue biopsies and 44 bronchial biopsies. AL amyloid was found in 183 cases (88.4%). AL amyloid of λ-light-chain origin (ALλ) was found in 141 (68.1% of all cases) and AL amyloid of κ-light-chain origin (ALκ) in 27 (13%) cases. In 15 (7.2%) cases with AL amyloidosis, the subclassification of amyloid was impossible and the cases were categorized as AL amyloidosis nos. ATTR was harboured in 20 (9.6%) cases and AA amyloid in 3 (1.4%) cases. Mixed amyloid, showing clearly positive staining with more than one antibody against different amyloid precursor proteins, was found in a single biopsy (0.5%). The amyloid deposits showed staining with antibodies directed against κ-light chain and TTR. In one patient with two biopsies, the amyloid was first classified as AL amyloid nos, as the light-chain subtype remained obscure. In the second biopsy, the amyloid could get diagnosed as ALλ amyloid.
Amyloid-positive biopsies from other organs additional to lung and bronchi were available from 10 patients. In all cases with biopsies obtained from different anatomical regions (six cardiac biopsies, three in combination with kidney biopsies and one of these with a gastrointestinal tract biopsy, two lymph node biopsies, one rectum biopsy and one breast biopsy), the amyloid type classified in the different anatomical sites by immunohistochemistry was identical with the amyloid type found in the lung and bronchial specimens, respectively.
Patient demographics
First, we examined the distribution of the different types of amyloid and correlated the results with patient age and gender. The overall median age at diagnosis was 67 years (range 24 to 88 years). The highest median age was found in ATTR amyloidosis (79 years), AL nos (69), ALλ (67) and ALκ (63). The difference in median age was found to be significant between AA and ATTR amyloidosis (p = 0.005) and between AL and ATTR amyloidosis (p < 0.001, respectively). No difference in patient age was found between AA and AL amyloidosis.
One hundred seventeen (57.1%) patients were male and 88 (42.4%) female. The gender difference between AL (54.7% male) and ATTR amyloidosis (80% male) was significant (p = 0.033) as well as the difference between ALκ (40.7% male) and ATTR amyloidosis (p = 0.009). Also, we examined the gender distribution in local and systemic form of AL amyloidosis, but no significant difference was found (see below).
Histoanatomical distribution of amyloid
Next, we assessed the distribution patterns of the different types of amyloid and the presence of an inflammatory infiltrate, giant cells and ossifications (Table 1). The examined distribution patterns are alveolar septal, tumour-like, nodular, vascular and interstitial nos (Fig. 1).

Amyloid deposition patterns in lung specimens. Overview of alveolar septal ATTR amyloid (a, d), tumour-like ALλ amyloid (b, e), nodular ALλ amyloid (c), vascular ATTR amyloid (f) and interstitial nos ALλ amyloid deposits (g). Giant cells (arrowheads) surrounding amyloid nodule (h) and an ossification in tumour-like ALλ amyloid (i). Haematoxylin eosin staining (a, b), immunostaining with antibodies directed against transthyretin (c, f) and λ-light chain (d, e). Original magnifications: 10-fold (a), 6-fold (b, d), 70-fold (c), 120-fold (e) and 140-fold (f)
In nearly all biopsies, interstitial nos (99.0%) and vascular (99.0%) amyloid deposits were found. Alveolar septal deposits were found in 105 (50.7%) of all cases. Interestingly, the alveolar septal deposits were found in 18 (90.0%) of the ATTR amyloidosis cases. In comparison to the other amyloid types, a significant difference of the appearance of alveolar septal amyloid was found between ATTR and AL amyloidosis (p < 0.001).
Nodular deposits were found in 83 cases (40.1%). Tumour-like amyloid deposits were present in 116 cases (56.0%). Tumour-like deposits were only found in cases with AL amyloidosis. Within these cases, tumour-like deposition was found in 63.4%.
