
Abstract
In recent years, numerous clinical trials for disease modification in Parkinson’s disease (PD) have failed, possibly because of a “one-size-fits all” approach. Alternatively, a precision medicine approach, which customises treatments based on patients’ individual genotype, may help reach disease modification. Here, we review clinical trials that target genetic forms of PD, i.e., GBA-associated and LRRK2-associated PD. In summary, six ongoing studies which explicitely recruit GBA-PD patients, and two studies which recruit LRRK2-PD patients, were identified. Available data on mechanisms of action, study design, and challenges of therapeutic trials are discussed.
Similar content being viewed by others
Parkinson’s disease (PD) is the second most common neurodegenerative disorder, affecting more than 6 million people worldwide [1]. Numerous drugs for the treatment of PD are avilable on the market. While drugs targeting the dopaminergic pathway treat motor symptoms, there is no evidence that they modify disease progression. This “one-size-fits all” approach may very well explain why clinical trials for disease modification in PD have failed. Treatments that target the underlying pathophysiology are required. Since the pathophysiology of PD may be different in different patients, studies should be designed that assess PD treatment on a more individual basis. Therefore, a precision medicine approach in PD is very timely.
Precision medicine—a conceptual framework
Precision medicine—also referred to as personalized medicine or individualized medicine—aims to tailor the specific treatment for the right person at the right time. To achieve this, it uses diagnostic tools to identify specific biomarkers, often genetic, to help assess which medical treatments will be best for each patient [2]. By combining data from these tests with a patient’s medical history and important factors influencing health status, targeted prevention and treatment plans can hopefully be developed in the future [2]. Thus, precision medicine is not a new concept. It is a conceptual framework which became a hot topic beyond the medical sphere when President Obama announced a research initiative that aims to accelerate progress toward a new era of precision medicine in 2015 [3]. There are various benefits of precision medicine, including the detection of disease onset at the earliest moment and thereby shifting the emphasis in medicine from reaction to prevention (Table 1).
In the context of neurodegenerative disorders, at the time of clinical manifestation (and certainly at the time of diagnosis), a substantial number of neuros has been permanently lost. Hence, early detection of at-risk individuals will be instrumental for early treatments (which will ideally protect the cells from neuronal death). Sucessful precision medicine will thus move from current reactive approaches to early detection, protection and prevention. Early detection of individuals at-risk to develop neurodegenerative disorders is a major challenge. However, in the case of genetic subforms, early detection is feasible by confirming their genetic status with a minimal-invasive test.
The genetic architecture of Parkinson’s disease
In the past decade, we have seen incredible progress in elucidating the genetic architecture of PD: over a dozen Mendelian loci are known to cause familial PD. In addition, multiple loci have been identified by genome-wide association studies (GWAS) that are mostly associated with a small increase in risk of PD. These latter genetic variations of weak effect strength may occur as commonly as 40% in the general population, but convey only a mildly (up to ~ 1.5-fold) increased disease risk [4]. Even when combining all risk factors, the odds ratio is only 3–4 (i.e. there is a 3 to 4 times increased risk of developing the disease) [5]. Notably, alteration in the same gene may lead to different variants and mutations with differrent risk association with PD [6]. For example, some point mutations in LRRK2 are causative for PD, while coding polymorphisms in the gene are strong risk factors and additional higher frequency variants at the LRRK2 locus contribute to a small increase in risk of developing PD [7].
A recent meta-analysis suggested that the detectible heritable component of PD (based on genome-wide SNPs and less significant SNPs included in a polygenic risk score) is around 20% [8]. There is compelling evidence of yet-to-be-discovered additional genetic factors that contribute to the etiology of PD. In addition, environmental risk factors are yet to be discovered.
This tremendous progress in understanding the genetic architecture has set the ground for the development of treatments based on disease mechanism rather than symptoms. Here, we will review clinical trials which target genetic forms of PD, i.e., explicitly recruit (or enrich for) patients with a genetic form of PD.
Parkinsonism associated with GBA mutations
Homozygous mutations in the glucocerebrosidase (GBA) gene cause Gaucher disease (GD), the most common autosomal recessive lysosomal storage disease, with an estimated annual incidence of 1/60,000 and an estimated carrier frequency [9] of 0.7–0.8% in the general population. Some ethnicities show higher mutation rates; specifically, in the Ashkenazi Jewish (AJ) population, there is an annual incidence is 1/1,000 and carrier frequencies as high as 6% in all AJ.
