
Abstract
The development of disease-modifying drugs and differential diagnostic agents is an urgent medical need in Parkinson’s disease. Despite the complex pathophysiological pathway, the misfolding of alpha-synuclein has been identified as a putative biomarker for detecting the onset and progression of the neurodegeneration associated with Parkinson’s disease. Identifying the most appropriate alpha-synuclein-based diagnostic modality with clinical translation will revolutionize the diagnosis of Parkinson’s. Likewise, molecules that target alpha-synuclein could alter the disease pathway that leads to Parkinson’s and may serve as first-in class therapeutics compared to existing treatment options such as levodopa and dopamine agonist that do not necessarily modify the disease pathway. Notwithstanding the promising benefits that alpha-synuclein presents to therapeutics and diagnostics development for Parkinson’s disease, finding ways to address potential challenges such as inadequate preclinical models, safety and efficacy will be paramount to achieving clinical translation. In this comprehensive review paper, we described the role of alpha-synuclein in the pathogenesis of Parkinson’s disease, as well as how its structure and function relationship delineate disease onset and progression. We further discussed different alpha-synuclein-based diagnostic modalities including biomolecular assays and molecular imaging. Finally, we presented current small molecules and biologics that are being developed as disease-modifying drugs or positron emission tomography imaging probes for Parkinson’s disease.
Similar content being viewed by others
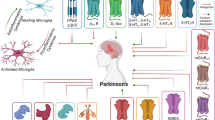

Parkinson’s disease
Parkinson’s disease (PD) is ranked the second most common neurodegenerative disorder after Alzheimer’s disease (AD), and more than ten million people worldwide are diagnosed with PD [1]. Approximately 1% and 4% of the general population over the ages of 60 and 80 years, respectively, are diagnosed with PD [2]. PD is a chronic neurodegenerative disorder that is associated with the progressive loss of dopaminergic neurons in the substantia nigra pars compacta (SNpc) (Fig. 1) [3]. It is both chronic and progressive because it persists over a long period of time and the symptoms get worse as more dopaminergic neurons die, respectively [4]. Depending on the level of dopaminergic cell death, PD can be diagnosed based on non-motor symptoms such as anosmia, depression, cognitive dysfunction and autonomic failure and/or motor symptoms such as bradykinesia, akinesia, tremors and muscle rigidity [5, 6]. The non-motor symptoms are usually present at the early stage of the disease, but the motor symptoms are observed at the later stage of the disease [4, 7]. Because of the overlap in the clinical, neurocognitive and pathological profile of PD with other neurodegenerative disorders such as PD with dementia (PDD), dementia with Lewy bodies (DLB) and multiple system atrophy (MSA), accurate diagnosis is challenging especially at the early stage of the disease [7]. Indeed, a study by Parkinson’s UK [8] found that 1 in 4 PD cases were initially misdiagnosed. Similarly, a previous study found that at least 15% of PD patients do not strictly meet the clinical criteria for the disease [9]. This highlights the need to develop accurate diagnostic agents for early detection of the onset and progression of dopaminergic neuronal death.

A schematic diagram of substantia nigra pars compacta region of healthy brain and Parkinson’s brain (Figure was created with biorender)
PD is the most common form of Parkinsonism. It is sometimes called “idiopathic Parkinsonism,” meaning Parkinsonism with no recognizable cause [10, 11]. Parkinsonism or Parkinsonian syndrome is a condition with a similar clinical appearance as in PD but different etiology; for instance, some patients have Parkinsonism secondary to antidopaminergic drugs without the pathology of the SNpc [4]. Neurotrophic viruses including West Nile virus, herpes viruses, lethargic encephalitis, influenza A virus, human immunodeficiency virus, severe acute respiratory syndrome coronavirus 2 are also associated with Parkinsonism [12,13,14,15,16,17,18,19]. With the increasing prevalence of PD among the elderly population, as well as the continuous evolution of neurotrophic viruses, it is important to develop disease-modifying drugs that will serve this patient population.
Currently, there is no cure for PD, and the only definitive diagnostic approach is postmortem examination of the diseased brain. Therefore, the development of disease-modifying treatments and differential diagnostic agents are important aspects of research in PD. Although alpha-synuclein (AS) has been identified as one of the putative biological targets for the development of therapeutic and diagnostic agents for PD, none of these AS-targeting molecules have been moved from bench to bedside. A major challenge with developing effective therapeutics and diagnostics for PD is the high possibility of clinical development failures due to safety and efficacy issues partly contributed by inadequate in vitro and in vivo models used for preclinical studies.
Herein, we described the role of AS in the pathogenesis of PD, as well as how its structure and function relationship delineate disease onset and progression. We further discussed different AS-based diagnostic modalities including biomolecular assays and molecular imaging. Finally, we presented current small molecules and biologics that are being developed as disease-modifying drugs or positron emission tomography (PET) imaging probes for PD.
Structure–function relationship of alpha-synuclein
Synucleins are a group of small soluble proteins which include alpha-, beta- and gamma-synuclein [20]. AS is encoded by the SNCA gene and there are at least four known isoforms of AS encoded by the same SNCA gene in humans and produced through alternative splicing [21]. These variants are characterized by their unique number of amino acids and aggregation potentials. Furthermore, the high molecular weight isoforms (containing about 140 amino acids) retain all the sites responsible for post-translational modifications [22] while the low molecular weight isoforms (containing 112,126 or 98 amino acids) do not retain these sites and may be predisposed to abnormal aggregation [22, 23]. This indicates that post-translational modification may be a predisposing factor to amyloidosis. AS is mainly expressed in the brain at the presynaptic terminals where it is thought to play a significant role in the mobilization of synaptic vesicles for the exocytotic release of neurotransmitters into the synaptic cleft (Fig. 2) [24,25,26]. Although AS is mostly an intracellular protein, it has also been found in extracellular fluids such as cerebrospinal fluid (CSF), blood and plasma [27,28,29,30,31].

A schematic diagram of alpha-synuclein-mediated mobilization of synaptic vesicles for exocytotic release of neurotransmitters into the synaptic cleft (Figure was created with biorender)
The structure of the major variant (AS-140) can be divided into two functionally distinct regions, namely the N and C terminuses. The N-terminal region ranges from 1 to 103 amino acid residues and contains amphipathic apolipoprotein binding helical motifs, which are responsible for binding lipids such as phospholipids [32]. This binding to phospholipids induces AS to adopt a helical conformation, [32] which is critical for its role in neurotransmission. The N-terminal also includes a hydrophobic region, the non-amyloid-β-component which is responsible for protein–protein interactions [32]. This protein–protein interaction induces AS to adopt a beta sheet conformation which is a critical event in the prion-like aggregation phenomena associated with PD [32]. A unique feature of this prion-like aggregation phenomena is permissive templating, in which the misfolded AS interacts with a normal AS and converts it to an amyloidogenic form [33,34,35]. The aggregation of the misfolded AS results in the formation of insoluble fibrils and their subsequent inclusion in Lewy bodies (Fig. 3) [36]. The C-terminal region has been suggested to be involved in mediating interactions of AS with other cytosolic or membrane-bound proteins [32, 33].

A schematic representation of the pathological hallmark of Parkinson’s disease
The role of alpha-synuclein in the diagnosis of Parkinson’s disease
Clinical diagnosis of PD is mostly based on the triad motor symptoms: resting tremors, rigidity and bradykinesia, non-motor symptoms such as constipation, anosmia, cognitive dysfunction, depression and dysautonomia [6, 37, 38], as well as postmortem pathological examination. The clinical diagnosis of PD can be less challenging when obvious signs and symptoms are present, as well as good response to levodopa treatment; however, diagnosis becomes more challenging at the onset of the disease due to similar signs and symptoms associated with PD and other neurodegenerative disorders such as PDD, DLB and MSA [38]. This implies that the only definitive confirmation of PD remains postmortem pathological examination where progressive degeneration of dopaminergic neurons at the SNpc along with AS-rich Lewy bodies are observed [6, 39, 40]. Consequently, about 15% of patients diagnosed with PD do not strictly meet the clinical criteria for the disease [9] and postmortem pathological examination of the diseased brain show a different diagnosis in about 35% of PD patients [41,42,43,44]. The misdiagnosis of PD can be explained by the overlapping neuropathological, cognitive and clinical profile of PD with several other neurodegenerative disorders [7]. For example, depending on the stage of the disease, dementia is present in 10% to 80% of PD patients [45, 46] and so the cognitive profile of PDD overlaps with DLB and MSA.
Accurate diagnosis is clearly a challenge in PD, and current research has focused on developing accurate diagnostic methods that would be valuable for early detection and tracking of disease progression. Early detection of the disease will allow administration of disease-modifying treatments and most likely circumvent the onset of motor symptoms. To develop a putative biomarker, an understanding of the molecular and biochemical mechanism of the disease will be an inevitable step. There is also a good chance that such a putative biomarker will provide a novel target for drug discovery and development in PD.
Given the implication of AS in the pathophysiology of PD, AS might be a good candidate for diagnosis of PD. Furthermore, the fact that the misfolding of AS is a critical event in the pathogenesis of PD implies that aberrant AS may be present in the early stages of the disease. In addition, a point mutation in the SNCA gene has been implicated in familial PD [47, 48]. Also, genetic risk factors involving relatively rare duplications or triplications of the SNCA gene locus result in elevated AS in both brain and blood [49]. Hence, AS in plasma, serum or CSF may serve as a useful diagnostic marker for PD. Several studies have identified pathological AS in CSF, blood and saliva [50,51,52].
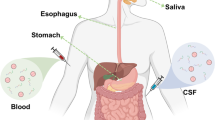
Assay of alpha-synuclein in peripheral tissues and body fluids
As previously discussed, the non-motor symptoms of PD are usually present at the onset of the disease before the classical clinical symptoms appear [26]. Several analytical techniques such as western blot, enzyme-linked immunosorbent assay (ELISA), luminex assay and mass spectrometry have been used to assay for total AS, as well as oligomeric, and phosphorylated AS in body fluids such as CSF, plasma and serum. The variation in the specificity, sensitivity and precision of these analytical techniques account for variations in the assay results. However, not one method appears to be entirely better than the other as each has its advantages and disadvantages.
Alpha-synuclein in peripheral tissues
Antemortem and postmortem tissue samples have been studied for the presence of pathological AS. These tissues include cardiac plexus [53], sympathetic ganglia [53, 54], gastric myenteric plexus [55], colonic tissue [56,57,58,59,60], gastrointestinal tract (GIT) [54], cardiac sympathetic nervous system [61], heart [62, 63], salivary gland [64,65,66] and vagus nerve [66, 67]; however, the outcome of assays using these tissues is quite variable. These variabilities can be caused by different methods of AS analysis, the severity of PD between studies, site of biopsy collection, specificity and sensitivity of the applied analytical method and cohort size [7, 68]. To overcome these challenges associated with the measurement of pathological AS in peripheral tissues, standardized protocols for sample collection as well as validated analytical techniques are needed [7].
Alpha-synuclein in body fluids
Cerebrospinal fluid
There is evidence indicating that CSF AS may be the most consistent, sensitive and specific marker for the diagnosis of PD [7]. Assays of total CSF AS have been reported to be 61–94% sensitive and 25–64% specific for distinguishing PD from controls [30, 68,69,70,71].
It has been observed that while monomeric AS is not affected in PD [27, 72], both oligomeric [70, 73, 74] and phosphorylated [71, 75] AS increased in PD patients compared with the control group. Several studies have shown that CSF AS may be useful in the differential diagnosis of parkinsonism caused by several other neurodegenerative disorders [30, 76, 77]. Although CSF seems to be the best specimen for diagnosis of PD, the relatively invasive nature by which it is obtained makes the procedure almost impracticable [68].
Plasma and serum
Given the less invasive nature by which plasma and serum are collected as well as its availability, plasma and serum may be the most appropriate clinical specimen for assay of pathological AS. Assays of the levels of serum AS showed conflicting results finding either that they are unaffected in PD patients [78] or are decreased compared with control groups in a larger cohort [79]. Similarly, several conflicting results have been reported for the measurement of total plasma AS using ELISA [29, 73, 80, 81], western blot [82], mass spectrometry [81] and luminex assay [68, 83].
