
Abstract
Background
Niemann Pick type C is an autosomal recessive lysosomal storage disorder caused by mutations in NPC1 and NPC2 genes. It is a neuro-visceral disease with a heterogeneous phenotype. Clinical features depend on the age at onset. Visceral manifestations are more prominent in the early onset (infantile) form, while neuro-psychiatric symptoms are more prominent in the late disease onset (juvenile and adult forms).
Methods
A total number of 150 patients have been screened for changes in NPC1 and NPC2 gene at the Neurology Clinic, University Clinical Centre of Serbia in the period 2012–2020. Clinical data were extracted for patients with biallelic mutations.
Results
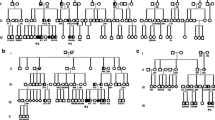
Fifteen patients carried biallelic mutations in the NPC1. Out of eight different reported NPC1 variants, four are novel (c.1204_1205TT>GC, p.F402A; c.2486T>G, p.L829R; c.2795+5 G>C; c.3722T>A, p.L1241*). The mean age at the disease onset was 20.3 ± 11.9 years with the average diagnostic delay of 7.7 ± 4.3 years. Movement disorders and psychiatric or cognitive disturbances were the most common initial symptoms (in 33% and 28% patients, respectively). The average age at the first neurological manifestation was 21 ± 12.0 years. At the last examination, eye movement abnormalities (vertical slow saccades or vertical supranuclear gaze palsy), and ataxia were present in all patients, while dystonia was common (in 78.6% of patients). Presence of c.2861C>T, p.S954L mutation in homozygous state was associated with older age at the neurological symptom onset.
Conclusions
Clinical findings were in line with the expected, but the diagnostic delay was common. We hypothesize that the presence of c.2861C>T, p.S954L mutation may contribute to the phenotype attenuation.