The clinical presentation of GD can be divided into systemic, which are present in all forms of GD, and include hepatosplenomegaly, painful skeletal disorders and pancytopenia, and neurological manifestations, which are present in the more severe types of GD, GD-II and GD-III. Both GD patients and healthy heterozygous carriers are at increased risk for PD [10] and longterm follow-up showed worsening in motor and non-motor prodromal PD features [11]. GBA mutations are a common risk for PD and are present in 7–10% of PD patients worldwide. Among Ashkenazi Jews, around 20% of PD patients carry a GBA mutation [12]. High prevalences have also been reported in the Netherlands, where 15% of PD patients carry a GBA mutation (oral communication, Dana Hilt). The lowest carrier frequency was reported to be 2.3% in Norwegian Parkinson's disease patients [13]. Notably, there is considerable reduction of penetrance in that only about 10% of GBA carriers will develop PD (which is however considerably higher compared to the global PD prevalence of 1–2% of the general population aged 65 years or older) and studies suggest that penetrance is age-dependent [14].
Clinically, GBA heterozygotes may be indistinguishable from iPD. However, they may have an earlier age at onset, more prevalent cognitive impairment and may not respond to levodopa as well as iPD patients [15, 16]. GBA mutations are also associated with other alpha-synucleinopathies, including DLB [17] (pathologically confirmed) and in some, but not all studies, with MSA [18,19,20,21,22]. In contrast, there was no association with essential tremor or AD (Alzheimer’s disease).
More than 300 mutations in GBA have been reported [23], some with milder (e.g., the N370S mutation), others with more severe (e.g., the L444P mutation) biological consequences and clinical presentations (e.g., age at onset or progression rate). The encoded protein, glucocerebrosidase, is a lysosomal enzyme which plays a role in the breakdown of glucocerebroside into glucose and ceramide. In GD, there is lysosomal build-up of the substrate glucocerebroside in the reticulo-endothelial system with reduced clearance capacities.
Pathologically, the brains of patients with heterozygous GBA mutations strongly resemble those from patients with iPD. However, there is also widespread cortical Lewy body involvement in GBA mutation carriers [16, 22]. A few studies showed a reciprocal relationship between levels of glucocerebrosidase (Gcase; the enzyme encoded by GBA) and levels of the aggregate-prone protein alpha-synuclein [24]. Notably, iPD patients also have reduced GCase activity (about 33% decrease) in the substantia nigra and cerebellum, making treatments that target GBA relevant for iPD and patients with PD dementia as well [21].
While the PD field can benefit from decades of research done for GD, the underlying mechanisms of how exactly GBA leads to the development of PD are not fully understood. One of the hypotheses is that there is a self-propagating bidirectional feedback loop between GCase and a-synuclein. On the one hand, loss of GCase activity causes a-synuclein accumulation and oligomerization, resulting in neurotoxicity through aggregate-dependent mechanisms [25]. Furthermore, elevated a-synuclein inhibits lysosomal maturation and normal GCase activity. a-synuclein hinders GCase transport from the endoplasmic reticulum to the lysosome. This continues over time until the threshold for neurodegeneration is reached [25].
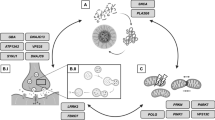
Based on this, targeted treatments can take different approaches including modulation of gylcosphingolipid turnover and restoration of enzyme function (Table 2; Fig. 1).

Treatment approaches for GBA-associated PD include modulation of gylcosphingolipid turnover and restoration of enzyme function
Treatment directed at modulation of gylcosphingolipid turnover
Substrate reduction therapy inihibits the biosynthesis of lipid subtrates and thereby prevents their accumulation. While this approach does not target the mutant gene or dysfunctional enzyme itself, it is an effective FDA approved treatment of the systemic symptoms of Gaucher disease. However, the approved inhibitors show no effective CNS concentration and do not affect the neurological symptoms of Gaucher disease (i.e., Gaucher type II and III). However, new glucosylceramide synthase inhibitors, show good brain penetration and improved a-synuclein processing and behavioural outcomes in mouse models [26, 27]. Based on these findings, Sanofi launched the MOVES-PD study, a randomized, double-blind, placebo-controlled trial, to assess the efficacy and safety of the glycosylsynthase inhibitor Venglustat (GZ/SAR402671). Initial results from a phase I study were recently published [28]. Briefly, 17 GBA-PD were enrolled (13 on venglustat, 4 on placebo; mean age 58 years, mean disease duration 7 years) into this 36-week randomized placebo-controlled double-blind sequential cohort study of once daily venglustat at three escalating doses. No serious adverse events occurred. Side effects included psychological, neurological and gastrointestinal-related symptoms. Plasma and cerebrospinal fluid (CSF) glucosylceramide levels decreased in a dose-dependent manner (up to 75%). This favorable safety and tolerability profile of venglustat at all doses led the company to advance to a phase II, a 52-week trial which is currently ongoing [28].