Furthermore, a recent study has shown that total plasma AS measured with ELISA decreased in both familial and sporadic PD patients compared with the control groups [84]. The authors suggested that the lack of significance could be attributed to the small cohorts in the familial group [84]. The conflicting results in studies assessing plasma AS could be caused by factors such as hemolysis, contamination of platelets in plasma, inadequate age-matched controls and variation in the specificity and sensitivity of the analytical techniques used [68].
A way to overcome some of these confounders may be to assay exosomal AS [7]. Measurement of exosomal AS has shown diagnostic sensitivity and specificity comparable to those determined by CSF AS [85], making it potentially valuable in the diagnosis of PD and determination of disease severity [7]. Another possible way to minimize the confounding factors associated with measurement of plasma AS is to assay for pathological variants of AS [7]. For example, oligomeric AS has been reported to either increase [28, 80] or be unaffected [73, 81, 86] in PD patients compared with controls while phosphorylated AS is increased in PD patients compared with controls [81]. However, the interpretation of these reports should be handled with caution as the studies involved only a small cohort.
Blood
As red blood cells (RBC) are a significant source of AS, accounting for about 99% of its blood levels, RBC may serve as a potential specimen for diagnosis of PD [87]. Several studies involving the assay of total RBC AS have shown controversial results [49, 88]. Differences in the analytical technique utilized and cohort size could account for the discrepancy in results. To validate the potential of blood as a specimen for accurate diagnosis of PD, further studies should be conducted to determine the effect of PD on blood AS and RBC count which could cause alterations in AS levels [7].
Saliva
As saliva is a readily available specimen compared to other body fluids already discussed, salivary AS may be a good candidate for diagnosis of PD; however, assays of unstimulated salivary AS using western blot, luminex or mass spectrometry showed no significant difference in either the cellular component, supernatant [89] or cellular pellet lysate [90]. Previous studies have shown conflicting results of either an increase in salivary AS in PD patients compared with the control group [91] or no alterations in salivary AS [89, 90]. Further studies are required to provide a standardized protocol for the collection of saliva, as well as the most appropriate method of analysis [7]. The advantages and disadvantages of the different assays for PD diagnosis are summarized in Table 1.
Given the current challenges with the diagnosis of PD by estimation of pathological AS from body fluids or tissues, recent research has focused on developing molecular imaging probes for PD diagnosis.
Molecular imaging probes for diagnosis of Parkinson’s disease
Molecular imaging is one of the most widely used technologies in clinical and preclinical studies that help researchers to understand the pathophysiology, and drug treatment monitoring of various diseases including neurodegenerative disorders such as PD, by providing a real-time visualization for in vivo characterization and qualification of biological processes at the molecular and cellular level [92]. Molecular imaging consists of the imaging probe and imaging modalities. A molecular imaging probe is an agent used to visualize, characterize and quantify biological processes in living systems [93, 94].
The molecular imaging probe exists in a number of forms such as radiotracers, contrast agents or molecular beacons [95]. A molecular imaging probe consists of a linker, a signal moiety and a target moiety. The signal agent usually produces a signal that can be detected in an in vivo model to image different tissues. The type of imaging probe used for a given imaging modality can be determined by the physical property of the signal moiety; for example, a PET imaging probe requires a positron-emitting radionuclide as the signal agent, whereas a single photon emission computed tomography (SPECT) imaging probe employs a gamma-emitting radionuclide [95].
The targeting moiety interacts with a biomarker in a specific biological process, and such targeting ligands include but are not limited to small molecules, peptides, proteins, antibodies and its fragments and nanoparticles [95]. The linker used in a molecular imaging probe can couple the targeting moiety with the signal agent, minimize the interaction between the targeting moiety and the signal agent and, most importantly, modify the pharmacokinetics of the imaging probe [95]. Because many aspects need to be optimized to obtain the best imaging outcome, in some cases not all three components—the signal agent, the linker and the targeting moiety—are simultaneously present in a molecular imaging probe [95].
A molecular imaging probe with clinical translation is expected to have high binding affinity to the target, high specificity to target, high sensitivity, high contrast ratio, high in vivo stability, low immunogenicity and toxicity [95]. Several emerging molecular neuroimaging modalities have made it possible to noninvasively identify the fundamental biological processes involved in several degenerative disorders [92]. These molecular imaging modalities include magnetic resonance imaging, X-ray computed tomography, PET and SPECT. The advantages of molecular imaging reside in the real-time elucidation of the complex biological and metabolic pathways of various diseases at the molecular and cellular levels [94] compared to the quantitative estimation of the biological target which can be challenged by both variations in analytical techniques and specimen. Additionally, molecular imaging provides useful information that makes it possible to diagnose diseases at an early stage as well as conduct therapeutic trials of various disease conditions [96]. Of all the molecular imaging modalities, PET and SPECT have been used extensively in clinical neuroscience for the diagnosis of neurodegenerative disorders such as PD (Table 1) and AD; however, we will focus on the application of PET in the diagnosis of PD.
PET molecular imaging
PET is a high-performance molecular imaging technique that has found relevant applications in medical sciences due to its excellent sensitivity of 10–11–10–12 mol/L and limitless depth of penetration [92]. The modus operandi of PET involves recording pairs of high-energy γ-rays emitted indirectly from the decay of radioisotopes which are introduced into the subject [92]. Most commonly used PET radioisotopes include 11C, 13N, 15O, 18F, 64Cu, 68Ga, 82Rb and 166Ho. The positrons emitted from the radioactive decay of these imaging probes travel a few millimeters through the surrounding tissue, then they lose their kinetic energy rapidly [92]. The positrons move slowly and annihilate with electrons to generate two 511 keV γ-rays (Fig. 4), which travel in opposite directions [97]. Given that these radioisotopes have short half-lives (t1/2) such as 18F: t1/2 = 109.8 min, 11C: t1/2 = 20.3 min and 15O: t1/2 = 2.04 min, they need to be made at the site and introduced into the subject quickly [98]. Of all the radioisotopes, 11C, 15O and 18F are the most frequently used radioisotopes for brain imaging [92].

A schematic diagram of PET imaging principle
PET scanning with the metabolic tracer 18F-2-Fluoro-2-deoxy-D-glucose ([18F]FDG) is widely used for detecting tumors, staging cancers and drug treatment monitoring because cancer cells are actively metabolizing glucose [92]. [18F] FDG is a structural analog of glucose and when injected into the subject, it is transported into the cell by glucose transporters where it is phosphorylated by hexokinase; however, the phosphorylated [18F] FDG does not have a transporter; hence, it is trapped in the cell. The idea is that trapping the intact radiolabeled imaging probe provides information about the target cell.
Although PET has played a critical role in clinical research, a recent report has suggested that PET can be used in preclinical research involving small animal models; for instance, the Rat Conscious Animal PET (RatCAP) is a microPET instrument with a spatial resolution of 1–2 mm and sensitivity of 10–11–10–12 mol/L that has been constructed to allow small animal free of anesthesia to be scanned [92]. Despite the benefits associated with PET, there are two major limitations of the technique and these are the use of high-cost cyclotron [99] for the development of radioisotopes and the short t1/2 of the radioisotopes [92].
Several PET imaging probes have been reported to be used for diagnosing several central nervous system (CNS) diseases such as 1-(6-[(2-[18F]fluoroethyl)(methyl)amino]-2-naphthyl)ethylidene) malononitrile ([18F]-FDDNP) for AD [100],18F-dihydroxyphenylalanine ([18F]-DOPA) for PD [101], N-methyl-[11C]2-(4′-methylaminophenyl)-6-hydroxybenzothiazole termed Pittsburgh Compound-B (PIB) ([11C]-PIB) for AD [102, 103], [11C]-raclopride for PD, schizophrenia and depression [104, 105], [11C]-1-(2-chrorophynyl)-N-methylpropyl)-3 isoquinoline carboxamide ([11C]-PK11195) for AD, multiple sclerosis and huntington’s disease [106, 107], [11C]-flumazenil for epilepsy [108] and [11C]-nicotine for AD [109] (Fig. 5).

PET imaging probes for CNS diseases
PET molecular imaging in Parkinson’s disease
It is important to emphasize that PET imaging probes are chosen based on their ability to interact with a given molecular target involved in the etiology of a disease condition. Of all molecular agents, small molecules have been the most useful in diagnosing CNS disorders because of their ability to cross the blood–brain barrier (BBB) and be cleared from the tissue at a very fast rate [92]. Some of these small molecules have a high affinity for specific transporters, ion channels or specific receptors such as peripheral benzodiazepine receptor (PBR) while others can reflect the enzymatic or metabolic activity of a given biochemical pathway involved in various disease conditions [92] such as the glycolytic pathway.
The development of [11C]PIB (Fig. 6), an analog of Thioflavin-T facilitated the in vivo imaging of amyloid beta (Aβ) plaques with PET, providing a breakthrough in clinical evaluation of patients suspected of having AD [102].

Aβ plaques (Aβ1-42) PET imaging probes for AD
There are currently three food and drug administration (FDA)-approved 18F-PET imaging radiotracers: florbetaben, florbetapir and flutemetamol (Fig. 6) for in vivo imaging of Aβ plaques in AD; however, there have been no report/approval of clinically relevant 18F-PET imaging radiotracers for in vivo imaging of AS aggregates in PD [110]. The development of PET probes for monitoring the pathology of PD will be useful in evaluation of therapeutics designed for slowing down the disease [111].
The success of PET probes for Aβ plaques (Aβ1-42) and Tau fibrils has prompted relevant research into development of PET probes for AS fibrils in PD. Due to the similarity in beta-sheet structure of Aβ and AS after aggregation, [11C]PIB was evaluated against AS fibrils. [11C]PIB exhibited a similar in vitro binding affinity (Kd = 4 nM) for AS fibrils compared to Aβ1-42 [112] but failed to bind to Lewy bodies in brain homogenates [113]. 18F-BF227 (Fig. 7), a benzoxazole known to bind to Aβ1-42 in AD displayed lower binding affinity for AS fibrils (Kd = 9.63 nM) and also failed to bind to Lewy bodies in brain homogenates [114]. In order to develop compounds with good and selective binding affinity for AS fibrils, Yu et al. [115] explored a phenothiazine pharmacophore (SIL compounds) as probes for the fibrils. Among the different SIL compounds synthesized, SIL26 (Fig. 7) was synthesized as an 18F-radiolabel compound. Ex vivo biodistribution studies in Sprague–Dawley rats indicate that SIL26 is able to cross the BBB with high initial uptake percentage injected dose per gram (%ID/g) of 0.76 ± 0.01 while showing good affinity for AS aggregates (Thio-T comp Ki = 49.0 ± 4.9 nM, 125I comp Ki = 19.9 nM) [116] and moderate selectivity for AS fibrils (6.6- and 8.1-fold selectivity versus amyloid-beta and tau aggregates) [110, 116]. Indolinone and indolinone-diene analogs have been reported as new generation lead compounds for AS fibrils imaging. Among these series of compounds, 46a (indolinone-diene pharmacophore) (Fig. 7) displayed the highest affinity for AS fibrils (Thio-T comp Ki = 2.1 nM) and favorable selectivity for AS fibrils (70- and 840-fold selectivity versus Aβ1-42 and tau aggregates) [117].

Structures of promising radiotracers for AS fibrils in PD
In a search for new scaffolds, Watanabe et al. [110] synthesized new radioiodinated benzimidazole derivatives as probes for AS fibrils; despite the moderate affinity (Kd = 99.5 ± 20.8 nM) and good selectivity (7.3-fold selectivity versus amyloid-beta aggregates), the brain uptake and clearance of [125I]BI-2 (Fig. 7) did not meet the criteria for in vivo imaging of AS fibrils. While several PET/SPECT tracers targeting dopamine system have been developed, the current clinical diagnosis with dopamine system occurs at the later stage of disease progression involving severe loss of dopaminergic neurons and as a result, there is urgent need to develop probes for imaging early pathology which includes the formation of AS fibrils [118]. There is still an unmet need in the development of PET/SPECT probes for AS fibrils in diagnosis and monitoring of disease progression.
As previously mentioned, PD is characterized by the progressive loss of dopaminergic neurons in the SNpc [119, 120]. By imaging the dopaminergic system, several functional and neurochemical changes can be used for early diagnosis, as well as differential diagnosis of PD.
