
Abstract
Sneddon syndrome is a rare disorder affecting small and medium-sized blood vessels that is characterized by the association of livedo reticularis and stroke. We performed whole-exome sequencing (WES) in 2 affected siblings of a consanguineous family with childhood-onset stroke and identified a homozygous nonsense mutation within the epidermal growth factor repeat (EGFr) 19 of NOTCH3, p.(Arg735Ter). WES of 6 additional cases with adult-onset stroke revealed 2 patients carrying heterozygous loss-of-function variants in putative NOTCH3 downstream genes, ANGPTL4, and PALLD. Our findings suggest that impaired NOTCH3 signaling is one underlying disease mechanism and that bi-allelic loss-of-function mutation in NOTCH3 is a cause of familial Sneddon syndrome with pediatric stroke.
Similar content being viewed by others
Introduction
Sneddon syndrome (SS) is a rare disorder (about 4 patients per million), affecting mainly young and predominately female adults [1,2]. It is characterized by recurrent strokes and livedo reticularis, a violaceous, netlike patterning of the skin [3]. Skin biopsies often display distinct histopathological findings consisting in sequential stage-specific changes in small to medium- sized arteries at the border between dermis and subcutis such as a possibly short-lived endotheliitis, followed by inflammatory obstruction, subendothelial cell proliferation and fibrosis of the occluded artery and shrinkage of the vessel [3,4,5]. Nevertheless, several cases with SS have also been described showing inconspicuous histopathological results [6]. Apart from cerebrovascular events as the prominent clinical manifestation, the range of associated pathologies varies from migraine and seizures to spontaneous abortion or cardiac and renal involvement [2]. The pathogenesis of SS is still unresolved and a matter of discussion [5]. An association with the occurrence of antiphospholipid antibodies and cofactors is described, although the reported frequencies show a vast range also including cases without any antibodies [7]. One explanatory model proposes that the presence of antiphospholipid antibodies might point towards a thrombotic process causing the disease whereas skin biopsies of antibody negative patients suggest a primary inflammatory process with migration and proliferation of smooth muscle cells leading to the narrowing and occlusion of the vessel [3,8]. Furthermore, it has been suggested that genetic factors also contribute to disease development [9,10]. In 2014, a compound heterozygous mutation in the adenosine deaminase 2 (ADA2) gene was identified in a large Portuguese family, who presented with livedo reticularis, stroke during early adulthood, leg ulcerations and intermittent fever [11]. More recently, a homozygous NOTCH3 nonsense mutation was identified in a patient who exhibited livedo reticularis from birth and childhood-onset cavitating leukoencephalopathy with multiple deep lacunar infarcts, disseminated microbleeds and two saccular aneurysms of middle cerebral arteries [12,13].
NOTCH3 encodes a transmembrane receptor predominantly expressed in mural cells of small blood vessels that plays a critical role in their integrity [14]. Dominant mutations in NOTCH3 cause CADASIL, a small vessel disease of the brain that manifests in mid-adulthood with leukoencephalopathy and subcortical ischemic events, progressively leading to disability, cognitive decline and premature death (MIM#125,310) [15]. In our study, we performed whole–exome sequencing (WES) in a consanguineous family with SS and 6 additional unrelated patients to analyze the genetic background of this disease.
Methods
Study participants
The diagnosis of SS was made based on the clinical criteria for SS, the occurrence of generalized livedo reticularis and the history of cerebrovascular events.
Sequence analyses
Whole exome data were generated from individuals III:2 and III:3 of family 1 and from the 6 other SS cases. Exomes were enriched with the SureSelect Human All Exon v6 kit (Agilent Technologies, Santa Clara, USA) and DNA libraries were sequenced on a HiSeq 4000 instrument (2 × 100 cycles, Illumina, San Diego, USA). The average exome coverages ranged from 115 × to 197 × and 100% of the NOTCH3 region was covered with at least 25 ×. Variants were filtered on the minor allele frequency (MAF < 0.001), which was estimated using the in-house database of the Helmholtzzentrum (> 20.000 exomes) and confirmed by the Genome Aggregation Database (gnomAD). NOTCH3, PALLD and ANGPTL4 sequence variants were confirmed by Sanger sequencing using standard protocols.
Differential gene expression of GSE58368 and GSE55203
To find out differentially expressed genes in Notch3 knock-out (KO) mouse models we analyzed 2 microarray datasets (GSE58368 and GSE55203) derived from the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/gds/). Using the GEO2R web tool(https://www.ncbi.nlm.nih.gov/geo/geo2r/) samples from the same cell type were analyzed comparing either heterozygous vs. homozygous (GSE58368) or homozygous vs. wildtype (GSE55203) mice.