Treatment directed at restoration of enzyme function
Other therapies focus on restoration of enzyme function, thus increasing glucocerebrosidase activity, especially in the brain. This can be achieved by (1) enzyme-replacement therapy (ERT) with recombinant glucocerebrosidase. This treatment is available for patients with Gaucher disease. However, as in the case of the currently approved substrate reduction therapies, ERT does not cross the blood–brain barrier and does cannot affect the neurological symptoms found in GD Type II and III. Of note, there are no data on the use of ERT in PD.
Another approach would be (2) gene therapy using adeno-associated virus vectors to deliver engineered DNA to human cells [27, 29]. As for GBA, preclinical studies in mice demonstrated that adeno-associated virus-mediated expression of glucocerebrosidase corrected the aberrant accumulation of the toxic lipid glucosylsphingosine and reduced the levels of ubiquitin, tau, and alpha-synuclein aggregates [30]. Prevail Therapeutics, a new company launched in 2017, aims to start clinical trials with PR001 in 16 GBA-PD patients in late 2019 [31]. The company will also test the compound in children with neuropathic Gaucher disease starting in the first half of 2020.
Furthermore, (3) small molecules have attracted attention [32]. Early glucocerebrosidase chaperones that underwent clinical trials for Gaucher disease included isofagomine (afegostat‐tartrate, AT2101). This treatment did not lead to significant clinical improvement, and further clinical development for this indication was discontinued [27].
Ambroxol, which is a promising small molecule chaperone widely used in Europe as a mucolytic agent, may potentially facilitate the transit of the misfolded GCase protein to the lysosome [33]. Ambroxol has been shown to improve lysosomal function and increase enzyme activity in in-vitro studies utilizing dermal fibroblasts with GBA1 mutations [34] as well as in studies performed on non-human primates (i.e., cynomolgus monkeys) with GBA1 mutations [35]. The effects of ambroxol at high doses are currently being studied in the AiM-PD study, sponsored by UCL and the Cure Parkinson’s Trust, UK [36]. Twenty PD patients (10 GBA-positive & 10 GBA-negative status) are treated with up to 420 mg/day (which is considerably higher compared to 30–120 mg used for the treatment of respiratory disease) in order to evaluate the safety, tolerability and pharmacodynamics of ambroxol at five escalating doses. Outcome measures include clinical assessments of motor and cognitive function as well as blood and CSF biomarkers. As mentioned above, GCase activity is also reduced in iPD patients’ brains (SN) [37], making the therapy potentially relevant for iPD. The effect of ambroxol in non-GBA-PD [36] and non-GBA-PD dementia [38] will be better understood once results from the two ongoing studies become available which include ten non-GBA-PD and 75 PDD patients [38, 39] (treated at a daily doses of 420 mg / day or 525–1050 mg/day, respectively).
The effects of the activator of the GCase enzyme LTI-291 were studied in a one-month phase 1b trial conducted in the Netherlands, where the rate of GBA mutations are reported to be around 15%. Around 40 GBA-PD patients participated. There were no safety events and data showed a good dose-dependent brain penetration (personal communication). The company, Lysosomal Therapeutics, Inc. (LTI), is a Massachusetts-based biotech venture, which plans to develop therapies for Gaucher disease and other lysosome-based neurodegenerative diseases.
Small molecules have also targeted PD by modifying GBA-independent pathways. One such example is RTB101, which is an inhibitor of target of rapamycin complex 1 (TORC1) [40]. Rapamycin, which is known for its immunosuppressant properties, prolongs lifespan by 15–25% in various non-mammalian organisms, even when given late in life; it has also been found to increase health-span. A five year-study in dogs is planned to test geroprotective effects of RTB101 in mammals [41]. Rapamycin reached public attention when Bloomberg magazine publicized it as the potential “forever pill” on its cover in 2015, which reflects the great desire of rejuvenation. TORC1 plays a role in cell growth, aging and is the switch between fasting and feeding states [40]. Mutations in TORC1 cause focal cortical dysplasia, an established cause of epilepsy. The role of mTORC1 in regulating autophagy may also have implications for neurodegenerative diseases. In preclinical models, TORC1 inhibition induces autophagy and prevents neuronal loss [41, 42]. It improves motor function in multiple PD models including a-synuclein transgenic mice and MPTP models of PD [43]. In oncology cell cultures, treatment with RTB101 reduced the levels of glucosylceramide, the main substrate of GCase. A current phase 1b/2a trial of RTB101 in combination with sirolimus involved 45 PD patients with or without GBA mutation. The study was initiated in early 2019; data are expected in 2020.