Previous PET studies with 11C-PK11195 for PBR have been used to understand the progression of the neurodegenerative process and disease state in PD patients [92]. Furthermore, several studies suggest that there may be a correlation between the activation of the microglia and the loss of dopaminergic neurons in the SNpc [121,122,123,124,125]. Microglia are known to be involved in the modulation of immune responses in the intact brain and become activated in response to inflammation, trauma, ischemia, tumor and neurodegeneration [126, 127]. Two PET studies have measured microglial activation using 11C-PK11195, as well as the availability of the presynaptic dopamine active transporter (DAT) using 11C-2β-carbomethoxy-3β-(4-fluorophenyl)tropane ([11C]-CFT) [127] and [18F]-DOPA [128]. DAT is a membrane-bound protein that is responsible for the high-affinity uptake of dopamine from the synaptic cleft back into the presynaptic neuron. The decrease in DAT may be indicative of loss of dopaminergic nerve terminals [127]. Ouchi et al. [127] suggested that these in vivo imaging methods play dual roles in monitoring the progressive degeneration of dopaminergic neurons and these roles are: alterations in neuroinflammatory reactions on the cell body side and the resulting deletion of nerve terminals in the striatum.
Ouchi et al., studied the binding potential (BP) of [11C]-PK11195 and [11C]-CFT in ten early-stage drug-naïve PD patients and ten age-matched healthy subjects and showed that [11C]-PK11195 BP in the midbrain is inversely related with [11C]-CFT BP in the putamen, which regulates movement, and directly related to motor severity. This inverse relationship was also reported to be positively correlated with the severity of motor symptoms [127]. This study suggests that the oxidative stress triggered by microglia-mediated immune response contributes to the loss of dopaminergic nerve terminals indicating the importance of early therapeutic intervention with neuroprotective drugs [127].
Another study examined [11C](R)-PK11195 BP and [18F]-DOPA BP using 18 PD patients and 11 healthy subjects [128]. Their study showed a significant increase in mean levels of [11C](R)-PK11195 binding in the pons, basal ganglia, as well as frontal and temporal cortical regions compared to the healthy subjects [128]. Furthermore, the longitudinal study conducted on 8 PD patients showed that their [11C](R)-PK11195 signal remained stable for 2 years [128]. The authors suggested that the absence of changes during the longitudinal study indicates that microglia are activated early in the disease and their levels remain relatively static, possibly driving the disease through cytokine release [128]. The conclusion of their study agrees with that of Ouchi et al. [127, 128] that the activation of microglia is associated with the pathogenesis of PD. However, Gerhard et al. [128] reported that there is no positive correlation between the levels of microglia activation and the clinical severity of PD or putamen [18F]-DOPA uptake. [18F]-DOPA PET was the first neuroimaging technique suited for measuring the integrity of dopaminergic nerve terminals [119, 129].
These previous studies indicate that PET imaging provides useful information about the biochemical and neurological changes associated with PD. Not only can this information be used for diagnosis, but also for the discovery of neurotherapeutic agents for PD. Given the benefits associated with PET, further studies are underway to identify imaging probes for differential diagnosis, early diagnosis and drug treatment monitoring of PD.
Management of Parkinson’s disease
Although several drugs can help provide relief from the symptoms of PD, there is currently no cure for PD. Furthermore, the gold standard treatment depends on the phase of the disease; therefore, PD patients are usually on several treatment regimens. These treatment regimens can be grouped into pharmacological and non-pharmacological therapies.
Non-pharmacological therapies include psychosocial intervention methods that can augment clinical improvement in PD patients [4]. These methods include multiple forms of physical exercise such as tai chi or Lee Silverman Voice Treatment (LSVT Global, Inc, Tucson, AZ, USA) and speech therapy with the LSVT [130]. Furthermore, exercise, physical therapy, speech and/or occupational therapy have been reported to have a sustainable effect for PD patients by improving their quality of life [130]. Despite the importance of these non-pharmacological therapies, its relevance becomes useless without an augmentation of medical treatments.
Pharmacological therapies include drugs that provide symptomatic relief of the motor and non-motor deficits in PD patients [4]. Ayano has previously reported the three different categories of medications for PD [4]. The first category includes drugs that increase the level of dopamine in the brain such as levodopa. The second category involves drugs that affect other neurotransmitters in the body with the overall aim of easing the symptoms of the disease; for example, anticholinergic drugs interfere with the production or uptake of acetylcholine and are effective in reducing tremors. The third category includes drugs that help control the non-motor symptoms of PD; for example, PD patients with depression may be prescribed antidepressants.
Additionally, there are several emerging new therapeutic options such as continuous pump therapies; for example, with apomorphine or parenteral levodopa, or the implantation of electrodes for deep brain stimulation [130].
Promising agents for preventing alpha-synuclein mediated toxicity in Parkinson’s disease
Although the misfolding of AS is a critical event in the pathogenesis of PD, the exact cause of dopaminergic cell death is not well understood [36]. However, it has been suggested that increasing the activity of the clearance pathways for the misfolded protein might provide improved therapies [131,132,133]. Additionally, since the misfolding and prion-like aggregation of AS is a crucial event in the pathophysiology of PD, an emerging therapeutic target might be the prevention of the misfolding pathway [134].
Previous studies suggest that caffeine, nicotine, 1-aminoindan and metformin might be neuroprotective [135,136,137,138,139,140,141,142,143,144,145]; however, it is not exactly clear how these compounds exert their neuroprotective effects. It was previously hypothesized that compounds which bind to AS at the N and C terminuses and induce it to adopt a loop conformation could be neuroprotective, whereas compounds which cause more compact structure could be neurotoxic [36]. In view of this hypothesis, Kakish et al. [134] used nanopore analysis and isothermal calorimetry (ITC) to show that nicotine, caffeine and 1-aminoindan all bind to AS at the N- and C-terminal. Their research showed that caffeine, nicotine and 1-aminoindan induce AS to adopt a loop conformation and that the stoichiometry of drug to AS in the complex is 1:1 [134]. They also reported that since metformin does not interact with the N terminus, it may exert its neuroprotective effect by inhibiting C-terminal cleavage of AS [36]. It was previously reported that C-terminal cleavage of AS increases the rate of aggregation and aggravates the neurodegeneration and propagation of PD in mouse models [146, 147].
In light of this new evidence, Kakish et al. [36] reasoned that modifications to these neuroprotective compounds might increase their efficacy without necessarily increasing their toxicity. To do this, they prepared bifunctional compounds which are a linked combination of any two neuroprotective compounds such as caffeine–nicotine (C8-6-N), caffeine–caffeine (C8-6-C8) and caffeine–1-aminoindan (C8-6-I) [36] (with structures shown in Fig. 8). These neuroprotective bifunctional compounds all had a caffeine scaffold and were linked through a six-carbon alkyl chain to minimize solubility problems and yet retain enough flexibility to allow both moieties to bind simultaneously [36]. The bifunctional compounds were screened by nanopore analysis and ITC. Finally, a yeast model of PD which expresses an AS-green fluorescent protein (AS-GFP) construct under the control of a galactose promoter was used to test the ability of these compounds to interact with AS in a cell system [36]. Fluorescent microscopy revealed that in 5 mM galactose the yeast strain would not grow and large cytoplasmic foci were observed [36]. This implies that aberrant AS might have prevented the growth of the yeast strain. All the bifunctional compounds and monomers were tested; however, it was observed that two of the compounds, C8-6-I and C8-6-N at a concentration of 0.1 μM were the most effective at rescuing yeast from AS-mediated cell death [36]. This shows that some of the bifunctional compounds are more effective at rescuing yeast growth compared to the monomers, either alone or in combination [36]. The study concluded that C8-6-I and C8-6-N are the only bifunctional compounds that did not cause AS to adopt a more compact structure at high concentrations making these compounds the most promising candidates for preventing the progression of PD by prion-like aggregation [36]. Given that the binding constants were in the order of 105/M, the authors suggested that other factors might be responsible for the protective effects of these compounds and these factors include: increased clearance of the compound/AS complexes, inhibition of vesicle clustering, decreased expression of AS and inhibition of multimer formation by binding to key intermediates [36].


Structures of some of the small molecules investigated for inhibiting AS fibrillation or facilitating AS fibril clearance pathway in PD
Since these novel bifunctional compounds can bind AS, they can be developed as PET imaging probes for PD. To achieve this goal, preclinical studies such as metabolism was conducted, and metabolic studies of these compounds provided useful information about their in vitro lifetime as well as the most appropriate position to include a signal moiety on the bifunctional compounds. Nwabufo et al., developed an appropriate analytical method for the qualitative and quantitative analysis of these novel bifunctional compounds [148] and in vitro phase 1 metabolism of these compounds in mouse, rat and human liver microsomes showed that C8-6-N undergoes rapid metabolism, C8-6-I undergoes moderate metabolism, and C8-6-C8 remained intact after 60 min incubation time [149, 150].
Isorhynchophylline (Fig. 8) is another alkaloid that has been investigated for its ability to degrade AS fibrils and protect neuronal cells via autophagy–lysosome pathways [151, 152]. Similarly, acetylcorynoline (Fig. 8), an alkaloid from Corydalis Bungeana has been reported to reduce AS aggregation and dopaminergic degenerations in animal models of PD [152]. The neuroprotective property of acetylcorynoline is due to its antioxidant, and antiapoptotic activity and due to the mediation of the proteasome system. Previous research has shown that the neuroprotective effect of acetylcorynoline may be linked to increase in expression of rpn5, a subunit of 19S proteasome [153]. Leem et al. [154] reported that the opium alkaloid, papaverine (Fig. 8) can modulate AS aggregation by inhibiting the phosphorylation of AS at serine 129 amino acid residue. Papaverine is also able to prevent nigrostriatal dopaminergic degeneration and neuroinflammation in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)/P-treated mice. Jayaraj et al. [155] reported that the benzylisoquinoline alkaloid, noscapine (Fig. 8), exhibited neuroprotective effects in rotenone-induced PD rat model by suppression of rotenone-induced oxidative stress, suppression of microglia and astrocytes inflammation activity, and regulation of the autophagy pathway promoting lysosomal degradation of AS. Recently, Ghanem et al., have reported a study that suggests that synephrine, trigonelline, cytisine, harmine, koumine, hupehenine and peimisine may act as strong inhibitors of AS seeded aggregation in accordance with thioflavin-s assay and electron microscopy. However, all except hupehenine was successful in reducing the neurotoxic effect provoked by AS fibril seeding process as demonstrated by WT SH-SY5Y cell viability studies [156].
The flavonoid polyphenol, epigallocatechin gallate (EGCG; Fig. 8) is probably the most studied small molecule for inhibition of AS aggregation into structured and toxic amyloid fibrils [118]. Andersen et al., utilized liquid-state nuclear magnetic resonance (NMR) spectroscopy to show that EGCG binds to monomeric and oligomeric AS and is able to promote the formation of AS oligomers that are non-toxic and non-structured. Utilizing liquid-state NMR, the authors indicated that a critical concentration of EGCG is important to induce rapid and complete sequestration of monomeric AS. Extrapolation led Andersen et al. [157] to conclude that a 54:1 ratio of EGCG molecules to monomeric AS is critical for the formation of non-toxic oligomers during oligomerization. Using molecular simulations, Yao et al., have also shown that EGCG disrupts AS fibril β-sheet structures by reducing the stability of two regions: region 1 (residues 45–55) and region 2 (residues 86–96). In addition, the authors also stated that EGCG destroys a E46–K80 salt bridge, a bridge which helps in stabilization of the Greek-key-like structure and inter-protofibril interface [158]. In MPTP-intoxicated PD monkeys, Chen et al., showed that oral administration of tea polyphenols improves motor function, and neurochemical, immunohistochemical and cell viability studies indicate neuroprotection is due to rescue of dopaminergic neurons in the substantia nigra from 1-methyl-4-phenylpyridinium (MPP+)-induced metabolic toxicity. Additionally, their studies indicate that neuroprotection from MPTP toxicity is also a result of inhibiting toxic AS oligomers in the striatum [159]. It is also believed that the neuroprotective property of EGCG is a result of the polyphenol accelerating the removal of active oligomers which could disrupt cell membrane and cause cellular degeneration. Yang et al. [160] indicate that this process is made possible as EGCG converts active oligomers into amyloid fibrils. Low concentrations of baicalein and its oxidized form have been reported to inhibit AS aggregation and disrupt existing AS fibrils [161].