An inflammatory infiltrate was present in 198 cases (95.7%). Giant cells were present in 87 cases (42.0%). Nearly all cases presenting giant cells in the specimens were AL amyloidosis (n = 85; 98.8%). Significant differences were found between AL and ATTR amyloidosis (p < 0.001); 29.5% of the cases (61) showed ossifications and were assigned to nodular, tumour-like, alveolar septal and vascular deposits, respectively. No ossifications were found in AA amyloidosis.
Clinical information
Clinical information was available from 118 patients and was obtained from general practitioners and clinicians. One hundred seventeen were diagnosed with AL amyloidosis (65.2% of AL amyloidosis patients) and one patient was diagnosed with a mixed-type amyloidosis of ALκ and ATTR amyloid.
Systemic and local AL amyloidosis
For 111 patients with AL amyloidosis (61.3% of AL amyloidosis patients), we received information on whether the clinical form of AL amyloidosis was local or systemic. One hundred patients (90.1%) had a localised and 11 (9.9%) a systemic form. In systemic AL amyloidosis, organ involvement was confirmed histologically for the gastrointestinal tract (2 cases), heart (2), lymph nodes (1), adipose tissue (1), breast (1), rectum (1), thyroid gland (1) and bone marrow (1). In one case with kidney, one with skin and one with lymph node involvement the histological verification is not known.
Next, we compared the two forms of AL amyloidosis (local vs. systemic) with the histoanatomical distribution patterns of bronchial and pulmonary amyloid deposits. All cases with tumour-like amyloid deposits (73 valid cases) were clinically diagnosed as local AL amyloidosis. In eight of these cases, nodular deposits were also found. Of the 38 patients without tumour-like amyloid in the biopsy specimens, 27 were local and 11 systemic forms. The differences were statistically significant (p < 0.001). Nodular amyloid deposits were found in 44 of the patients with clinical information. Thirty-five of these patients were diagnosed with a local form of AL amyloidosis. Within the 67 patients without nodular deposits, 65 had a local and 2 a systemic AL amyloidosis. This difference was significant (p = 0.006).
Presence of MGUS in serum immunofixation electrophoresis
Information about an MGUS in serum immunofixation electrophoresis was available from 109 patients. Thirty patients (27.5%) had an MGUS and for one patient, it was suspected. For 25 of these patients, we got further information about the MGUS type. Nine patients had an IgGλ MGUS, five a pure light chain MGUS (two of κ- and three of λ-light chain type), three an IgGκ MGUS, two an IgMκ MGUS, one an IgMλ MGUS and one an IgAλ MGUS. In 16 of the latter, the MGUS light chain matched the amyloid light chain origin. In three cases, the amyloid was AL nos and in two cases, the MGUS light chain was not matching the amyloid light chain origin. For one patient, it was only known that it was an IgA MGUS and in three patients, a biclonal gammopathy was present, the latter presenting with IgMκ/IgMλ, IgMλ/IgGκ, and IgGλ/IgGκ.
Bone marrow examinations
Data from bone marrow specimens were obtained from 71 (60.1%) patients. In 60 patients, the bone marrow biopsies showed no evidence of a plasma cell dyscrasia. In two patients, an increase of λ-positive plasma cells was found, in one biopsy, an increase of κ-positive plasma cells and in one biopsy, a general increase of polyclonal plasma cells had been reported. Five patients were diagnosed with multiple myeloma and two with marginal zone lymphoma. For one patient, no further information on the plasma cell dyscrasia was available. In nine cases, the amyloid light chain origin was matching the expressed light chain type.
Lymphoma in patient history
Ten patients had a lymphoma diagnosis in their history (8.5%). Three patients had the diagnosis of an extranodal marginal zone lymphoma of mucosa-associated lymphoid tissue (MALT lymphoma) and two patients of multiple myeloma. A diffuse large B cell lymphoma (NHL), a follicular B-NHL, a classical Hodgkin lymphoma, an indolent B-NHL with an immunophenotype of a chronic lymphocytic leukaemia and a lymphoplasmacytic lymphoma (Waldenström’s macroglobulinemia) were reported each in a single patient.