LRRK2-associated Parkinsonism: kinase inhibitors are a promosing target
LRRK2 mutations are the most common cause of autosomal dominant PD accounting for 5–15% of dominant familial PD and 1–3% of sporadic PD (Fig. 2). The International LRRK2 Consortium study estimated that the most common mutation in LRRK2, G2019S, alone accounts for 1% of sporadic and 4% of familial PD patients [44].

World map of LRRK2-associated Parkinsonism. 533 cases have been reported. Circles reflect frequency per region. Data and image were retrieved from the MDSGene Website [47]
Similar to GBA, mutations in LRRK2 are more common in certain ethnicities. North African Arabs (mutation-positive: 36% in familial, 39% in sporadic PD) and Ashkenazi Jews (28% in familial, 10% in sporadic PD) have the highest frequencies. As in GBA, different mutations and variants confer different levels of risk for PD. For example, the G2019S mutation, which is common in Whites, confers a higher risk for PD than the common Asian variants, G2385R and R1628P [11, 13]. These latter two variants are detected in around 5–10% of Asian PD patients [45]. The G2385R variant is associated with an odds ratio of 2.24. Penetrance in LRRK2 is age-dependent and estimations in the general population are widely variable, ranging between 30 and 74% [46, 47].
Additionally, non-coding polymorphisms close to the LRRK2 locus act as risk factors for sporadic disease [48] Furthermore, LRRK2 interacts with the protein products of at least two GWAS hits, RAB7L1 and GAK, linking PD-related genes with monogenic and complex forms [49, 50].
LRRK2 is a large gene with 51 exons, spanning a genomic region of 144 kb. It contains five functional domains including a leucine rich repeat domain. More than 80 missense mutations have been reported, but only around one dozen are pathogenic [48]. The mechanisms by which mutations cause PD have not been completely disentangled yet, but there is increasing evidence of increased LRRK2 kinase function in PD. The G2019S mutation, for example, results in a direct two-to-threefold increase in kinase activity [51, 52]. Others studies have focused on the GTP domain which may also play an important role. Loss of function, on the other hand, i.e., by haploinsufficiency of LRRK1 or LRRK2, appears to be neither a cause of nor protective against PD [53].
The potential gain-of-function effect would be an attractive target for treatment because inhibition is easier to achieve than improvement of reduced protein activity (as in GBA). Furthermore, kinase inhibitors are widely used in oncology, and the PD field can benefit from such achievements in other fields. Since the first generation of LRRK2 inhibitors, newer compounds have progressively improved in potency, selectivity, and brain penetrance. However, efficacy and safety remain a concern. That is because other tissues, particularly the kidney, lung, and a subtype of peripheral immune cells, robustly express LRRK2. For example, the kidney has a ~ 6.2-fold higher expression of LRRK2 compared to the brain [52]. This is a potential source of peripheral side effects, which can include abnormal accumulations, a-syn aggregations, and impaired autophagy-lysosomal function induced by LRRK2 inhibitors [53,54,55]. More recent data, however, suggest that compounds that only partially downregulate LRRK2 levels or kinase activity, i.e., by 50% or less, are unlikely to produce major side effects related to on-target safety [56] and lipid droplets in lamelar bodies are absored after the drug is withdrawn. One alternative to avoid systemic toxicity is to find a way to specifically modify LRRK2 activity in the brain without modifying activity peripherally. Several strucutrally different LRRK2 inhibitors from Genentech, GSK, Merck and Pfizer are in the pipeline (Table 3) [27]. The compound developed by Denali is already in clinical trial. A phase 1b trial in healthy individuals has been completed, which included pulmonary and renal safety parameters. The company is advancing DNL201 (GNE-7915) to a Phase 1b safety and biomarker study in LRRK2-linked PD and iPD. 30 mild to moderately affected PD patients with or without LRRK2 mutation will be randomized to low or high dose DNL201 or placebo in this 28-day randomized placebo-controlled trial. The first patient was reported in December 2018; data readout is expected for the end of 2019. To facilitate recruitment, a “direct-to-consumer” approach for testing and counselling will be available [57]–a strategy that proved successful in genetic testing with the PPMI initiative. Most recently, Denali has announced a strategic collaboration with a gene diagnostic lab, Centogene, in order to globally identify and recruit LRRK2 mutation carriers, further characterize this genetic subtype, and build a source for patient recruitment for future studies [58]. However, such strategies of a commercial diagnostic lab in concert with a drug company to offer directed to consumer diagnostic testing is viewed critically by some.