Results
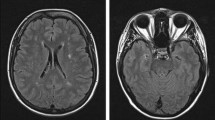
In the present study, we performed WES in 2 patients of a family with SS (Fig. 1a, subject III:2 and III:3). Clinical details of patient III:3 were previously reported [10]. Briefly, of 5 siblings 4 are affected with SS presenting livedo reticularis and a history of early onset stroke in childhood. Brain MRI from subjects III:2 and III:3 showed severe periventricular and subcortical white matter lesions and also multiple microbleeds predominantly in the white matter in sibling III:3 (Fig. 1b and Table 1). Laboratory results of sibling III:3 were negative for antiphospholipid antibodies and thrombophilia in general. No data were available on the antibody profile of the second sibling (III:2). The mother was reported to be healthy and the father, who died from a myocardial infarction at the age of 54, was reported to have had livedo reticularis but no signs of cerebrovascular disease (Fig. 1a). Exome-data analysis revealed in both siblings a large homozygous region of 9 Megabases on chr.19p13., indicating consanguinity. This region harbored only two homozygous variants shared by both siblings. First, a missense variant was detected in the KANK2 gene p.(Met278Lys), which is not present in any publicly available database. This variant was discarded since mutations in this gene have been associated with a distinct recessive disease characterized by a nephrotic syndrome and palmoplantar keratoderma with woolly hair (OMIM*614610). Second, a homozygous nonsense variant was identified in exon 14 of the NOTCH3 gene, p.(Arg735Ter) (Fig. 1b), which lies in the Epidermal Growth Factor repeat (EGFr) 19 of NOTCH3. This variant is predicted to result in a premature stop codon and elicit nonsense-mediated mRNA decay [16]. The variant is present heterozygously in 2 out of 61.000 individuals in the gnomAD database. (https://gnomad.broadinstitute.org/). The healthy mother was tested to be heterozygous for the p.(Arg735Ter) variant (Fig. 1c, II:3). No other family member was available for genetic testing either because they lived in another region of the country and were not able to transfer to the clinic for testing or were not interested in participating in the study. Based on the severity of the mutation and the known role of NOTCH3 in cerebrovascular disease, we believe that this mutation is most probably the cause of the disease in this family. To analyze whether NOTCH3 loss-of-function (lof) mutations are a more frequent cause for SS, we performed WES in 6 additional unrelated patients (Table 2). We found none of the patients to carry a rare variant (MAF < 0.01) in the NOTCH3 or in the ADA2 gene. Moreover, high coverage sequencing allowed us to also exclude copy number variations in both genes. We next hypothesized that genetic variations in genes involved in the NOTCH3 pathway might be plausible disease candidates. To explore this possibility, we made use of two microarray datasets of Notch3 KO mouse models from which RNA expression data of brain microvascular fragments (GSE55203) or brain-derived smooth muscle cells (GSE58368) were deposited. Using the GEO2R tool we searched for genes which were significantly downregulated in KO cells compared to wildtype or heterozygous cells (p value < 0.01), assuming that these genes are likely downstream in the NOTCH3 signaling pathway. We then intersected these 445 genes with the 85 genes carrying lof variants in our 6 patients. We found two patients with lof variants in putative NOTCH3 downstream genes. Patient 895 carried a heterozygous nonsense variant in the Palladin (PALLD) gene, p.(Arg287Ter) and patient 898 carried a heterozygous frameshift variant in the Angiopoietin-like 4 gene (ANGPTL4), p.(Gly313AlafsTer49). Both variants are present heterozygously in the gnomAD database (PALLD: 10/277212 alleles, ANGPTL4: 57/280350 alleles) (Fig. 2, Table 2).