In 2019, Daniels et al., reported that interaction of AS with nordihydroguaiaretic acid (NDGA; Fig. 8) and its cyclized analogs inhibited the aggregation of AS into fibrils. The authors note that previous investigation into the neuroprotective properties of EGCG have focused on the formation of oligomers in the presence of EGCG. While AS aggregation in the presence of EGCG and NDGA does produce oligomers, the authors report that monomers are the predominant product in solution [162].
Two steroid polyamines, squalamine and trodusquemine, are molecules investigated as therapeutics for PD [163]. Polyamines can interact with the negatively charged C terminus of AS [164]. Perni et al., showed that squalamine can interfere with the initial events in AS aggregation by displacing AS from lipid membranes [165] and in 2018, the same group reported that trodusquemine is able to inhibit fibril-dependent secondary pathways in addition to the suppression of initial AS aggregation events [166].
Another class of small molecules currently being investigated for their neuroprotective activity is antibiotics. In vitro studies have shown that rifampicin can inhibit AS fibrillation [167] process and prevent MTPT-induced PC12 cellular toxicity [168]. However, in a clinical trial of MSA, rifampicin failed to demonstrate efficacy in slowing down progression of the disease [169, 170]. Dominguez-Meijide et al. [171] reported that doxycycline may inhibit AS fibrillation by conversion of AS oligomers into non-toxic and off-pathway, high molecular weight species. Their studies show doxycycline is also able to reduce the levels of reactive oxygen species produced by mitochondria. Ceftriaxone has been reported to demonstrate neuroprotective effects including modulation of glutamatergic activity in MTPT-induced rat model and treatment of AS and beta-amyloid related disorders [172].
A basic helix–loop–helix transcription factor EB (TFEB) has been recognized as an important protein involved in the regulation of several clearance pathways, a master regulator of autophagy and lysosomal biogenesis [173, 174]. TFEB is identified to play a role either in physiological or pathological conditions. Under physiological conditions, dephosphorylated TFEB translocates into the nucleus and activates the transcription of target genes [175]; however, under pathological conditions, TFEB is colocalized with Lewy bodies AS of substantia nigra dopaminergic cells in postmortem human brains [176].
In 2020, Liu et al. [174] reported a highly efficient TFEB-targeted nanoscavenger of AS and its mechanisms in models of PD. The authors previously demonstrated that a curcumin analog (CA) compound specifically binds to TFEB at the N terminus promoting TFEB nuclear translocation without inhibiting mechanistic targets of rapamycin (MTOR) activity [173]. MTOR plays a role in TFEB subcellular localization by phosphorylating key serine residues such as S142 and S211 and inhibition of MTOR has a preventive effect on dopaminergic loss [173]; however, due to the important role of MTOR in cell metabolism and growth, TFEB activators that are independent of MTOR are more appropriate as therapeutics for PD [174]. In the authors’ previous study, they found that curcumin although a natural inhibitor of MTOR, does not efficiently promote TFEB nuclear translocation. However, a CA (Fig. 8) was found to activate and promote TFEB-mediated clearance pathways independent of MTOR activity. Further investigation revealed that CA binds directly and specifically to the N-terminal Gly and Ala-rich domain of TFEB.
Apart from small compounds targeting AS, there has been a lot of work with macromolecules such as antibodies targeting AS for treatment of PD [177]. Several antibodies targeting the N terminus or C terminus of AS were able to alleviate neurodegeneration as well as attenuate behavioral deficits in preclinical models of PD [146, 177,178,179,180,181,182,183,184]. Protein immunization can be achieved by “passive immunization” or “active immunization.” In passive immunization, a protein-targeting antibody is administered while in active immunization, the immune response is induced to produce specific antibodies. The advantage of passive immunization is the specificity of the antibodies for a protein target while the advantage of active immunization is the sustained effect and need for less doses [177]. There are currently five AS-targeting antibodies in clinical trials.
Prasinezumab (PRX002/R07046015/RG7935) is a humanized IgG1 monoclonal antibody directed against the C terminus of aggregated AS. Preclinical studies in a mouse model of PD and DLB showed that the mouse version (PRX002) of the antibody was able to reduce a truncated C-terminal form of AS and also reduce AS cell to cell propagation. Two phase I/II trials (NCT03100149) indicate that the antibody was safe, well tolerated in healthy volunteers and PD patients, and able to reduce plasmatic concentration of AS up to 4% in a dose-dependent manner [182, 185,186,187].
ABBV-0805 (BAN0805) is another humanized monoclonal antibody targeting oligomeric/protofibrillar AS with nanomolar affinity. ABBV-0805 decreased AS aggregates in a dose-dependent manner in mice model expressing human AS [188, 189]. ABBV-0805 moved into phase I clinical trial, but in July 2020, it was withdrawn for strategic reasons (NCT04127695).
Cinpanemab (BIIB054), a human-derived monoclonal antibody, binds to AS amino acid residues 1–10 with 800-fold higher affinity for aggregated AS over monomeric AS. In vitro cell assays revealed that cinpanemab is able to reduce AS proliferation and able to relieve motor symptoms in a mice model [190]. Cinpanemab underwent a phase I single ascending dose trial and was found to be safe and well tolerated in healthy volunteers and PD patients [191]; however, a phase II SPARK (NCT03318523) clinical trial was discontinued because primary and secondary endpoints were not met [192]. MEDI134 (TAK-341); a monoclonal antibody with high affinity for the C terminus of AS has been found to block uptake of aggregated AS into cells and mitigate spreading of AS in mouse brain [193]. There is currently an ongoing multiple ascending dose Phase I (NCT04449484) clinical study for MEDI134 [177]. LU AF82422, a humanized monoclonal antibody, also targets the C-terminal of AS. In vitro and in vivo models revealed that LU AF82242 inhibits AS seeding and this antibody has been found to be safe in mice and monkeys [194]. A phase I trial (NCT03611569) in PD patients has been completed, and a phase II clinical trial (NCT05104476) to assess efficacy, safety and tolerability in MSA patients has recently been initiated [177]. A summary of these investigational drugs for the treatment of PD is provided in Table 2.
The translation of these compounds from bench to bedside is primarily dependent on their ability to demonstrate sufficient clinical efficacy and safety profile. Drug metabolizing enzymes and membrane-associated drug transporters are important modulators of the efficacy and safety profile of drugs and their effect on the disposition of these potential neuroprotective agents should be investigated at the early discovery stage to avoid costly clinical development failures [195,196,197].
P-glycoprotein (P-gp) and breast cancer resistance protein (BCRP) are two important efflux transporters that play a critical role in limiting the CNS activity of some neurotherapeutic agents by preventing their penetration across the BBB [197, 198]. In fact, P-gp and BCRP are also implicated in the neuropathology of PD [199, 200]. Hence, it is important to ascertain the effect of these transporters in the disposition of these potential neuroprotective agents using a PD-pathological BBB model, as well as a non-PD-pathological BBB model. There are several case studies implicating either P-gp, BCRP or both in the low CNS activity of neurotherapeutic agents and a typical example is the novel anti-PD candidate drug FLZ (N-2-(4-hydroxy-phenyl)-ethyl]-2-(2,5-dimethoxy-phenyl)-3-(3-methoxy-4-hydroxy-phenyl)-acrylamide) which showed low BBB penetration in rat brain [198]. Given the role of P-gp and BCRP in both efflux of substrates across the BBB, as well as neuropathology of PD, a study was conducted to determine whether P-gp and BCRP play a role in the low BBB permeability of FLZ and also to examine the influence of PD-pathological BBB in the transport of FLZ [201]. The study found greater expression of P-gp and BCRP in the PD-pathological BBB model associated with reduced in vitro BBB permeability of FLZ compared to the non-PD-pathological BBB model. The study concluded that P-gp but not BCRP is responsible for the low BBB permeability of FLZ [201]. One way to address this challenge would be to co-administer P-gp inhibitors such as haloperidol or the use of novel drug delivery P-gp bypass systems and pharmaceutical excipients such as chemosensitizers, natural and synthetic polymers, and formulation excipients used as P-gp inhibitors [202].
Conclusion
The pathophysiology and pathogenesis of PD is a complex process and not well understood. However, current clinical signs and symptoms appear at the late stage of the disease and therapeutic interventions at this stage only alleviate those symptoms without necessarily inhibiting the pathological pathway of the disease. AS misfolding and fibrillation has been implicated in early stages of Parkinson’s and this makes it a suitable target for both diagnosis and treatment of PD. The breakthrough in molecular imaging of Aβ1-42 biomarker for diagnosis of AD has prompted more research into finding the suitable molecule that can target AS fibrils. A challenge currently being encountered is finding molecules with good affinity for AS fibrils while maintaining moderate to good selectivity for AS fibrils compared to Aβ1-42 and Tau fibrils. Moreover, lead candidates for AS fibrils should have high initial brain uptake and favorable in vivo pharmacokinetics. Metabolic stability, plasma clearance, pKa, solubility, etc. are some of the pharmacokinetic and physicochemical parameters to take into consideration when designing molecules for AS fibrils.
Despite the low success rate in finding the best imaging probe for AS fibrils, there has been tremendous progress in the area of finding neuroprotective molecules. One of the reasons for the accelerated progress is the availability of natural products such as polyphenols, alkaloids, etc. for evaluation of their ability to inhibit AS misfolding or enhance AS fibril clearance pathway. The availability of suitable models and specimen for many of the assays is a huge challenge. It is still largely unknown how accurate our in vitro assays model AS fibrillation in the brain and as a result small molecule that have shown potentials to slow the fibrillation pathway in cell assays may fail in further preclinical studies or clinical trials.
Despite the difficulties in identifying the best probe(s) or neuroprotective compounds, the AS fibrillation pathway remains an important target for the diagnosis and treatment of PD. A better understanding of the pathogenesis, as well as the development of better PD models, may provide us with better opportunities to identify the best lead compounds for accurate diagnosis and treatment of PD.