Reasons for initial examination
The initial reason for medical consultation and probe excision was reported in 30 patients. Twelve patients had initial findings of multiple pulmonary nodular lesions in one patient in combination with cysts that lead to further examination. One patient had a solitary nodular mass. Four patients presented with dyspnoea and one of them combined with physical deterioration. Three patients were explored with suspected lung cancer and two with cough. Two patients had haemoptysis, one of them in combination with ground glass opacities and one in combination with recurring infiltrates and bronchitis. Further indications were physical deterioration, infiltrative pulmonary changes, pleural effusions, suspected chondromalacia, suspected mycobacteriosis, suspected sarcoidosis and surgery of lung cancer. Interestingly, clinically amyloid was not within the differential diagnoses of any of these cases.
Computed tomography scan findings
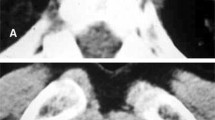
From 59 patients, we retrieved computer tomography scan results. For 32 patients, we could examine the CT scan pictures (Fig. 2, Table 2). Interestingly, systemic AL amyloidosis was more commonly associated with micronoduli, ground glass and reticular opacities, septal thickening, lymphadenopathy with calcifications, pleural thickening and lung cysts. Local AL amyloidosis predominantly showed a nodular pattern (Table 2).

Computed tomography scan. Comparison of CT scans of a patient with a localised AL amyloidosis (a, c) and a patient with systemic AL amyloidosis (b, d). The CT scan of the patient with a localised AL amyloidosis is presenting with a solitary nodule in the left upper lobe with an adjacent satellite nodule. The CT-guided biopsy of this nodule revealed a local ALλ amyloidosis. The CT scan of the patient with proven systemic AL amyloidosis is presenting with diffuse ground glass opacities, areas of consolidations, small nodules with perilymphatic distribution and interlobular thickening of the basal zones
Comparing histopathology with CT scans showed that 24 (96%) of 25 patients with nodular findings > 5 mm in the CT scan also had either tumour-like (13 patients) or nodular (11 patients) amyloid deposits on histology. However, one (4%) of these patients suffered from systemic AL amyloidosis and showed histologically alveolar septal, interstitial nos and vascular deposits but no nodular or tumour-like deposits, probably due to a sampling error, as only a biopsy specimen was obtained in this patient. Micronodules (< 5 mm diameter) on CT scan were associated either with nodular amyloid deposits (9 out of 13 patients (69.2%)) or tumour-like deposits (4 out of 13 patients (30.8%)). The mass-like lesions > 3 cm present on the CT scans were in in 6 (50%) of 12 patients associated with tumour-like and in 6 patients with nodular amyloid deposits. In seven cases, neither nodular nor micronodular findings were present in the CT scans. All of these patients had histologically vascular and interstitial nos amyloid deposits. Five (71%) of these seven patients also had histological findings of nodular amyloid deposits. Three of these additionally showed alveolar septal deposits. Two patients presented with tumour-like amyloid.
For the other 27 patients, we received reports: Fifteen presented with multiple pulmonary nodules, one of them in combination with cysts and one in combination with reticular opacities. Five patients had findings of consolidations, three of septal thickening and two of solitary round focuses. Signs of chondromalacia and calcifications were found each in a single patient.
Patients treated with amyloidosis targeting therapy
Fifteen patients received an amyloidosis targeting therapy (Supplement Table 1). Eight of these patients had a systemic form, six a local form of AL amyloidosis and from one patient the clinical form of AL amyloidosis was unknown, as he did not want further staging examinations. Twelve patients were treated with chemotherapy. Eight of these had a systemic and three a localised amyloidosis. Of these three patients, two (patients #12 and #14) had an underlying haematological disease. The other three patients with localised amyloidosis were treated with external beam radiation therapy. Patient #6 already got published as a case report [29].
Survival data
Follow-up information was available from 99 patients with AL amyloidosis (Fig. 3). Due to missing information about the cause of death, four patients were not included in the disease-specific survival analysis. The 10-year overall survival was 75.6% and the disease-specific survival 90.6%.