Finally, Biogen is currently recruiting LRRK2 patients into one arm of a phase 1 trial. These patients will receive a single intrathecal injection of the compound BIIB094, an antisense oligomere (ASO), on multiple days. Recently, ASOs have produced a lot of interst in a variety of disorders, including spinal muscular atrophy, Huntington’s disease or non-neurological disorders such as cancers [59, 60]. ASOs reduce the expression of a mutated gene by binding to target mRNAs and blocking the translation of the abnormal protein or inducing its degradation [60]. ASOs can also promote splicing around mutations. For disorders due to toxic gain-of-function such as LRRK2, further investigation regarding ASOs is warranted. In a preclinical study, administration of LRRK2 ASOs to the brains of mice reduced LRRK2 protein levels and fibril-induced α-syn inclusions [61]; data from humans are not yet available.
Interestingly, most recent studies found an mechanistic and therapeutic convergence of LRRK2 and GCase with reduced GCase activity in dopaminergic neurons derived from PD patients with LRRK2 mutations and increased GCase activity induced by inhibition of LRRK2 kinase activity [62]. Rab10 was identified as a key mediator of LRRK2 regulation of GCase activity and may be an interesting target for future studies [62].
Final remarks
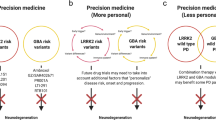
Recent failures in large Phase III clinical trials for PD suggest that disease modification would be difficult to achieve as long as we treat PD as one disease one pathophysiology. Therefore, we believe that precision medicine in PD may be a promising alternative.
As summarized in this review, several gene-targeted therapies are being tested in clinical trials and numerous more are in the pipeline. These are exciting times. However, the process of bringing a drug into the clinic is cumbersome [63]. Pharmaceutical Research and Manufacturers of America (PhRMA) estaimate that for every 5000–10,000 compounds screened, only 250 enter preclinical testing, five enter human clinical trials, and one is approved by the Food and Drug Administration, with only two in ten drugs generating enough revenue to recoup their research and development costs [64]. Thus, set-backs will not come unexpected.
In addition to the hurdles of all clinical trials, precision medicine trials may be more complicated. It is unclear who may benefit from precision medicine drugs. Would these be useful only for mutation carriers (and therefore require an orphan drug assignment) or would they be beneficial for the larger group of idiopathic PD or atypical parkinsonian disorders? It seems unlikely that all these patients will respond to the same drug. Indeed, even within the group of GBA mutation carriers, one may have to differentiate due to the effect that a specific mutation has on the protein. For example, the affinity of chaperones to a mutated enzyme may be different depending on the mutation. Furthermore, a new drug that facilitates protein function may fail in patients with null mutations who do not express any protein. The term “superprecision medicine” has been used to capture this phenomenon.
There are challenges that remain that need to be overcome when planning or conducting a clinical trial (Table 4) [65]. A major challenge will be to recruit a large enough number of study participants. Genotyping significantly larger proportions of PD patients would be required. Different strategies have been developed for this purpose, including the Parkinson’s Foundation effort called PD GENE and the “direct-to-consumer” approach, which may allow identification of eligible individuals even if they do not live close to a movement disorders unit. Raising awareness and educating the community, including physicians, patients and caregivers, will be an important step to reach critical numbers. We are hopeful that the treatment for PD will drastically change in the next decade and evolve beyond dopaminergic or surgical treatments.
Another challenge is the relative lack of biomarkers that reliably reflect disease progression and response to treatment. This applies to genetic subtypes as well as the larger group of iPD in general. Concerted efforts are being made to identify a biospecimen-based (i.e., blood, urine, CSF or biopsy), imaging, or other (e.g., electrophyiological) biomarker of PD or disease progression. Among these, the PPMI initiative is a valuable source that brings together longitudinal data and specimen collection from more than 1200 volunteers with PD [66].
Advancing precision medicine will further encourage and support the next generation of scientists to develop creative new approaches for detecting, measuring, and analyzing a wide range of biomedical information—including molecular, genomic, cellular, clinical, behavioral, physiological, and environmental parameters [3].