Abbreviations
- [11C]-CFT:
-
11C-2β-carbomethoxy-3β-(4-fluorophenyl) tropane
- [11C]-PIB:
-
Pittsburgh Compound-B
- [11C]-PK11195:
-
[11C]-1-(2-chrorophynyl)-N-methylpropyl)-3 isoquinoline carboxamide
- [18F]-DOPA:
-
18F-dihydroxyphenylalanine
- [18F]-FDDNP:
-
1-(6-[(2-[18F]fluoroethyl)(methyl)amino]-2-naphthyl)ethylidene) malononitrile
- [18F]FDG:
-
18F-2-Fluoro-2-deoxy-d-glucose
- %ID/g:
-
Percentage injected dose per gram
- AADC:
-
Aromatic amino acid decarboxylase
- AD:
-
Alzheimer’s disease
- AS:
-
Alpha-Synuclein
- AS-GFP:
-
AS-green fluorescent protein
- Aβ:
-
Amyloid beta
- BBB:
-
Blood–brain barrier
- BCRP:
-
Breast cancer resistance protein
- BP:
-
Binding potential
- C8-6-C8 :
-
Caffeine–caffeine
- C8-6-I:
-
Caffeine–aminoindan
- C8-6-N:
-
Caffeine–nicotine
- CA:
-
Curcumin analog
- cNDGA:
-
Cyclic NDGA
- CNS:
-
Central nervous system
- CSF:
-
Cerebrospinal fluid
- DAT:
-
Dopamine active transporter
- DLB:
-
Dementia with Lewy bodies
- EGCG:
-
Epigallocatechin gallate
- ELISA:
-
Enzyme-linked immunosorbent assay
- FDA:
-
Food and drug administration
- FLZ:
-
(N-2-(4-Hydroxy-phenyl)-ethyl]-2-(2,5-dimethoxy-phenyl)-3-(3-methoxy-4-hydroxy-phenyl)-acrylamide)
- GIT:
-
Gastrointestinal tract
- ITC:
-
Isothermal titration calorimetry
- LSVT:
-
Lee Silverman Voice Treatment
- MPP+ :
-
1-Methyl-4-phenylpyridinium
- MPTP:
-
1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- MSA:
-
Multiple system atrophy
- MTOR:
-
Mechanistic targets of rapamycin
- NDGA:
-
Nordihydroguaiaretic acid
- NMR:
-
Nuclear magnetic resonance
- PBR:
-
Peripheral benzodiazepine receptor
- PD:
-
Parkinson’s disease
- PDD:
-
PD with dementia
- PET:
-
Positron emission tomography
- P-gp:
-
P-glycoprotein
- RatCAP:
-
Rat Conscious Animal PET
- RBC:
-
Red blood cells
- SNpc:
-
Substantia nigra pars compacta
- SPECT:
-
Single photon emission computed tomography
- t 1/2 :
-
Half-life
- TFED:
-
Transcription factor EB
- Thio-T comp K i :
-
Thioflavin-T competitive Ki
- VMAT2:
-
Vesicular monoamine transporter 2
References
Parkinson’s Foundation (2022) Statistics. https://www.parkinson.org/Understanding-Parkinsons/Statistics
de Lau LML, Breteler MMB (2006) Epidemiology: Parkinson’s disease. Lancet Neurol 5:525. https://doi.org/10.1016/S1474-4422(06)70471-9
Klockgether T (2004) Parkinson’s disease: clinical aspects. Cell Tissue Res 318:115–120. https://doi.org/10.1007/s00441-004-0975-6
Ayano G (2016) Parkinson’s disease: a concise overview of etiology, epidemiology, diagnosis, comorbidity and management. J Neurol Disord. https://doi.org/10.4172/2329-6895.1000298
Jankovic J (2008) Parkinson’s disease: clinical features and diagnosis. J Neurol Neurosurg Psychiatry 79:368–376. https://doi.org/10.1136/jnnp.2007.131045
Shulman JM, De Jager PL, Feany MB (2011) Parkinson’s disease: genetics and pathogenesis. Annu Rev Pathol 6:193–222. https://doi.org/10.1146/annurev-pathol-011110-130242
Atik A, Stewart T, Zhang J (2016) Alpha-synuclein as a biomarker for Parkinson’s disease. Brain Pathol 26:410–418. https://doi.org/10.1111/bpa.12370
Parkinson’s UK (2022) https://www.parkinsons.org.uk/news/poll-finds-quarter-people-parkinsons-are-wrongly-diagnosed. Accessed 26 Jun 2022
Schrag A, Ben-Shlomo Y, Quinn N (2002) How valid is the clinical diagnosis of Parkinson’s disease in the community? J Neurol Neurosurg Psychiatry 73:529–534
Samii A, Nutt JG, Ransom BR (2004) Parkinson’s disease. Lancet 363:1783–1793. https://doi.org/10.1016/S0140-6736(04)16305-8
Schrag A (2007) Epidemiology of movement disorders. Lippincott Williams & Wilkins, Hagerstown
Limphaibool N, Iwanowski P, Holstad MJV et al (2019) Infectious etiologies of Parkinsonism: pathomechanisms and clinical implications. Front Neurol 10:1–11. https://doi.org/10.3389/fneur.2019.00652
Zheng KS, Dorfman BJ, Christos PJ et al (2012) Clinical characteristics of exacerbations in Parkinson disease. Neurologist 18:120–124. https://doi.org/10.1097/NRL.0b013e318251e6f2
Cohen ME, Eichel R, Steiner-Birmanns B et al (2020) A case of probable Parkinson’s disease after SARS-CoV-2 infection. Lancet Neurol. https://doi.org/10.1016/S1474-4422(20)30305-7
Méndez-Guerrero A, Laespada-García MI, Gómez-Grande A et al (2020) Acute hypokinetic-rigid syndrome following SARS-CoV-2 infection. Neurology. https://doi.org/10.1212/WNL.0000000000010282
Faber I, Brandão PRP, Menegatti F et al (2020) Coronavirus disease 2019 and parkinsonism: a non-post-encephalitic case. Mov Disord 35:1721–1722. https://doi.org/10.1002/mds.28277
Makhoul K, Jankovic J (2021) Parkinson’s disease after COVID-19. J Neurol Sci 422:117331. https://doi.org/10.1016/j.jns.2021.117331
Akilli NB, Yosunkaya A (2021) Part of the Covid19 puzzle: acute parkinsonism. Am J Emerg Med 47:333.e1-333.e3. https://doi.org/10.1016/j.ajem.2021.02.050
Morassi M, Palmerini F, Nici S et al (2021) SARS-CoV-2-related encephalitis with prominent parkinsonism: clinical and FDG-PET correlates in two patients. J Neurol 268:3980–3987. https://doi.org/10.1007/s00415-021-10560-3
Lavedan C (1998) The synuclein family. Genome Res 8:871–880. https://doi.org/10.1101/gr.8.9.871
McLean PJ, Hyman BT (2002) An alternatively spliced form of rodent alpha-synuclein forms intracellular inclusions in vitro: role of the carboxy-terminus in alpha-synuclein aggregation. Neurosci Lett 323:219–223
Levitan K, Chereau D, Cohen SI et al (2011) Conserved C-terminal charge exerts a profound influence on the aggregation rate of alpha-synuclein. J Mol Biol 411:329–333. https://doi.org/10.1016/j.jmb.2011.05.046
Beyer K, Ariza A (2013) alpha-Synuclein posttranslational modification and alternative splicing as a trigger for neurodegeneration. Mol Neurobiol 47:509–524. https://doi.org/10.1007/s12035-012-8330-5
Iwai A, Masliah E, Yoshimoto M et al (1995) The precursor protein of non-A beta component of Alzheimer’s disease amyloid is a presynaptic protein of the central nervous system. Neuron 14:467–475
Nakajo S, Shioda S, Nakai Y, Nakaya K (1994) Localization of phosphoneuroprotein 14 (PNP 14) and its mRNA expression in rat brain determined by immunocytochemistry and in situ hybridization. Brain Res Mol Brain Res 27:81–86
Braak H, Del Tredici K, Rub U et al (2003) Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 24:197–211
Borghi R, Marchese R, Negro A et al (2000) Full length alpha-synuclein is present in cerebrospinal fluid from Parkinson’s disease and normal subjects. Neurosci Lett 287:65–67
El-Agnaf OM, Salem SA, Paleologou KE et al (2006) Detection of oligomeric forms of alpha-synuclein protein in human plasma as a potential biomarker for Parkinson’s disease. FASEB J 20:419–425. https://doi.org/10.1096/fj.03-1449com
Lee PH, Lee G, Park HJ et al (2006) The plasma alpha-synuclein levels in patients with Parkinson’s disease and multiple system atrophy. J Neural Transm (Vienna) 113:1435–1439. https://doi.org/10.1007/s00702-005-0427-9
Mollenhauer B, Locascio JJ, Schulz-Schaeffer W et al (2011) alpha-Synuclein and tau concentrations in cerebrospinal fluid of patients presenting with parkinsonism: a cohort study. Lancet Neurol 10:230–240. https://doi.org/10.1016/S1474-4422(11)70014-X
Tokuda T, Salem SA, Allsop D et al (2006) Decreased alpha-synuclein in cerebrospinal fluid of aged individuals and subjects with Parkinson’s disease. Biochem Biophys Res Commun 349:162–166. https://doi.org/10.1016/j.bbrc.2006.08.024
Eliezer D, Kutluay E, Bussell R Jr, Browne G (2001) Conformational properties of alpha-synuclein in its free and lipid-associated states. J Mol Biol 307:1061–1073. https://doi.org/10.1006/jmbi.2001.4538
Aguzzi A, Heikenwalder M, Polymenidou M (2007) Insights into prion strains and neurotoxicity. Nat Rev Mol Cell Biol 8:552–561. https://doi.org/10.1038/nrm2204
Eisenberg D, Jucker M (2012) The amyloid state of proteins in human diseases. Cell 148:1188–1203. https://doi.org/10.1016/j.cell.2012.02.022
Sacino AN, Thomas MA, Ceballos-Diaz C et al (2013) Conformational templating of alpha-synuclein aggregates in neuronal-glial cultures. Mol Neurodegener 8:17. https://doi.org/10.1186/1750-1326-8-17
Kakish J, Allen KJ, Harkness TA et al (2016) Novel dimer compounds that bind alpha-synuclein can rescue cell growth in a yeast model overexpressing alpha-synuclein. A possible prevention strategy for Parkinson’s disease. ACS Chem Neurosci 7:1671–1680. https://doi.org/10.1021/acschemneuro.6b00209
Jankovic J (2008) Parkinson’s disease and movement disorders: moving forward. Lancet Neurol 7:9–11. https://doi.org/10.1016/S1474-4422(07)70302-2
Tolosa E, Wenning G, Poewe W (2006) The diagnosis of Parkinson’s disease. Lancet Neurol 5:75–86. https://doi.org/10.1016/S1474-4422(05)70285-4
Moore DJ, West AB, Dawson VL, Dawson TM (2005) Molecular pathophysiology of Parkinson’s disease. Annu Rev Neurosci 28:57–87. https://doi.org/10.1146/annurev.neuro.28.061604.135718
Sollinger AB, Goldstein FC, Lah JJ et al (2010) Mild cognitive impairment in Parkinson’s disease: subtypes and motor characteristics. Parkinsonism Relat Disord 16:177–180. https://doi.org/10.1016/j.parkreldis.2009.11.002
Hughes AJ, Daniel SE, Lees AJ (2001) Improved accuracy of clinical diagnosis of Lewy body Parkinson’s disease. Neurology 57:1497–1499
Litvan I, MacIntyre A, Goetz CG et al (1998) Accuracy of the clinical diagnoses of Lewy body disease, Parkinson disease, and dementia with Lewy bodies: a clinicopathologic study. Arch Neurol 55:969–978
Rajput AH, Rozdilsky B, Rajput A (1991) Accuracy of clinical diagnosis in parkinsonism—a prospective study. Can J Neurol Sci 18:275–278
Hughes AJ, Ben-Shlomo Y, Daniel SE, Lees AJ (1992) What features improve the accuracy of clinical diagnosis in Parkinson’s disease: a clinicopathologic study. Neurology 42:1142–1146
Aarsland D, Kurz MW (2010) The epidemiology of dementia associated with Parkinson disease. J Neurol Sci 289:18–22. https://doi.org/10.1016/j.jns.2009.08.034
Aarsland D, Zaccai J, Brayne C (2005) A systematic review of prevalence studies of dementia in Parkinson’s disease. Mov Disord 20:1255–1263. https://doi.org/10.1002/mds.20527
Polymeropoulos MH, Lavedan C, Hollmann M et al (1997) Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 276:2045–2048. https://doi.org/10.1126/science.276.5321.2045
Ahn TB, Kim SY, Kim JY et al (2008) alpha-Synuclein gene duplication is present in sporadic Parkinson disease. Neurology 70:43–49. https://doi.org/10.1212/01.wnl.0000271080.53272.c7
Miller DW, Hague SM, Clarimon J et al (2004) Alpha-synuclein in blood and brain from familial Parkinson disease with SNCA locus triplication. Neurology 62:1835–1838
Lee HJ, Patel S, Lee SJ (2005) Intravesicular localization and exocytosis of alpha-synuclein and its aggregates. J Neurosci 25:6016–6024. https://doi.org/10.1523/JNEUROSCI.0692-05.2005