Genealogical tree of the mutated family and representative brain MRI of patient III:3 a Pedigree of family 1: Unaffected family members are indicated by open symbols; affected members by closed symbols including livedo reticularis and cerebrovascular manifestations; half closed symbol (II:1) indicates partial phenotype of SS, with only livedo reticularis; diagonal bars through symbols denote deceased individuals. b Sanger-Sequence Pherograms showing the NOTCH3 variant in heterozygous form (II:3) and homozygous form (III:2, III:3). c Brain MRI from patient III:3 showing diffuse white matter hyperintensities on fluid-attenuated inversion recovery (FLAIR) images; lacunes on T1 and microbleeds on T2*-weighted images, lesions are depicted by an arrow

Gene expression profiles GSE58368 and GSE55203 of Palld and Angptl4. Datasets were downloaded from the Gene Expression Omnibus (GEO) database. Data derived from GSE58368, show Angptl4 gene expression in brain derived smooth muscle cells from from Notch3 heterozygote mice (Notch3 ±) compared to cells from Notch3 KO (Notch3 −/−) mice. Data derived from GSE55203 show Palld gene expression in brain microvascular fragments from Notch3 KO (Notch3 −/−) mice compared to those from wild-type (Notch3 + / +) mice
Discussion
Herein, we identified a homozygous nonsense mutation in the NOTCH3 gene in 2 affected siblings of a consanguineous SS family with pediatric stroke. Remarkably, a NOTCH3 null mutation was identified in another unrelated patient with similar clinical and MRI features, as well as childhood-onset, originally diagnosed with SS [12,13]. These findings, added to the fact that NOTCH3 plays a key role in small brain vessels, strongly suggests that loss of NOTCH3 signaling is one underlying disease mechanism for SS.
Although these three patients with SS and a NOTCH3 null mutation exhibit clinical and neuroimaging features that share similarities with those observed in CADASIL patients, we believe that this genetic form of SS and CADASIL are two distinct entities. First, stroke events started in these 3 SS patients in childhood whereas they occur in CADASIL patients in adulthood, at a mean age of 49 years [15]. Second, livedo reticularis is absent in CADASIL [15]. Third, CADASIL mutations are dominantly inherited and characteristically lead to the loss or gain of a cysteine residue in one of the EGFr of the extracellular domain of NOTCH3 [15]. Fourth, accumulating evidence indicates that CADASIL is not caused by a loss of NOTCH3 function [17,18,19], but by a neomorphic effect related to the abnormal vascular accumulation of NOTCH3 protein and possibly an increased activity of the mutant receptor [20,21]. Interestingly, patients with NOTCH3 lof mutations like our patient (III:2) and the previous described case [12,13] seem to differ from other SS patients. In addition to the childhood-onset and the more severe disease course, NOTCH3 lof mutation carriers showed negative serum antibody profiles and no extraneurological manifestations. Thus, one might hypothesize that NOTCH3 lof mutations lead to a distinct and probably more severe clinical subtype of SS. In an attempt to find other possible contributing genes in SS patients with adult-onset stroke, we searched for lof variants in genes downstream to NOTCH3. Hereby, we found 2 patients carrying heterozygous lof variants in the PALLD and ANGPTL4 genes. Interestingly, both genes show connections to vascular biology and stroke. PALLD, which is predominantly expressed in arterial smooth muscle cells in the brain (https://betsholtzlab.org/VascularSingleCells/database.html), was shown to be involved in the modulation of the actin cytoskeleton and plays a role in vascular remodeling [22,23]. In addition, PALLD gene polymorphisms were found to be associated with stroke [24]. ANGPTL4 was found to be involved in angiogenesis and vessel sprouting in a rat stroke model and was also shown to have a vasculoprotective effect in a mouse stroke model [25,26]. However, both variants PALLD-Arg287Ter and ANGPTL4- Gly313AlafsTer49 are, although rare, present in the general population with 1 in ~ 14,000 and 1 in ~ 2500 respectively and thus unlikely to cause SS on their own. Therefore the presented candidate genes must only be regarded as suggestions for follow up studies and caution in interpretation is advised. Alternatively, we suggest that they could increase disease susceptibility, possibly in combination with other genetic and/or environmental factors. However, one limitation of our study is that unfortunately other family members of patients 895 and 898 were lost to follow up and segregation of the variants could therefore not be investigated.
In conclusion, we propose NOTCH3 null mutations as a genetic cause for SS with childhood-onset stroke. We further suggest that impairment of NOTCH3 signaling may also contribute to SS pathogenesis in general.
Availability of data and material
All raw data and a complete list of all rare sequence variants (MAF < 0.001) generated by WES are available upon request.