Patient survival. Comparison of disease-specific survival of patients with systemic or localised AL amyloidosis (a) and patients with systemic or tumour-like AL amyloidosis (b)
Next, we examined the survival in dependency of systemic and localised form of AL amyloidosis. For this analysis, we had data of 93 patients (11 systemic, 82 local). Three patients with localised amyloidosis were excluded as they were treated with chemotherapy. Four patients with unknown cause of death were excluded for the disease-specific survival analysis. The 10-year overall survival was 45.7% (systemic) vs. 79.5% (local; p = 0.012) and the disease-specific 10-year survival was 51.9% (systemic) vs. 97.0% (local; p < 0.001).
As we could show, tumour-like amyloid deposits are only found in tissue specimens of patients with localised AL amyloidosis. Next, we examined the survival of patients with tumour-like amyloid and compared it with systemic AL amyloidosis. Data were available from 59 patients with tumour-like amyloid. One patient was excluded because of chemotherapy treatment in the tumour-like group and three patients with unknown cause of death were excluded for the disease-specific survival analysis. Patients with localised tumour-like amyloidosis had a significantly better 10-year overall survival (80.3%) and 10-year disease-specific survival (95.8%) compared to patients suffering from systemic AL amyloidosis (overall survival p = 0.007; disease-specific p < 0.001).
Discussion
Amyloidosis is a rare disease with an estimated prevalence in Western countries of 14 per million person years [30] and 8.9 per million person years for AL amyloidosis [31]. A centralized collection of tissue specimens offers the possibility to examine large patient series and gain insight in this otherwise rarely seen disease. This is the hitherto largest patient series with amyloidosis in lungs and bronchi.
Demographically, amyloidosis of lungs and bronchi is a disease of the elderly with more men affected than women. We found AA, ATTR and AL amyloid. ALλ amyloid was the most common type followed by ALκ, ATTR and AL nos amyloid. AA amyloid and mixed amyloid are rarities, at least in our series. This is confirmed by previous reports [12, 13, 32]. Generally, pulmonary involvement should be found in AA, ALys, AL, ATTR, AApoI and AApoIV amyloidosis. But only AL, ATTR and AA amyloidosis seems to be clinically relevant with regard to diagnostic tissue resection and biopsy sampling.
While most of our patients had a localised AL amyloidosis, Ussavarungsi et al. reported a majority of systemic AL amyloidosis. This difference might be because Ussavarungsi et al. examined autopsy cases [13] while we examined cases obtained in clinical diagnostics. So, our collective does not reflect the natural history of the diverse types of amyloid. However, it rather reflects the clinical reality of cases, where tissue specimens are obtained for histological examination and amyloid is one of several putative alternative diseases. These are including lung cancer, fibrosis, interstitial, granulomatous, autoimmune and infectious lung diseases. So, mostly, the indication for a lung biopsy or resection in our study is to clarify unclear symptoms or findings of lung diseases. In most cases of patients with already known systemic amyloidosis, e.g. AA amyloidosis often presenting primarily with renal disease, no lung biopsy or resection would be sought except in clarification of suspicious findings for lung cancer or one of the other far more common differential diagnoses. This may explain the high percentage of localised AL amyloidosis in our study population. These patients can just get diagnosed by obtaining a tissue specimen. They do not present with other findings that could help reaching a diagnosis of localized AL amyloidosis without histology.
Amyloidosis of lungs and bronchi is often an incidental finding in the differential diagnostics of pulmonary nodules, unclear parenchymal consolidations and opacities, found on x-rays and CT scans, and general unclear symptoms of lung diseases. In the analysis of CT scans, we found that most patients had findings of more than one nodule with a sharp and lobulated structure. Both the systemic and the localised AL amyloidosis have findings of multiple nodules. As shown here, mass-forming lesions on CT scan, i.e. noduli and micronoduli nicely match with the histological appearance of nodular and tumour-like amyloid deposits. However, we only got CT scan results from patients with AL amyloidosis and cannot comment on the radiological appearance of, e.g. pulmonary AA or ATTR amyloidosis.