Marques O, Outeiro TF (2012) Alpha-synuclein: from secretion to dysfunction and death. Cell Death Dis 3:e350. https://doi.org/10.1038/cddis.2012.94
Nakai M, Fujita M, Waragai M et al (2007) Expression of alpha-synuclein, a presynaptic protein implicated in Parkinson’s disease, in erythropoietic lineage. Biochem Biophys Res Commun 358:104–110. https://doi.org/10.1016/j.bbrc.2007.04.108
Iwanaga K, Wakabayashi K, Yoshimoto M et al (1999) Lewy body-type degeneration in cardiac plexus in Parkinson’s and incidental Lewy body diseases. Neurology 52:1269–1271
Beach TG, White CL 3rd, Hladik CL et al (2009) Olfactory bulb alpha-synucleinopathy has high specificity and sensitivity for Lewy body disorders. Acta Neuropathol 117:169–174. https://doi.org/10.1007/s00401-008-0450-7
Braak H, de Vos RA, Bohl J, Del Tredici K (2006) Gastric alpha-synuclein immunoreactive inclusions in Meissner’s and Auerbach’s plexuses in cases staged for Parkinson’s disease-related brain pathology. Neurosci Lett 396:67–72. https://doi.org/10.1016/j.neulet.2005.11.012
Lebouvier T, Chaumette T, Damier P et al (2008) Pathological lesions in colonic biopsies during Parkinson’s disease. Gut 57:1741–1743. https://doi.org/10.1136/gut.2008.162503
Lebouvier T, Neunlist M, Bruley des Varannes S et al (2010) Colonic biopsies to assess the neuropathology of Parkinson’s disease and its relationship with symptoms. PLoS ONE 5:e12728. https://doi.org/10.1371/journal.pone.0012728
Pouclet H, Lebouvier T, Coron E et al (2012) A comparison between colonic submucosa and mucosa to detect Lewy pathology in Parkinson’s disease. Neurogastroenterol Motil 24:e202–e205. https://doi.org/10.1111/j.1365-2982.2012.01887.x
Pouclet H, Lebouvier T, Coron E et al (2012) A comparison between rectal and colonic biopsies to detect Lewy pathology in Parkinson’s disease. Neurobiol Dis 45:305–309. https://doi.org/10.1016/j.nbd.2011.08.014
Shannon KM, Keshavarzian A, Mutlu E et al (2012) Alpha-synuclein in colonic submucosa in early untreated Parkinson’s disease. Mov Disord 27:709–715. https://doi.org/10.1002/mds.23838
Orimo S, Uchihara T, Nakamura A et al (2008) Axonal alpha-synuclein aggregates herald centripetal degeneration of cardiac sympathetic nerve in Parkinson’s disease. Brain 131:642–650. https://doi.org/10.1093/brain/awm302
Fujishiro H, Frigerio R, Burnett M et al (2008) Cardiac sympathetic denervation correlates with clinical and pathologic stages of Parkinson’s disease. Mov Disord 23:1085–1092. https://doi.org/10.1002/mds.21989
Ghebremedhin E, Del Tredici K, Langston JW, Braak H (2009) Diminished tyrosine hydroxylase immunoreactivity in the cardiac conduction system and myocardium in Parkinson’s disease: an anatomical study. Acta Neuropathol 118:777–784. https://doi.org/10.1007/s00401-009-0596-y
Beach TG, Adler CH, Dugger BN et al (2013) Submandibular gland biopsy for the diagnosis of Parkinson disease. J Neuropathol Exp Neurol 72:130–136. https://doi.org/10.1097/NEN.0b013e3182805c72
Cersosimo MG, Perandones C, Micheli FE et al (2011) Alpha-synuclein immunoreactivity in minor salivary gland biopsies of Parkinson’s disease patients. Mov Disord 26:188–190. https://doi.org/10.1002/mds.23344
Del Tredici K, Hawkes CH, Ghebremedhin E, Braak H (2010) Lewy pathology in the submandibular gland of individuals with incidental Lewy body disease and sporadic Parkinson’s disease. Acta Neuropathol 119:703–713. https://doi.org/10.1007/s00401-010-0665-2
Mu L, Sobotka S, Chen J et al (2013) Parkinson disease affects peripheral sensory nerves in the pharynx. J Neuropathol Exp Neurol 72:614–623. https://doi.org/10.1097/NEN.0b013e3182965886
Shi M, Zabetian CP, Hancock AM et al (2010) Significance and confounders of peripheral DJ-1 and alpha-synuclein in Parkinson’s disease. Neurosci Lett 480:78–82. https://doi.org/10.1016/j.neulet.2010.06.009
Hong Z, Shi M, Chung KA et al (2010) DJ-1 and alpha-synuclein in human cerebrospinal fluid as biomarkers of Parkinson’s disease. Brain 133:713–726. https://doi.org/10.1093/brain/awq008
Parnetti L, Chiasserini D, Persichetti E et al (2014) Cerebrospinal fluid lysosomal enzymes and alpha-synuclein in Parkinson’s disease. Mov Disord 29:1019–1027. https://doi.org/10.1002/mds.25772
Wang Y, Shi M, Chung KA et al (2012) Phosphorylated alpha-synuclein in Parkinson’s disease. Sci Transl Med 4:121ra20. https://doi.org/10.1126/scitranslmed.3002566
Jakowec MW, Petzinger GM, Sastry S et al (1998) The native form of alpha-synuclein is not found in the cerebrospinal fluid of patients with Parkinson’s disease or normal controls. Neurosci Lett 253:13–16
Park MJ, Cheon SM, Bae HR et al (2011) Elevated levels of alpha-synuclein oligomer in the cerebrospinal fluid of drug-naive patients with Parkinson’s disease. J Clin Neurol 7:215–222. https://doi.org/10.3988/jcn.2011.7.4.215
Tokuda T, Qureshi MM, Ardah MT et al (2010) Detection of elevated levels of alpha-synuclein oligomers in CSF from patients with Parkinson disease. Neurology 75:1766–1772. https://doi.org/10.1212/WNL.0b013e3181fd613b
Stewart T, Sossi V, Aasly JO et al (2015) Phosphorylated alpha-synuclein in Parkinson’s disease: correlation depends on disease severity. Acta Neuropathol Commun 3:7. https://doi.org/10.1186/s40478-015-0185-3
Shi M, Bradner J, Hancock AM et al (2011) Cerebrospinal fluid biomarkers for Parkinson disease diagnosis and progression. Ann Neurol 69:570–580. https://doi.org/10.1002/ana.22311
Toledo JB, Korff A, Shaw LM et al (2013) CSF alpha-synuclein improves diagnostic and prognostic performance of CSF tau and Abeta in Alzheimer’s disease. Acta Neuropathol 126:683–697. https://doi.org/10.1007/s00401-013-1148-z
Smith LM, Schiess MC, Coffey MP et al (2012) alpha-Synuclein and anti-alpha-synuclein antibodies in Parkinson’s disease, atypical Parkinson syndromes, REM sleep behavior disorder, and healthy controls. PLoS ONE 7:e52285. https://doi.org/10.1371/journal.pone.0052285
Besong-Agbo D, Wolf E, Jessen F et al (2013) Naturally occurring alpha-synuclein autoantibody levels are lower in patients with Parkinson disease. Neurology 80:169–175. https://doi.org/10.1212/WNL.0b013e31827b90d1
Duran R, Barrero FJ, Morales B et al (2010) Plasma alpha-synuclein in patients with Parkinson’s disease with and without treatment. Mov Disord 25:489–493. https://doi.org/10.1002/mds.22928
Foulds PG, Mitchell JD, Parker A et al (2011) Phosphorylated alpha-synuclein can be detected in blood plasma and is potentially a useful biomarker for Parkinson’s disease. FASEB J 25:4127–4137. https://doi.org/10.1096/fj.10-179192
Li QX, Mok SS, Laughton KM et al (2007) Plasma alpha-synuclein is decreased in subjects with Parkinson’s disease. Exp Neurol 204:583–588. https://doi.org/10.1016/j.expneurol.2006.12.006
Mata IF, Shi M, Agarwal P et al (2010) SNCA variant associated with Parkinson disease and plasma alpha-synuclein level. Arch Neurol 67:1350–1356. https://doi.org/10.1001/archneurol.2010.279
Gorostidi A, Bergareche A, Ruiz-Martinez J et al (2012) Alphalpha-synuclein levels in blood plasma from LRRK2 mutation carriers. PLoS ONE 7:e52312. https://doi.org/10.1371/journal.pone.0052312
Shi M, Liu C, Cook TJ et al (2014) Plasma exosomal alpha-synuclein is likely CNS-derived and increased in Parkinson’s disease. Acta Neuropathol 128:639–650. https://doi.org/10.1007/s00401-014-1314-y
Yanamandra K, Gruden MA, Casaite V et al (2011) alpha-synuclein reactive antibodies as diagnostic biomarkers in blood sera of Parkinson’s disease patients. PLoS ONE 6:e18513. https://doi.org/10.1371/journal.pone.0018513
Barbour R, Kling K, Anderson JP et al (2008) Red Blood Cells Are the Major Source of Alpha-Synuclein in Blood. Neurodegener Dis 5:55–59. https://doi.org/10.1159/000112832
Abd-Elhadi S, Honig A, Simhi-Haham D et al (2015) Total and proteinase K-resistant alpha-synuclein levels in erythrocytes, determined by their ability to bind phospholipids, associate with Parkinson’s Disease. Sci Rep 5:11120. https://doi.org/10.1038/srep11120
Devic I, Hwang H, Edgar JS et al (2011) Salivary alpha-synuclein and DJ-1: potential biomarkers for Parkinson’s disease. Brain 134:e178. https://doi.org/10.1093/brain/awr015
Stewart T, Sui Y-T, Gonzalez-Cuyar LF et al (2014) Cheek cell–derived α-synuclein and DJ-1 do not differentiate Parkinson’s disease from control. Neurobiol Aging 35:418–420. https://doi.org/10.1016/j.neurobiolaging.2013.08.008
Al-Nimer MS, Mshatat SF, Abdulla HI (2014) Saliva alpha-synuclein and a high extinction coefficient protein: a novel approach in assessment biomarkers of Parkinson’s disease. N Am J Med Sci 6:633–637. https://doi.org/10.4103/1947-2714.147980
Lu FM, Yuan Z (2015) PET/SPECT molecular imaging in clinical neuroscience: recent advances in the investigation of CNS diseases. Quant Imaging Med Surg 5:433–447. https://doi.org/10.3978/j.issn.2223-4292.2015.03.16
Massoud TF, Gambhir SS (2003) Molecular imaging in living subjects: seeing fundamental biological processes in a new light. Genes Dev 17:545–580. https://doi.org/10.1101/gad.1047403
Weissleder R, Mahmood U (2001) Molecular imaging. Radiology 219:316–333. https://doi.org/10.1148/radiology.219.2.r01ma19316
Chen K, Chen X (2010) Design and development of molecular imaging probes. Curr Top Med Chem 10:1227–1236
Kim E, Howes OD, Kapur S (2013) Molecular imaging as a guide for the treatment of central nervous system disorders. Dialog Clin Neurosci 15:315–328
Phelps ME (2000) Positron emission tomography provides molecular imaging of biological processes. Proc Natl Acad Sci USA 97:9226–9233. https://doi.org/10.1073/pnas.97.16.9226
Halldin C, Gulyas B, Langer O, Farde L (2001) Brain radioligands–state of the art and new trends. Q J Nucl Med 45:139–152
Strijckmans K (2001) The isochronous cyclotron: principles and recent developments. Comput Med Imaging Graph 25:69–78
Shoghi-Jadid K, Small GW, Agdeppa ED et al (2002) Localization of neurofibrillary tangles and beta-amyloid plaques in the brains of living patients with Alzheimer disease. Am J Geriatr Psychiatry 10:24–35
Brooks DJ, Ibanez V, Sawle GV et al (1990) Differing patterns of striatal 18F-dopa uptake in Parkinson’s disease, multiple system atrophy, and progressive supranuclear palsy. Ann Neurol 28:547–555. https://doi.org/10.1002/ana.410280412
Klunk WE, Engler H, Nordberg A et al (2004) Imaging brain amyloid in Alzheimer’s disease with Pittsburgh compound-B. Ann Neurol 55:306–319. https://doi.org/10.2466/15.17.23.PMS.111.5.485-495
Mintun MA, Larossa GN, Sheline YI et al (2006) [11C]PIB in a nondemented population: potential antecedent marker of Alzheimer disease. Neurology 67:446–452. https://doi.org/10.1212/01.wnl.0000228230.26044.a4