References
Dorsey ER, Elbaz A, Nichols E, Abd-Allah F, Abdelalim A, Adsuar JC et al (2018) Global, regional, and national burden of Parkinson’s disease, a systematic analysis for the global burden of disease study. Lancet Neurol 17:939–953
Abrahams E, President | personalized medicine coalition personalized medicine: The changing landscape of health care; key note lecture, The 2nd biomarker meeting in personalized reproductive medicine valencia, Spain [http://www.comtecmed.com/biomarker/2014/Uploads/Editor/PDF/ppt/Edward%20Abrahams_Key%20Note%20Lecture.pdf]. Accessed 16 Jan 2020
Collins FS, Varmus H (2015) A new initiative on precision medicine. N Engl J Med 372:793–795
Gasser T (2015) Usefulness of genetic testing in PD and PD trials: a balanced review. J Park Dis 5:209–215
Nalls MA, Pankratz N, Lill CM et al (2014) Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson's disease. Nat Genet 46:989–993
van der Brug MP, Singleton A, Gasser T, Lewis PA (2015) Parkinson’s disease: from human genetics to clinical trials. Sci Transl Med 7:205ps220
Paisan-Ruiz C, Lewis PA, Singleton AB (2013) LRRK2: cause, risk, and mechanism. J Parkinsons Dis 3:85–103
Nalls MA, Blauwendraat C, Vallerga CL et al (2019) Identification of novel risk loci, causal insights, and heritable risk for Parkinson's disease: a meta-analysis of genome-wide association studies. Lancet Neurol 18:1091–1102
Zuckerman S, Lahad A, Shmueli A et al (2007) Carrier screening for Gaucher disease: lessons for low-penetrance, treatable diseases. JAMA 298:1281–1290
Sidransky E, Nalls MA, Aasly JO et al (2009) Multicenter analysis of glucocerebrosidase mutations in Parkinson's disease. N Engl J Med 361:1651–1661
Mullin S, Beavan M, Bestwick J et al (2019) Evolution and clustering of prodromal parkinsonian features in GBA1 carriers. Mov Disord 34:1365–1373
Gan-Or Z, Giladi N, Rozovski U et al (2008) Genotype-phenotype correlations between GBA mutations and Parkinson disease risk and onset. Neurology 70:2277–2283
Toft M, Pielsticker L, Ross OA, Aasly JO, Farrer MJ (2006) Glucocerebrosidase gene mutations and Parkinson disease in the Norwegian population. Neurology 66:415–417
Rana HQ, Balwani M, Bier L, Alcalay RN (2013) Age-specific Parkinson disease risk in GBA mutation carriers: information for genetic counseling. Genet Med 15:146–149
Clark LN, Ross BM, Wang Y et al (2007) Mutations in the glucocerebrosidase gene are associated with early-onset Parkinson disease. Neurology 69:1270–1277
Clark LN, Kartsaklis LA, Wolf Gilbert R et al (2009) Association of glucocerebrosidase mutations with dementia with lewy bodies. Arch Neurol 66:578–583
Geiger JT, Ding J, Crain B et al (2016) Next-generation sequencing reveals substantial genetic contribution to dementia with Lewy bodies. Neurobiol Dis 94:55–62
Mitsui J, Matsukawa T, Sasaki H et al (2015) Variants associated with Gaucher disease in multiple system atrophy. Ann Clin Transl Neurol 2:417–426
Sklerov M, Kang UJ, Liong C, Marder K, Pauciulo M, Nichols WC, Chung WK, Honig LS, Cortes E, Vonsattel JP (2017) Frequency of GBA variants in autopsy-proven multiple system atrophy. Mov Disord Clin Pract 4(4):574–581
Segarane B, Li A, Paudel R et al (2009) Glucocerebrosidase mutations in 108 neuropathologically confirmed cases of multiple system atrophy. Neurology 72:1185–1186
Nishioka K, Ross OA, Vilarino-Guell C et al (2011) Glucocerebrosidase mutations in diffuse Lewy body disease. Parkinsonism Relat Disord 17:55–57
Goker-Alpan O, Giasson BI, Eblan MJ et al (2006) Glucocerebrosidase mutations are an important risk factor for Lewy body disorders. Neurology 67:908–910
Hruska KS, LaMarca ME, Scott CR, Sidransky E (2008) Gaucher disease: mutation and polymorphism spectrum in the glucocerebrosidase gene (GBA). Hum Mutat 29:567–583
Mazzulli JR, Xu YH, Sun Y et al (2011) Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 146:37–52