When the diagnosis of AL amyloidosis in lungs and bronchi is made, the classification and differentiation between localised and systemic AL amyloidosis is important as therapy and prognosis differ [7, 33]. With our study, we could now show that in clinical practice the localised AL amyloidosis of lungs and bronchi is far more common than the systemic form and is having an excellent prognosis (97.0% 10-year disease-specific survival). As only patients with localised amyloidosis and without chemotherapy were included in the survival analysis, we suggest that a restrictive and supportive treatment of the localised AL amyloidosis should be chosen [34].
In correlating histopathological and clinical findings, we observed that only localised AL amyloidosis forms tumour-like amyloid deposits in lungs and bronchi. Thus, over 60% of the AL amyloidosis patients of our study population could be classified as localised AL amyloidosis directly by the surgical pathologist. This might help in finding fast the right diagnosis and spare the patients of more and invasive staging examinations.
As also described by Merlini et al. [14], we found an association between AL amyloidosis and the appearance of MGUS in serum immunofixation electrophoresis and haematological disorders like multiple myeloma. Also, we found in most of the biopsies an inflammatory infiltrate. As Xiang et al. [35] and Gilmore et al. [36] reported, there is sometimes a local plasma cell dyscrasia found next to the amyloid deposits that is the putative source of the amyloid protein. This will need further studies on the nature of the infiltrate regarding clonality and possible affiliation to systemic haematological disorders. Alternatively, the inflammatory infiltrate and histiocytic giant cells may also be involved in the regression of amyloid. In some cases with a strong inflammatory infiltrate the amyloid deposits showed only sparse areas of congophilia.
The ossifications, found histologically, are also present in CT scans which is in line with previous reports [35, 37, 38]. Thus, pulmonary ossifications, especially in combination with pulmonary nodules in CT scans, should raise suspicion of amyloidosis.
ATTR amyloidosis most commonly presented with alveolar septal, vascular and interstitial nos deposits. The median age of the predominantly male patients was 79 years most likely representing wild-type ATTR amyloidosis, although genetic testing was beyond the scope of this study. Currently, the clinical significance of pulmonary ATTR amyloidosis remains obscure in our series and no data was available with regard to cardiac involvement or an association with carpal tunnel syndrome. However, we always recommended that cardiac manifestation should be ruled out.
In conclusion, amyloidosis of lungs and bronchi is a rare disease that is more prevalent in elderly men. As it can appear clinically in multiple ways, it should be thought of in the differential diagnosis of the most pulmonary diseases. As systemic and local form of AL amyloidosis have a completely different prognosis and therapy, a precise classification and differentiation is needed. For this, the histopathological findings can help making the right decisions.
References
Merlini G, Bellotti V (2003) Molecular mechanisms of amyloidosis. N Engl J Med 349:583–596
Sipe JD, Benson MD, Buxbaum JN, Ikeda S, Merlini G, Saraiva MJM, Westermark P (2016) Amyloid fibril proteins and amyloidosis: chemical identification and clinical classification International Society of Amyloidosis 2016 nomenclature guidelines. Amyloid 23:209–213
Puchtler H, Sweat F, Levine M (1962) On the binding of Congo red by amyloid. J Histochem Cytochem 10:355–364