References
Bottin L, Frances C, de Zuttere D, Boelle PY, Muresan IP, Alamowitch S (2015) Strokes in Sneddon syndrome without antiphospholipid antibodies. Ann Neurol 77(5):817–829. https://doi.org/10.1002/ana.24382
Bersano A, Morbin M, Ciceri E, Bedini G, Berlit P, Herold M, Saccucci S, Fugnanesi V, Nordmeyer H, Farago G, Savoiardo M, Taroni F, Carriero M, Boncoraglio Giorgio B, Perucca L, Caputi L, Parati Eugenio A, Kraemer M (2016) The diagnostic challenge of Divry van Bogaert and Sneddon Syndrome: Report of three cases and literature review. J Neurol Sci 364:77–83. https://doi.org/10.1016/j.jns.2016.03.011
Zelger B, Sepp N, Schmid KW, Hintner H, Klein G, Fritsch PO (1992) Life history of cutaneous vascular lesions in Sneddon's syndrome. Hum Pathol 23(6):668–675. https://doi.org/10.1016/0046-8177(92)90323-u
Stockhammer G, Felber SR, Zelger B, Sepp N, Birbamer GG, Fritsch PO, Aichner FT (1993) Sneddon's syndrome: diagnosis by skin biopsy and MRI in 17 patients. Stroke 24(5):685–690. https://doi.org/10.1161/01.str.24.5.685
Samanta D, Cobb S, Arya K (2019) Sneddon syndrome: a comprehensive overview. J Stroke Cerebrovasc Dis 28(8):2098–2108. https://doi.org/10.1016/j.jstrokecerebrovasdis.2019.05.013
Legierse CM, Canninga-Van Dijk MR, Bruijnzeel-Koomen CA, Kuck-Koot VC (2008) Sneddon syndrome and the diagnostic value of skin biopsies—three young patients with intracerebral lesions and livedo racemosa. Eur J Dermatol 18(3):322–328. https://doi.org/10.1684/ejd.2008.0397
Caldas CA, de Carvalho JF (2011) Primary antiphospholipid syndrome with and without Sneddon's syndrome. Rheumatol Int 31(2):197–200. https://doi.org/10.1007/s00296-009-1310-z
Frances C, Papo T, Wechsler B, Laporte JL, Biousse V, Piette JC (1999) Sneddon syndrome with or without antiphospholipid antibodies. A comparative study in 46 patients. Medicine (Baltimore) 78(4):209–219. https://doi.org/10.1097/00005792-199907000-00001
Szmyrka-Kaczmarek M, Daikeler T, Benz D, Koetter I (2005) Familial inflammatory Sneddon's syndrome-case report and review of the literature. Clin Rheumatol 24(1):79–82. https://doi.org/10.1007/s10067-004-0981-9
Llufriu S, Cervera A, Capurro S, Chamorro A (2008) Neurological picture. Familial Sneddon's syndrome with microbleeds in MRI. J Neurol Neurosurg Psychiatry 79(8):962. https://doi.org/10.1136/jnnp.2007.131912
Bras J, Guerreiro R, Santo GC (2014) Mutant ADA2 in vasculopathies. The New England journal of medicine 371(5):478–480. https://doi.org/10.1056/NEJMc1405506
Pippucci T, Maresca A, Magini P, Cenacchi G, Donadio V, Palombo F, Papa V, Incensi A, Gasparre G, Valentino ML, Preziuso C, Pisano A, Ragno M, Liguori R, Giordano C, Tonon C, Lodi R, Parmeggiani A, Carelli V, Seri M (2015) Homozygous NOTCH3 null mutation and impaired NOTCH3 signaling in recessive early-onset arteriopathy and cavitating leukoencephalopathy. EMBO Mol Med 7(6):848–858. https://doi.org/10.15252/emmm.201404399
Parmeggiani A, Posar A, De Giorgi LB, Sangiorgi S, Mochi M, Monari L, Patrizi A, Rossi PG (2000) Sneddon syndrome, arylsulfatase A pseudodeficiency and impairment of cerebral white matter. Brain Dev 22(6):390–393
Domenga V, Fardoux P, Lacombe P, Monet M, Maciazek J, Krebs LT, Klonjkowski B, Berrou E, Mericskay M, Li Z, Tournier-Lasserve E, Gridley T, Joutel A (2004) Notch3 is required for arterial identity and maturation of vascular smooth muscle cells. Genes Dev 18(22):2730–2735. https://doi.org/10.1101/gad.308904
Chabriat H, Joutel A, Dichgans M, Tournier-Lasserve E, Bousser MG (2009) Cadasil. The Lancet Neurology 8(7):643–653. https://doi.org/10.1016/S1474-4422(09)70127-9
Maquat LE (2004) Nonsense-mediated mRNA decay: splicing, translation and mRNP dynamics. Nat Rev Mol Cell Biol 5(2):89–99. https://doi.org/10.1038/nrm1310