Similar content being viewed by others


Data availability
Not applicable.
Code availability
Not applicable.
References
Vanier MT (2010) Niemann-Pick disease type C review. Orphanet J Rare Dis 5:16. https://doi.org/10.1186/1750-1172-5-16
Stenson PD, Ball EV, Mort M et al (2003) Human Gene Mutation Database (HGMD): 2003 update. Hum Mutat 21(6):577–581. https://doi.org/10.1002/humu.10212
Patterson MC, Hendriksz CJ, Walterfang M et al (2012) Recommendations for the diagnosis and management of Niemann-Pick disease type C: an update. Mol Genet Metab 106(3):330–344. https://doi.org/10.1016/j.ymgme.2012.03.012
Chamova T, Kirov A, Guergueltcheva V et al (2016) Clinical spectrum and genetic variability in Bulgarian patients with Niemann-Pick disease type C. Eur Neurol 75(3–4):113–123. https://doi.org/10.1159/000444480
Mengel E, Klünemann HH, Lourenço CM et al (2013) Niemann-Pick disease type C symptomatology: an expert-based clinical description. Orphanet J Rare Dis 8:166. https://doi.org/10.1186/1750-1172-8-166
Geberhiwot T, Moro A, Dardis A et al (2018) Consensus clinical management guidelines for Niemann-Pick disease type C. Orphanet J Rare Dis 13(1):50. https://doi.org/10.1186/s13023-018-0785-7
Wraith JE, Guffon N, Rohrbach M et al (2009) Natural history of Niemann-Pick disease type C in a multicentre observational retrospective cohort study. Mol Genet Metab 98(3):250–254. https://doi.org/10.1016/j.ymgme.2009.06.009
Wijburg FA, Sedel F, Pineda M et al (2012) Development of a suspicion index to aid diagnosis of Niemann-Pick disease type C. Neurology 78(20):1560–1567. https://doi.org/10.1212/WNL.0b013e3182563b82
Schwarz JM, Rödelsperger C, Schuelke M, Seelow D (2010) MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods 7(8):575–576. https://doi.org/10.1038/nmeth0810-575
Pejaver V, Urresti J, Lugo-Martinez J et al (2020) Inferring the molecular and phenotypic impact of amino acid variants with MutPred2. Nat Commun 11(1):5918. https://doi.org/10.1038/s41467-020-19669-x
Rentzsch P, Schubach M, Shendure J, Kircher M (2021) CADD-Splice-improving genome-wide variant effect prediction using deep learning-derived splice scores. Genome Med 13(1):31. https://doi.org/10.1186/s13073-021-00835-9
Desmet FO, Hamroun D, Lalande M, Collod-Béroud G, Claustres M, Béroud C (2009) Human splicing finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res 37(9):e67. https://doi.org/10.1093/nar/gkp215
Karczewski KJ, Francioli LC, Tiao G et al (2020) The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581(7809):434–443. https://doi.org/10.1038/s41586-020-2308-7
Database of Single Nucleotide Polymorphisms (dbSNP). Bethesda (MD): National Center for Biotechnology Information, National Library of Medicine. (dbSNP Build ID:155). http://www.ncbi.nlm.nih.gov/SNP/. Accessed July 2021
Dardis A, Zampieri S, Gellera C et al (2020) Molecular genetics of Niemann-Pick type C disease in Italy: an update on 105 patients and description of 18 NPC1 novel variants. J Clin Med 9(3):679. https://doi.org/10.3390/jcm9030679
Jahnova H, Dvorakova L, Vlaskova H et al (2014) Observational, retrospective study of a large cohort of patients with Niemann-Pick disease type C in the Czech Republic: a surprisingly stable diagnostic rate spanning almost 40 years. Orphanet J Rare Dis 9:140. https://doi.org/10.1186/s13023-014-0140-6
Macías-Vidal J, Rodríguez-Pascau L, Sánchez-Ollé G et al (2011) Molecular analysis of 30 Niemann-Pick type C patients from Spain. Clin Genet 80(1):39–49. https://doi.org/10.1111/j.1399-0004.2010.01504.x
Millat G, Marçais C, Rafi MA et al (1999) Niemann-Pick C1 disease: the I1061T substitution is a frequent mutant allele in patients of Western European descent and correlates with a classic juvenile phenotype. Am J Hum Genet 65(5):1321–1329. https://doi.org/10.1086/302626
Sévin M, Lesca G, Baumann N et al (2007) The adult form of Niemann-Pick disease type C. Brain 130(Pt 1):120–133. https://doi.org/10.1093/brain/awl260
Koens LH, Kuiper A, Coenen MA et al (2016) Ataxia, dystonia and myoclonus in adult patients with Niemann-Pick type C. Orphanet J Rare Dis 11(1):121. https://doi.org/10.1186/s13023-016-0502-3
Nadjar Y, Hütter-Moncada AL, Latour P et al (2018) Adult Niemann-Pick disease type C in France: clinical phenotypes and long-term miglustat treatment effect. Orphanet J Rare Dis 13(1):175. https://doi.org/10.1186/s13023-018-0913-4
Ganos C, Crowe B, Stamelou M et al (2016) The clinical syndrome of dystonia with anarthria/aphonia. Parkinsonism Relat Disord 24:20–27. https://doi.org/10.1016/j.parkreldis.2016.01.022
Kresojević N, Mandić-Stojmenović G, Dobričić V et al (2020) Very late-onset Niemann Pick type C disease: example of progressive supranuclear palsy look-alike disorder. Mov Disord Clin Pract 7(2):211–214. https://doi.org/10.1002/mdc3.12892
Kim R, Yoo D, Park S et al (2020) A rare case of late adult-onset Niemann-Pick disease type C. J Mov Disord 13(2):163–165. https://doi.org/10.14802/jmd.19077
Wu M, Ceponiene R, Bayram E, Litvan I (2020) Two patients with Niemann Pick disease type C diagnosed in the seventh decade of life. Mov Disord Clin Pract 7(8):961–964. https://doi.org/10.1002/mdc3.13085
Abela L, Plecko B, Palla A et al (2014) Early co-occurrence of a neurologic-psychiatric disease pattern in Niemann-Pick type C disease: a retrospective Swiss cohort study. Orphanet J Rare Dis 9:176. https://doi.org/10.1186/s13023-014-0176-7
Patterson MC, Vecchio D, Prady H, Abel L, Wraith JE (2007) Miglustat for treatment of Niemann-Pick C disease: a randomised controlled study. Lancet Neurol 6(9):765–772. https://doi.org/10.1016/S1474-4422(07)70194-1
Sitarska D, Tylki-Szymańska A, Ługowska A (2021) Treatment trials in Niemann-Pick type C disease. Metab Brain Dis. https://doi.org/10.1007/s11011-021-00842-0 (Epub ahead of print)
Seker Yilmaz B, Baruteau J, Rahim AA, Gissen P (2020) Clinical and molecular features of early infantile Niemann Pick type C disease. Int J Mol Sci 21(14):5059. https://doi.org/10.3390/ijms21145059
Funding
This study was supported by the Ministry of Education and Science of the Republic of Serbia (Grant #175090 to VK).
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Material preparation, data collection and analysis were performed by AT, IgP, ND, IvP, AM, MB, MJ, IN, MS, VSK. The first draft of the manuscript was written by NK, VD and MJL. All authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflicts of interest
Nikola Kresojević and Marina Svetel have received speaker honoraria from Actavis. Igor Petrović has received speaker honoraria from Actavis and Salveo. Vladimir S. Kostić has received speaker honoraria from Roche and Alkaloid and receives research supports from the Swiss Pharm and Serbian Ministry of Education, Science, and Development and Serbian Academy of Sciences and Art. Valerija Dobričić, Milica Ječmenica Lukić, Aleksandra Tomić, Nataša Dragašević, Ivana Perović, Ana Marjanović, Marija Branković, Milena Janković, Ivana Novaković: report no disclosures.
Ethical approval
This study was approved by the Local Ethics Committees on human studies and has, therefore, been performed in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments. All patients/caregivers provided written informed consent prior to study participation.
Rights and permissions
About this article

Cite this article
Kresojević, N., Dobričić, V., Lukić, M.J. et al. Genetic and phenotypic variability in adult patients with Niemann Pick type C from Serbia: single-center experience. J Neurol 269, 3167–3174 (2022). https://doi.org/10.1007/s00415-021-10918-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-021-10918-7