Barkhuizen M, Anderson DG, Grobler AF (2016) Advances in GBA-associated Parkinson's disease–Pathology, presentation and therapies. Neurochem Int 93:6–25
Sardi SP, Viel C, Clarke J et al (2017) Glucosylceramide synthase inhibition alleviates aberrations in synucleinopathy models. Proc Natl Acad Sci USA 114:2699–2704
Sardi SP, Cedarbaum JM, Brundin P (2018) Targeted Therapies for Parkinson's Disease: From Genetics to the Clinic. Mov Dis 33:684–696
Peterschmitt M, Gasser T, Isaacson S et al (2019) Safety, tolerability and pharmacokinetics of oral venglustat in Parkinson disease patients with a GBA mutation. Mol Genet Metabol Rep 126:S117
Hitti FL, Yang AI, Gonzalez-Alegre P, Baltuch GH (2019) Human gene therapy approaches for the treatment of Parkinson’s disease: an overview of current and completed clinical trials. Parkinsonism Relat Dis 66:16–24
Sardi SP, Clarke J, Viel C et al (2013) Augmenting CNS glucocerebrosidase activity as a therapeutic strategy for parkinsonism and other Gaucher-related synucleinopathies. Proc Natl Acad Sci USA 110:3537–3542
Prevail therapeutics: Prevail is developing a pipeline of potentially disease-modifying AAV9-based gene therapies for the treatment of genetically-defined neurodegenerative diseases: PR001. By: Prevail therapeutics. https://www.prevailtherapeutics.com/programs/#pr001
McMahon B, Aflaki E, Sidransky E (2016) Chaperoning glucocerebrosidase: a therapeutic strategy for both Gaucher disease and Parkinsonism. Neural Regen Res 11:1760–1761
Maegawa GH, Tropak MB, Buttner JD et al (2009) Identification and characterization of ambroxol as an enzyme enhancement agent for Gaucher disease. J Biol Chem 284:23502–23516
McNeill A, Magalhaes J, Shen C et al (2014) Ambroxol improves lysosomal biochemistry in glucocerebrosidase mutation-linked Parkinson disease cells. Brain 137:1481–1495
Migdalska-Richards A, Ko WKD, Li Q, Bezard E, Schapira AHV (2017) Oral ambroxol increases brain glucocerebrosidase activity in a nonhuman primate. Synapse. https://doi.org/10.1002/syn.21967
Trust hcginNsbSUaCPs. identification number: NCT02914366; sponsored by Sponsor: Lawson Health Research Institute. https://clinicaltrials.gov
Gegg ME, Burke D, Heales SJ et al (2012) Glucocerebrosidase deficiency in substantia nigra of parkinson disease brains. Ann Neurol 72:455–463
Silveira CRA, MacKinley J, Coleman K et al (2019) Ambroxol as a novel disease-modifying treatment for Parkinson's disease dementia: protocol for a single-centre, randomized, double-blind, placebo-controlled trial. BMC Neurol 19:20
Institute hcginNsbSLHR. Available
Saxton RA, Sabatini DM (2017) mTOR signaling in growth, metabolism, and disease. Cell 169:361–371
Savage N (2017) New tricks from old dogs join the fight against ageing. Nature 552:S57–S59
Spilman P, Podlutskaya N, Hart MJ et al (2010) Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-beta levels in a mouse model of Alzheimer's disease. PLoS ONE 5:e9979
Decressac M, Bjorklund A (2013) mTOR inhibition alleviates L-DOPA-induced dyskinesia in parkinsonian rats. J Parkinsons Dis 3:13–17
Healy DG, Falchi M, O'Sullivan SS et al (2008) Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson's disease: a case-control study. Lancet Neurol 7:583–590
Lim SY, Tan AH, Ahmad-Annuar A et al (2019) Parkinson's disease in the Western Pacific Region. Lancet Neurol 18(9):865–879
Ozelius LJ, Senthil G, Saunders-Pullman R et al (2006) LRRK2 G2019S as a cause of Parkinson's disease in Ashkenazi Jews. N Engl J Med 354:424–425
MDS Gene (2019) PARK-LRRK2 data summary. International parkinson and movement disorder society. University of Lübeck. Retrieved on April 18, 2019. https://www.mdsgene.org/d/1/g/1?action=plot_map&fc=0&_mu=1&_country=1
West AB (2015) Ten years and counting: moving leucine-rich repeat kinase 2 inhibitors to the clinic. Mov Dis 30:180–189
MacLeod DA, Rhinn H, Kuwahara T et al (2013) RAB7L1 interacts with LRRK2 to modify intraneuronal protein sorting and Parkinson’s disease risk. Neuron 77:425–439