Cognat E, Baron-Menguy C, Domenga-Denier V, Cleophax S, Fouillade C, Monet-Lepretre M, Dewerchin M, Joutel A (2014) Archetypal Arg169Cys mutation in NOTCH3 does not drive the pathogenesis in cerebral autosomal dominant arteriopathy with subcortical infarcts and leucoencephalopathy via a loss-of-function mechanism. Stroke 45(3):842–849. https://doi.org/10.1161/STROKEAHA.113.003339
Joutel A (2013) Loss-of-function mutation in the NOTCH3 gene: simply a polymorphism? Hum Mutat 34(11):v. https://doi.org/10.1002/humu.22198
Rutten JW, Boon EM, Liem MK, Dauwerse JG, Pont MJ, Vollebregt E, Maat-Kievit AJ, Ginjaar HB, Lakeman P, van Duinen SG, Terwindt GM, Lesnik Oberstein SA (2013) Hypomorphic NOTCH3 alleles do not cause CADASIL in humans. Hum Mutat 34(11):1486–1489. https://doi.org/10.1002/humu.22432
Baron-Menguy C, Domenga-Denier V, Ghezali L, Faraci FM, Joutel A (2017) Increased Notch3 activity mediates pathological changes in structure of cerebral arteries. Hypertension 69(1):60–70. https://doi.org/10.1161/HYPERTENSIONAHA.116.08015
Neves KB, Harvey AP, Moreton F, Montezano AC, Rios FJ, Alves-Lopes R, Nguyen Dinh Cat A, Rocchicciolli P, Delles C, Joutel A, Muir K, Touyz RM (2019) ER stress and Rho kinase activation underlie the vasculopathy of CADASIL. JCI Insight. https://doi.org/10.1172/jci.insight.131344
Liu SQ (1999) Biomechanical basis of vascular tissue engineering. Crit Rev Biomed Eng 27(1–2):75–148
Vanlandewijck M, He L, Mae MA, Andrae J, Ando K, Del Gaudio F, Nahar K, Lebouvier T, Lavina B, Gouveia L, Sun Y, Raschperger E, Rasanen M, Zarb Y, Mochizuki N, Keller A, Lendahl U, Betsholtz C (2018) A molecular atlas of cell types and zonation in the brain vasculature. Nature 554(7693):475–480. https://doi.org/10.1038/nature25739
Morrison AC, Bare LA, Luke MM, Pankow JS, Mosley TH, Devlin JJ, Willerson JT, Boerwinkle E (2008) Single nucleotide polymorphisms associated with coronary heart disease predict incident ischemic stroke in the atherosclerosis risk in communities study. Cerebrovasc Dis 26(4):420–424. https://doi.org/10.1159/000155637
Buga AM, Margaritescu C, Scholz CJ, Radu E, Zelenak C, Popa-Wagner A (2014) Transcriptomics of post-stroke angiogenesis in the aged brain. Front Aging Neurosci 6:44. https://doi.org/10.3389/fnagi.2014.00044
Bouleti C, Mathivet T, Coqueran B, Serfaty JM, Lesage M, Berland E, Ardidie-Robouant C, Kauffenstein G, Henrion D, Lapergue B, Mazighi M, Duyckaerts C, Thurston G, Valenzuela DM, Murphy AJ, Yancopoulos GD, Monnot C, Margaill I, Germain S (2013) Protective effects of angiopoietin-like 4 on cerebrovascular and functional damages in ischaemic stroke. Eur Heart J 34(47):3657–3668. https://doi.org/10.1093/eurheartj/eht153
Funding
Open access funding provided by Medical University of Vienna.
Author information
Authors and Affiliations
Contributions
EKG and AZ contributed to conception and design of the study. All authors contributed to material preparation, data collection and patient recruitment. EKG, WM, SL, SG, TMS, AJ and AZ analyzed the data. EKG, AJ and AZ wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflicts of interest
Nothing to report.
Ethical approval
The study-protocol was approved by the local ethics committee (EK1129/2010).
Informed consent
All patients gave their written informed consent for genetic analyses.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Greisenegger, E.K., Llufriu, S., Chamorro, A. et al. A NOTCH3 homozygous nonsense mutation in familial Sneddon syndrome with pediatric stroke. J Neurol 268, 810–816 (2021). https://doi.org/10.1007/s00415-020-10081-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-020-10081-5