Howie AJ, Brewer DB (2009) Optical properties of amyloid stained by Congo red: history and mechanisms. Micron 40:285–301
Röcken C, Eriksson M (2009) Amyloid und Amyloidosen. Pathologe 30:182–192
Röcken C, Sletten K (2003) Amyloid in surgical pathology. Virchows Arch 443:3–16
Mahmood S, Bridoux F, Venner CP, Sachchithanantham S, Gilbertson JA, Rowczenio D, Wagner T, Sayed R, Patel K, Fontana M, Whelan CJ, Lachmann HJ, Hawkins PN, Gillmore JD, Wechalekar AD (2015) Natural history and outcomes in localised immunoglobulin light-chain amyloidosis: a long-term observational study. Lancet Haematol 2:e241–e250
Röcken C (2015) Systemic and localised light-chain amyloidosis: two diseases. Lancet Haematol 2:e225–e226
Ando Y, Coelho T, Berk JL, Cruz MW, Ericzon BG, Ikeda S, Lewis WD, Obici L, Planté-Bordeneuve V, Rapezzi C, Said G, Salvi F (2013) Guideline of transthyretin-related hereditary amyloidosis for clinicians. Orphanet J Rare Dis 8:31
Plante-Bordeneuve V (2014) Update in the diagnosis and management of transthyretin familial amyloid polyneuropathy. J Neurol 261:1227–1233
Dubrey S, Ackermann E, Gillmore J (2015) The transthyretin amyloidoses: advances in therapy. Postgrad Med J 91(1078):439–448
Milani P, Basset M, Russo F, Foli A, Palladini G, Merlini G (2017) The lung in amyloidosis. Eur Respir Rev 26:170046
Ussavarungsi K, Yi ES, Maleszewski JJ, Kurtin PJ, Dasari S, Theis JD, Dispenzieri A, Ryu JH (2017) Clinical relevance of pulmonary amyloidosis: an analysis of 76 autopsy-derived cases. Eur Respir J 49:1602313
Merlini G, Seldin DC, Gertz MA (2011) Amyloidosis: pathogenesis and new therapeutic options. J Clin Oncol 29:1924–1933
Palladini G, Merlini G (2016) What is new in diagnosis and management of light chain amyloidosis? Blood 128:159–168
Kristen AV, Brokbals E, aus dem Siepen F, Bauer R, Hein S, Aurich M, Riffel J, Behrens HM, Krüger S, Schirmacher P, Katus HA, Röcken C (2016) Cardiac amyloid load. J Am Coll Cardiol 68:13–24
Scala R, Maccari U, Madioni C, Venezia D, La Magra LC (2015) Amyloidosis involving the respiratory system: 5-year’s experience of a multi-disciplinary group’s activity. Ann Thorac Med 10:212–216
Hagmeyer L, Stieglitz S, Röcken C, Randerath W (2012) Amyloidosis in pneumology. Pneumol Stuttg Ger 66:483–492
de Almeida RR, Zanetti G, Pereira E, Silva JL, Neto CA, Gomes AC, Meirelles GS, da Silva TK, Nobre LF, Hochhegger B, Escuissato DL, Marchiori E (2015) Respiratory tract amyloidosis. State-of-the-art review with a focus on pulmonary involvement. Lung 193:875–883
Renapurkar RD, Kanne JP (2013) Metabolic and storage lung diseases: spectrum of imaging appearances. Insights Imaging 4:773–785
Chung MJ, Lee KS, Franquet T, Müller NL, Han J, Kwon OJ (2005) Metabolic lung disease: imaging and histopathologic findings. Eur J Radiol 54:233–245
Schönland SO, Hegenbart U, Bochtler T, Mangatter A, Hansberg M, Ho AD, Lohse P, Röcken C (2012) Immunohistochemistry in the classification of systemic forms of amyloidosis: a systematic investigation of 117 patients. Blood 119:488–493
Kebbel A, Röcken C (2006) Immunohistochemical classification of amyloid in surgical pathology revisited. Am J Surg Pathol 30:673–683
Kuci H, Ebert MP, Röcken C (2007) Anti-lambda-light chain peptide antibodies are suitable for the immunohistochemical classification of AL amyloid. Histol Histopathol 22:379
Röcken C, Schwotzer EB, Linke RP, Saeger W (1996) The classification of amyloid deposits in clinicopathological practice. Histopathology 29:325–335
Gioeva Z, Urban P, Meliss RR, Haag J, Axmann H-D, Siebert F, Becker K, Radtke HG, Röcken C (2013) ATTR amyloid in the carpal tunnel ligament is frequently of wildtype transthyretin origin. Amyloid 20:1–6