Farde L, Wiesel FA, Stone-Elander S et al (1990) D2 dopamine receptors in neuroleptic-naive schizophrenic patients. A positron emission tomography study with [11C]raclopride. Arch Gen Psychiatry 47:213–219
Hirvonen J, Karlsson H, Kajander J et al (2008) Striatal dopamine D2 receptors in medication-naive patients with major depressive disorder as assessed with [11C]raclopride PET. Psychopharmacology 197:581–590. https://doi.org/10.1007/s00213-008-1088-9
Banati RB, Newcombe J, Gunn RN et al (2000) The peripheral benzodiazepine binding site in the brain in multiple sclerosis: quantitative in vivo imaging of microglia as a measure of disease activity. Brain 123(Pt 1):2321–2337
Groom GN, Junck L, Foster NL et al (1995) PET of peripheral benzodiazepine binding sites in the microgliosis of Alzheimer’s disease. J Nucl Med 36:2207–2210
Savic I, Thorell JO, Roland P (1995) [11C]flumazenil positron emission tomography visualizes frontal epileptogenic regions. Epilepsia 36:1225–1232
Kadir A, Almkvist O, Wall A et al (2006) PET imaging of cortical 11C-nicotine binding correlates with the cognitive function of attention in Alzheimer’s disease. Psychopharmacology 188:509–520. https://doi.org/10.1007/s00213-006-0447-7
Watanabe H, Ariyoshi T, Ozaki A et al (2017) Synthesis and biological evaluation of novel radioiodinated benzimidazole derivatives for imaging α-synuclein aggregates. Bioorg Med Chem 25:6398–6403. https://doi.org/10.1016/j.bmc.2017.10.010
Kotzbauer PT, Tu Z, Mach RH (2017) Current status of the development of PET radiotracers for imaging alpha synuclein aggregates in Lewy bodies and Lewy neurites. Clin Transl Imaging 5:3–14. https://doi.org/10.1007/s40336-016-0217-4
Ye L, Velasco A, Fraser G et al (2008) In vitro high affinity α-synuclein binding sites for the amyloid imaging agent PIB are not matched by binding to Lewy bodies in postmortem human brain. J Neurochem 105:1428–1437. https://doi.org/10.1111/j.1471-4159.2008.05245.x
Fodero-Tavoletti MT, Smith DP, McLean CA et al (2007) In vitro characterization of Pittsburgh compound-B binding to lewy bodies. J Neurosci 27:10365–10371. https://doi.org/10.1523/JNEUROSCI.0630-07.2007
Fodero-Tavoletti MT, Mulligan RS, Okamura N et al (2009) In vitro characterisation of BF227 binding to α-synuclein/Lewy bodies. Eur J Pharmacol 617:54–58. https://doi.org/10.1016/j.ejphar.2009.06.042
Yu L, Cui J, Padakanti PK et al (2012) Synthesis and in vitro evaluation of α-synuclein ligands. Bioorg Med Chem 20:4625–4634. https://doi.org/10.1016/j.bmc.2012.06.023
Zhang X, Jin H, Padakanti PK et al (2014) Radiosynthesis and in vivo evaluation of two PET radioligands for imaging α-synuclein. Appl Sci (Switz) 4:66–78. https://doi.org/10.3390/app4010066
Chu W, Zhou D, Gaba V et al (2015) Design, synthesis, and characterization of 3-(benzylidene)indolin-2-one derivatives as ligands for α-synuclein fibrils. J Med Chem 58:6002–6017. https://doi.org/10.1021/acs.jmedchem.5b00571
Xuming M, Ryan P, Rudrawar S et al (2020) Advances in the development of imaging probes and aggregation inhibitors for alpha-synuclein. Acta Pharmacol Sin 41:483–498. https://doi.org/10.1038/s41401-019-0304-y
Pavese N, Brooks DJ (2009) Imaging neurodegeneration in Parkinson’s disease. Biochim Biophys Acta Mol Basis Dis 1792:722–729. https://doi.org/10.1016/j.bbadis.2008.10.003
Wang L, Zhang Q, Li H, Zhang H (2012) SPECT molecular imaging in Parkinson’s disease. J Biomed Biotechnol 2012:412486. https://doi.org/10.1155/2012/412486
Gao HM, Jiang J, Wilson B et al (2002) Microglial activation-mediated delayed and progressive degeneration of rat nigral dopaminergic neurons: relevance to Parkinson’s disease. J Neurochem 81:1285–1297
Mirza B, Hadberg H, Thomsen P, Moos T (2000) The absence of reactive astrocytosis is indicative of a unique inflammatory process in Parkinson’s disease. Neuroscience 95:425–432
Sherer TB, Betarbet R, Kim JH, Greenamyre JT (2003) Selective microglial activation in the rat rotenone model of Parkinson’s disease. Neurosci Lett 341:87–90
Sugama S, Yang L, Cho BP et al (2003) Age-related microglial activation in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced dopaminergic neurodegeneration in C57BL/6 mice. Brain Res 964:288–294
Vila M, Jackson-Lewis V, Guegan C et al (2001) The role of glial cells in Parkinson’s disease. Curr Opin Neurol 14:483–489
Kreutzberg GW (1996) Microglia: a sensor for pathological events in the CNS. Trends Neurosci 19:312–318
Ouchi Y, Yoshikawa E, Sekine Y et al (2005) Microglial activation and dopamine terminal loss in early Parkinson’s disease. Ann Neurol 57:168–175. https://doi.org/10.1002/ana.20338
Gerhard A, Pavese N, Hotton G et al (2006) In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson’s disease. Neurobiol Dis 21:404–412. https://doi.org/10.1016/j.nbd.2005.08.002
Cumming P, Borghammer P (2012) Molecular imaging and the neuropathologies of Parkinson’s disease. Curr Top Behav Neurosci 11:117–148. https://doi.org/10.1007/7854_2011_165
Pedrosa DJ, Timmermann L (2013) Review: management of Parkinson’s disease. Neuropsychiatr Dis Treat 9:321–340. https://doi.org/10.2147/NDT.S32302
Feany MB, Pallanck LJ (2003) Parkin: a multipurpose neuroprotective agent? Neuron 38:13–16
Klein C, Westenberger A (2012) Genetics of Parkinson’s disease. Cold Spring Harb Perspect Med 2:a008888. https://doi.org/10.1101/cshperspect.a008888
Youle RJ, Narendra DP (2011) Mechanisms of mitophagy. Nat Rev Mol Cell Biol 12:9–14. https://doi.org/10.1038/nrm3028
Kakish J, Lee D, Lee JS (2015) Drugs that bind to alpha-synuclein: neuroprotective or neurotoxic? ACS Chem Neurosci 6:1930–1940. https://doi.org/10.1021/acschemneuro.5b00172
Bar Am O, Amit T, Youdim MB (2004) Contrasting neuroprotective and neurotoxic actions of respective metabolites of anti-Parkinson drugs rasagiline and selegiline. Neurosci Lett 355:169–172
Bar-Am O, Weinreb O, Amit T, Youdim MB (2010) The neuroprotective mechanism of 1-(R)-aminoindan, the major metabolite of the anti-parkinsonian drug rasagiline. J Neurochem 112:1131–1137. https://doi.org/10.1111/j.1471-4159.2009.06542.x
Chau KY, Cooper JM, Schapira AH (2010) Rasagiline protects against alpha-synuclein induced sensitivity to oxidative stress in dopaminergic cells. Neurochem Int 57:525–529. https://doi.org/10.1016/j.neuint.2010.06.017
Dimpfel W, Hoffmann JA (2011) Effects of rasagiline, its metabolite aminoindan and selegiline on glutamate receptor mediated signalling in the rat hippocampus slice in vitro. BMC Pharmacol 11:2. https://doi.org/10.1186/1471-2210-11-2
Patil SP, Jain PD, Ghumatkar PJ et al (2014) Neuroprotective effect of metformin in MPTP-induced Parkinson’s disease in mice. Neuroscience 277:747–754. https://doi.org/10.1016/j.neuroscience.2014.07.046
Postuma RB, Lang AE, Munhoz RP et al (2012) Caffeine for treatment of Parkinson disease: a randomized controlled trial. Neurology 79:651–658. https://doi.org/10.1212/WNL.0b013e318263570d
Prediger RDS (2010) Effects of caffeine in Parkinson’s disease: From neuroprotection to the management of motor and non-motor symptoms. J Alzheimer Dis. https://doi.org/10.3233/JAD-2010-091459
Quik M (2004) Smoking, nicotine and Parkinson’s disease. Trends Neurosci 27:561–568. https://doi.org/10.1016/j.tins.2004.06.008
Quik M, Perez XA, Bordia T (2012) Nicotine as a potential neuroprotective agent for Parkinson’s disease. Mov Disord 27:947–957. https://doi.org/10.1002/mds.25028
Ross GW, Petrovitch H (2001) Current evidence for neuroprotective effects of nicotine and caffeine against Parkinson’s disease. Drugs Aging 18:797–806. https://doi.org/10.2165/00002512-200118110-00001
Wahlqvist ML, Lee MS, Hsu CC et al (2012) Metformin-inclusive sulfonylurea therapy reduces the risk of Parkinson’s disease occurring with Type 2 diabetes in a Taiwanese population cohort. Parkinson Relat Disord 18:753–758. https://doi.org/10.1016/j.parkreldis.2012.03.010
Games D, Valera E, Spencer B et al (2014) Reducing C-terminal-truncated alpha-synuclein by immunotherapy attenuates neurodegeneration and propagation in Parkinson’s disease-like models. J Neurosci 34:9441–9454. https://doi.org/10.1523/JNEUROSCI.5314-13.2014
Dufty BM, Warner LR, Hou ST et al (2007) Calpain-cleavage of alpha-synuclein: connecting proteolytic processing to disease-linked aggregation. Am J Pathol 170:1725–1738. https://doi.org/10.2353/ajpath.2007.061232
Nwabufo CK, El-Aneed A, Krol ES (2019) Tandem mass spectrometric analysis of novel caffeine scaffold-based bifunctional compounds for Parkinson’s disease. Rapid Commun Mass Spectrom 33:1792–1803. https://doi.org/10.1002/rcm.8540
Nwabufo CK, Aigbogun OP, Allen KJH et al (2021) Employing in vitro metabolism to guide design of F-labelled PET probes of novel α-synuclein binding bifunctional compounds. Xenobiotica 51:885–900. https://doi.org/10.1080/00498254.2021.1943566
Nwabufo C, Krol E (2019) Unraveling the metabolic fate of potential therapeutic dimer compounds for Parkinson’s disease. Drug Metab Pharmacokinet 34:S59–S60
Hussain G, Rasul A, Anwar H et al (2018) Role of plant derived alkaloids and their mechanism in neurodegenerative disorders. Int J Biol Sci 14:341–357. https://doi.org/10.7150/ijbs.23247
Zhang H, Bai L, He J et al (2017) Recent advances in discovery and development of natural products as source for anti-Parkinson’s disease lead compounds. Eur J Med Chem 141:257–272. https://doi.org/10.1016/j.ejmech.2017.09.068
Fu RH, Wang YC, Chen CS et al (2014) Acetylcorynoline attenuates dopaminergic neuron degeneration and α-synuclein aggregation in animal models of Parkinson’s disease. Neuropharmacology 82:108–120. https://doi.org/10.1016/j.neuropharm.2013.08.007
Leem YH, Park JS, Park JE et al (2020) Papaverine inhibits α-synuclein aggregation by modulating neuroinflammation and matrix metalloproteinase-3 expression in the subacute MPTP/P mouse model of Parkinson’s disease. Biomed Pharmacother 130:110576. https://doi.org/10.1016/j.biopha.2020.110576
Jayaraj RL, Beiram R, Azimullah S et al (2021) Noscapine prevents rotenone-induced neurotoxicity: Involvement of oxidative stress, neuroinflammation and autophagy pathways. Molecules. https://doi.org/10.3390/molecules26154627
Ghanem SS, Fayed HS, Zhu Q et al (2021) Natural alkaloid compounds as inhibitors for alpha-synuclein seeded fibril formation and toxicity. Molecules 26:1–13. https://doi.org/10.3390/molecules26123736
Andersen CB, Yoshimura Y, Nielsen J et al (2021) How epigallocatechin gallate binds and assembles oligomeric forms of human alpha-synuclein. J Biol Chem 296:100788. https://doi.org/10.1016/j.jbc.2021.100788
Yao Y, Tang Y, Wei G (2020) Epigallocatechin gallate destabilizes α-synuclein fibril by disrupting the E46–K80 salt-bridge and inter-protofibril interface. ACS Chem Neurosci 11:4351–4361. https://doi.org/10.1021/acschemneuro.0c00598