Freudenthaler S, Hegenbart U, Schönland S, Behrens H-M, Krüger S, Röcken C (2016) Amyloid in biopsies of the gastrointestinal tract-a retrospective observational study on 542 patients. Virchows Arch 468:569–577
Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate—a practical and powerful approach to multiple testing. J R Stat Soc Ser B 57:289–300
Lang SM, Täuscher D, Füller J, Müller AH, Schiffl H (2015) Multifocal primary amyloidosis of the airways: case report and review of the literature. Respir Med Case Rep 15:115–117
Magy-Bertrand N, Dupond JL, Manny F, Dupond AS, Duchene F, Gil H, Kantelip B (2008) Incidence of amyloidosis over 3 years: the AMYPRO study. Clin Exp Rheumatol 26:1074
Kyle RA, Linos A, Beard CM, Linke RP, Gertz MA, O’Fallon WM, Kurland LT (1992) Incidence and natural history of primary systemic amyloidosis in Olmsted County, Minnesota, 1950 through 1989. Blood 79:1817–1822
Utz JP (1996) Pulmonary amyloidosis. The Mayo Clinic experience from 1980 to 1993. Ann Intern Med 124:407–413
Kourelis TV, Kyle RA, Dingli D, Buadi FK, Kumar SK, Gertz MA, Lacy MQ, Kapoor P, Go RS, Gonsalves WI, Warsame R, Lust JA, Hayman SR, Rajkumar SV, Zeldenrust SR, Russell SJ, Lin Y, Leung N, Dispenzieri A (2017) Presentation and outcomes of localized immunoglobulin light chain amyloidosis. Mayo Clin Proc 92:908–917
Gertz MA, Buadi FK, Hayman SR (2011) Treatment of immunoglobulin light chain (primary or AL) amyloidosis. Oncol Williston Park N 25:620–626
Xiang H, Wu Z, Wang Z, Yao H (2015) Nodular pulmonary amyloidosis and obvious ossification due to primary pulmonary MALT lymphoma with extensive plasmacytic differentiation: report of a rare case and review of the literature. Int J Clin Exp Pathol 8:7482
Gillmore J, Hawkins P (1999) Amyloidosis and the respiratory tract. Thorax 54:444–451
Ohdama S, Akagawa S, Matsubara O, Yoshizawa Y (1996) Primary diffuse alveolar septal amyloidosis with multiple cysts and calcification. Eur Respir J 9:1569–1571
Thompson PJ, Citron KM (1993) Amyloid and the lower respiratory tract. Thorax 38:84–87
Author information
Authors and Affiliations
Contributions
Study concept and design was done by Julius-Valentin Baumgart, Christiane Stuhlmann-Laeisz and Christoph Röcken. Clinical data including were provided by Ute Hegenbart, Stefan Schönland and Johanna Nattenmüller. Radiological data were provided and interpreted by Johanna Nattenmüller. Surgical pathological data were acquired by Julius-Valentin Baumgart, Christiane Stuhlmann-Laeisz, Sandra Krüger and Christoph Röcken. The data were analysed and interpreted by Julius-Valentin Baumgart, Christiane Stuhlmann-Laeisz and Christoph Röcken. Statistics were done by Hans-Michael Behrens. Administrative, technical and material support was provided by Sandra Krüger and Hans-Michael Behrens. The study was supervised by Christoph Röcken. Drafting and critical revision of the manuscript for important intellectual content was done by all authors. All authors are be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Corresponding author
Ethics declarations
This study was performed according to the Declaration of Helsinki. Ethical approval was obtained from the local ethical review board (D 581/15–585/15). All patient data were pseudonymized after study inclusion.
Conflict of interest
The authors declare that they have no competing interests.
Electronic supplementary material
Supplement Table 1
(DOCX 26 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Baumgart, JV., Stuhlmann-Laeisz, C., Hegenbart, U. et al. Local vs. systemic pulmonary amyloidosis—impact on diagnostics and clinical management. Virchows Arch 473, 627–637 (2018). https://doi.org/10.1007/s00428-018-2442-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00428-018-2442-x