Chen M, Wang T, Yue F et al (2015) Tea polyphenols alleviate motor impairments, dopaminergic neuronal injury, and cerebral α-synuclein aggregation in MPTP-intoxicated parkinsonian monkeys. Neuroscience 286:383–392. https://doi.org/10.1016/j.neuroscience.2014.12.003
Yang JE, Rhoo KY, Lee S et al (2017) EGCG-mediated protection of the membrane disruption and cytotoxicity caused by the “active oligomer” of α-synuclein. Sci Rep 7:1–10. https://doi.org/10.1038/s41598-017-18349-z
Zhu M, Rajamani S, Kaylor J et al (2004) The flavonoid baicalein inhibits fibrillation of α-synuclein and disaggregates existing fibrils. J Biol Chem 279:26846–26857. https://doi.org/10.1074/jbc.M403129200
Daniels MJ, Nourse JB, Kim H et al (2019) Cyclized NDGA modifies dynamic α-synuclein monomers preventing aggregation and toxicity. Sci Rep 9:1–17. https://doi.org/10.1038/s41598-019-39480-z
Pujols J, Peña-Díaz S, Pallarès I, Ventura S (2020) Chemical chaperones as novel drugs for Parkinson’s disease. Trends Mol Med 26:408–421. https://doi.org/10.1016/j.molmed.2020.01.005
Fernández CO, Hoyer W, Zweckstetter M et al (2004) NMR of α-synuclein-polyamine complexes elucidates the mechanism and kinetics of induced aggregation. EMBO J 23:2039–2046. https://doi.org/10.1038/sj.emboj.7600211
Perni M, Galvagnion C, Maltsev A, Meisl G, Müller MBD, Challa PK, Kirkegaard JB, Flagmeier P, Cohen SIA, Cascella R, Chen SW, Limbocker R, Sormanni P, Heller GT, Fra CMD (2017) A natural product inhibits the initiation of α-synuclein aggregation and suppresses its toxicity. Proc Natl Acad Sci USA 114:E2543. https://doi.org/10.1073/pnas.1701964114
Perni M, Flagmeier P, Limbocker R et al (2018) Multistep Inhibition of α-synuclein aggregation and toxicity in vitro and in vivo by trodusquemine. ACS Chem Biol 13:2308–2319. https://doi.org/10.1021/acschembio.8b00466
Li J, Zhu M, Rajamani S et al (2004) Rifampicin inhibits-synuclein fibrillation and disaggregates fibrils. Chem Biol 11:1513–1521
Xu J, Wei C, Xu C et al (2007) Rifampicin protects PC12 cells against MPP+-induced apoptosis and inhibits the expression of an α-Synuclein multimer. Brain Res 1139:220–225. https://doi.org/10.1016/j.brainres.2006.12.074
Low PA, Robertson D, Gilman S et al (2014) Efficacy and safety of rifampicin for multiple system atrophy: a randomised, double-blind, placebo-controlled trial. Lancet Neurol 13:268–275. https://doi.org/10.1016/S1474-4422(13)70301-6
Török N, Majláth Z, Szalárdy L, Vécsei L (2016) Investigational α-synuclein aggregation inhibitors: hope for Parkinson’s disease. Expert Opin Investig Drugs 25:1281–1294. https://doi.org/10.1080/13543784.2016.1237501
Dominguez-Meijide A, Parrales V, Vasili E et al (2021) Doxycycline inhibits α-synuclein-associated pathologies in vitro and in vivo. Neurobiol Dis. https://doi.org/10.1016/j.nbd.2021.105256
Tai CH, Bellesi M, Chen AC et al (2019) A new avenue for treating neuronal diseases: ceftriaxone, an old antibiotic demonstrating behavioral neuronal effects. Behav Brain Res 364:149–156. https://doi.org/10.1016/j.bbr.2019.02.020
Song JX, Sun YR, Peluso I et al (2016) A novel curcumin analog binds to and activates TFEB in vitro and in vivo independent of MTOR inhibition. Autophagy 12:1372–1389. https://doi.org/10.1080/15548627.2016.1179404
Liu J, Liu C, Zhang J et al (2020) A self-assembled α-synuclein nanoscavenger for Parkinson’s disease. ACS Nano 14:1533–1549. https://doi.org/10.1021/acsnano.9b06453
Sardiello M, Palmieri M, di Ronza A, Medina DL, Valenza M, Gennarino V, Di Malta A, Donaudy C, Polishchuk RS, Banfi S, Cattaneo PG, Ballabio EA (2009) A gene network regulating lysosomal biogenesis and function. Science 325:473–478
Decressac M, Mattsson B, Weikop P et al (2013) TFEB-mediated autophagy rescues midbrain dopamine neurons from α-synuclein toxicity. Proc Natl Acad Sci USA. https://doi.org/10.1073/pnas.1305623110
Lopez-Cuina M, Meissner WG (2022) Targeting alpha-synuclein or tau for treating neurodegenerative movement disorders. Revue Neurologique 178:460–471. https://doi.org/10.1016/j.neurol.2022.03.010
Tran HT, Chung CH-Y, Iba M et al (2014) α-Synuclein immunotherapy blocks uptake and templated propagation of misfolded α-synuclein and neurodegeneration. Cell Rep 7:2054–2065. https://doi.org/10.1016/j.celrep.2014.05.033
Masliah E, Rockenstein E, Adame A et al (2005) Effects of α-synuclein immunization in a mouse model of Parkinson’s disease. Neuron 46:857–868. https://doi.org/10.1016/j.neuron.2005.05.010
Spencer B, Valera E, Rockenstein E et al (2017) Anti-α-synuclein immunotherapy reduces α-synuclein propagation in the axon and degeneration in a combined viral vector and transgenic model of synucleinopathy. Acta Neuropathol Commun 5:7. https://doi.org/10.1186/s40478-016-0410-8
Valera E, Spencer B, Fields JA et al (2017) Combination of alpha-synuclein immunotherapy with anti-inflammatory treatment in a transgenic mouse model of multiple system atrophy. Acta Neuropathol Commun 5:2. https://doi.org/10.1186/s40478-016-0409-1
Masliah E, Rockenstein E, Mante M et al (2011) Passive immunization reduces behavioral and neuropathological deficits in an alpha-synuclein transgenic model of Lewy Body disease. PLoS ONE 6:e19338. https://doi.org/10.1371/journal.pone.0019338
Mandler M, Valera E, Rockenstein E et al (2015) Active immunization against alpha-synuclein ameliorates the degenerative pathology and prevents demyelination in a model of multiple system atrophy. Mol Neurodegeneration 10:10. https://doi.org/10.1186/s13024-015-0008-9
Mandler M, Valera E, Rockenstein E et al (2014) Next-generation active immunization approach for synucleinopathies: implications for Parkinson’s disease clinical trials. Acta Neuropathol 127:861–879. https://doi.org/10.1007/s00401-014-1256-4
Games D, Seubert P, Rockenstein E et al (2013) Axonopathy in an α-synuclein transgenic model of Lewy body disease is associated with extensive accumulation of c-terminal–truncated α-synuclein. Am J Pathol 182:940–953. https://doi.org/10.1016/j.ajpath.2012.11.018
Schenk DB, Koller M, Ness DK et al (2017) First-in-human assessment of PRX002, an anti-α-synuclein monoclonal antibody, in healthy volunteers: immunotherapy for Parkinson’s disease. Mov Disord 32:211–218. https://doi.org/10.1002/mds.26878
Jankovic J, Goodman I, Safirstein B et al (2018) Safety and tolerability of multiple ascending doses of PRX002/RG7935, an anti–α-synuclein monoclonal antibody, in patients with Parkinson disease: a randomized clinical trial. JAMA Neurol 75:1206. https://doi.org/10.1001/jamaneurol.2018.1487
Lindström V, Fagerqvist T, Nordström E et al (2014) Immunotherapy targeting α-synuclein protofibrils reduced pathology in (Thy-1)-h[A30P] α-synuclein mice. Neurobiol Dis 69:134–143. https://doi.org/10.1016/j.nbd.2014.05.009
Kallab M, Herrera-Vaquero M, Johannesson M et al (2018) Region-specific effects of immunotherapy with antibodies targeting α-synuclein in a transgenic model of synucleinopathy. Front Neurosci 12:452. https://doi.org/10.3389/fnins.2018.00452
Weihofen A, Liu Y, Arndt JW et al (2019) Development of an aggregate-selective, human-derived α-synuclein antibody BIIB054 that ameliorates disease phenotypes in Parkinson’s disease models. Neurobiol Dis 124:276–288. https://doi.org/10.1016/j.nbd.2018.10.016
Brys M, Fanning L, Hung S et al (2019) Randomized phase I clinical trial of anti–α-synuclein antibody BIIB054. Mov Disord 34:1154–1163. https://doi.org/10.1002/mds.27738
Biogen Annual Report (2020) Biogen
Schofield DJ, Irving L, Calo L et al (2019) Preclinical development of a high affinity α-synuclein antibody, MEDI1341, that can enter the brain, sequester extracellular α-synuclein and attenuate α-synuclein spreading in vivo. Neurobiol Dis 132:104582. https://doi.org/10.1016/j.nbd.2019.104582
Fjord-Larsen L, Thougaard A, Wegener KM et al (2021) Nonclinical safety evaluation, pharmacokinetics, and target engagement of Lu AF82422, a monoclonal IgG1 antibody against alpha-synuclein in development for treatment of synucleinopathies. MAbs 13:1994690. https://doi.org/10.1080/19420862.2021.1994690
Thakur A, Tan Z, Kameyama T et al (2021) Bioanalytical strategies in drug discovery and development. Drug Metab Rev 53:434–458. https://doi.org/10.1080/03602532.2021.1959606
Nwabufo CK (2021) Introduction to the mini special issue on next generation drug discovery and development: rethinking translational pharmacology for accelerated drug development. Drug Metab Rev 53:171–172. https://doi.org/10.1080/03602532.2021.1909614
Nwabufo CK (2022) Relevance of ABC transporters in drug development. CDM. https://doi.org/10.2174/1389200223666220621113524
Hou J, Qu F, Wu C et al (2012) Quantitative determination and pharmacokinetic study of the novel anti-Parkinson’s disease candidate drug FLZ in rat brain by high performance liquid chromatography-tandem mass spectrometry. J Pharm Biomed Anal 66:232–239. https://doi.org/10.1016/j.jpba.2012.03.001
Bartels AL, Willemsen ATM, Kortekaas R et al (2008) Decreased blood-brain barrier P-glycoprotein function in the progression of Parkinson’s disease, PSP and MSA. J Neural Transm 115:1001–1009. https://doi.org/10.1007/s00702-008-0030-y
Zlokovic BV (2008) The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron 57:178–201. https://doi.org/10.1016/j.neuron.2008.01.003
Liu Q, Hou J, Chen X et al (2014) P-glycoprotein mediated efflux limits the transport of the novel anti-Parkinson’s disease candidate drug FLZ across the physiological and PD pathological in vitro BBB models. PLoS ONE 9:e102442. https://doi.org/10.1371/journal.pone.0102442
Hoosain FG, Choonara YE, Tomar LK et al (2015) Bypassing P-glycoprotein drug efflux mechanisms: possible applications in pharmacoresistant schizophrenia therapy. Biomed Res Int 2015:484963. https://doi.org/10.1155/2015/484963
Zhu L, Ploessl K, Kung HF (2014) PET/SPECT imaging agents for neurodegenerative diseases. Chem Soc Rev 43:6683–6691. https://doi.org/10.1039/C3CS60430F
Funding
CKN is a recipient of the University of Saskatchewan College of Pharmacy and Nutrition Graduate Scholarship, Ontario Graduate Scholarship and Leslie Dan Faculty of Pharmacy Deans Fellowship. OPA is a recipient of the University of Saskatchewan Department of Chemistry Graduate Scholarship and Teacher—Scholar Doctoral Fellowship.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare no financial relationship to this work.
Rights and permissions
About this article

Cite this article
Nwabufo, C.K., Aigbogun, O.P. Diagnostic and therapeutic agents that target alpha-synuclein in Parkinson’s disease. J Neurol 269, 5762–5786 (2022). https://doi.org/10.1007/s00415-022-11267-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-022-11267-9