Manzoni C, Denny P, Lovering RC, Lewis PA (2015) Computational analysis of the LRRK2 interactome. PeerJ 3:e778
Jaleel M, Nichols RJ, Deak M et al (2007) LRRK2 phosphorylates moesin at threonine-558: characterization of how Parkinson’s disease mutants affect kinase activity. Biochem J 405:307–317
Atashrazm F, Dzamko N (2016) LRRK2 inhibitors and their potential in the treatment of Parkinson’s disease: current perspectives. Clin Pharmacol 8:177–189
Blauwendraat C, Reed X, Kia DA et al (2018) Frequency of loss of function variants in LRRK2 in Parkinson disease. JAMA Neurol 75:1416–1422
Fuji RN, Flagella M, Baca M et al (2015) Effect of selective LRRK2 kinase inhibition on nonhuman primate lung. Sci Transl Med 7:273ra215
Baptista MAS, Merchant K, Barrett T, Bryce DK, Ellis M, Estrada AA et al (2018) LRRK2 kinase inhibitors induce a reversible effect in the lungs of non-human primates with no measurable pulmonary deficits. BioRxiv. https://doi.org/10.1101/390815v1
Whiffin N, Armean IA, Kleinman A, Marshall JL, Minikel EV, Goodrich JK et al (2019) Human loss-of-function variants suggest that partial LRRK2 inhibition is a safe therapeutic strategy for Parkinson’s disease. BioRxiv. https://doi.org/10.1101/561472v2
Foroud T, Smith D, Jackson J et al (2015) Novel recruitment strategy to enrich for LRRK2 mutation carriers. Mol Genet Genomic Med 3:404–412
Centogene AG (2018): Centogene and denali therapeutics announce strategic collaboration to recruit LRRK2 patients for clinical trials. Hg. v. Centogene AG. https://www.centogene.com/company/article/centogene-and-denali-therapeutics-announce-strategic-collaboration-to-recruit-lrrk2-patients-for-cli.html
Coutinho MF, Matos L, Santos JI, Alves S (2019) RNA therapeutics: how far have we gone? Adv Exp Med Biol 1157:133–177
Platt FM, d'Azzo A, Davidson BL, Neufeld EF, Tifft CJ (2018) Lysosomal storage diseases. Nat Rev Dis Primers 4:27
Zhao HT, John N, Delic V et al (2017) LRRK2 Antisense oligonucleotides ameliorate alpha-synuclein inclusion formation in a parkinson's disease mouse model. Mol Ther Nucleic Acids 8:508–519
Ysselstein D, Nguyen M, Young TJ et al (2019) LRRK2 kinase activity regulates lysosomal glucocerebrosidase in neurons derived from Parkinson's disease patients. Nat Commun 10:5570
Analysis Group, Inc (2017) The biopharmaceutical pipeline: innovative therapies in clinical development. Unter mitarbeit von genia long. Analysis Group, Inc. https://www.analysisgroup.com/uploadedfiles/content/insights/publishing/the_biopharmaceutical_pipeline_report_2017.pdf
The Pharmaceutical Research and Manufacturers of America (PhRMA) (2009) PHarmaceutical Research and Manufacturers of America (PhRMA) SPECIAL 301 SUBMISSION 2009. http://phrmadocs.phrma.org/sites/default/files/pdf/phrma_special_301_submission_20092.pdf
Mathur S, DeWitte S, Robledo I, Isaacs T, Stamford J (2015) Rising to the challenges of clinical trial improvement in Parkinson’s disease. J Parkinsons Dis 5:263–268
Marek K, Chowdhury S, Siderowf A et al (2018) The Parkinson’s progression markers initiative (PPMI)-establishing a PD biomarker cohort. Ann Clin Transl Neurol 5:1460–1477
Acknowledgements
Open Access funding provided by Projekt DEAL. We thank Baccara Hizli for assistance with data collection and Mr. Joshua Halpern for his careful work editing the manuscript.
Funding
S. Schneider was supported by LMU Clinician Scientist Programme, the Ara Parseghian Medical Research Fund, and the Verum Stiftung. She received a publication honorium from Springer Publishers.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
Dr. Alcalay reports receiving consultation fees from Genzyme/Sanofi, Roche and ResTORbio.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Schneider, S.A., Alcalay, R.N. Precision medicine in Parkinson’s disease: emerging treatments for genetic Parkinson’s disease. J Neurol 267, 860–869 (2020). https://doi.org/10.1007/s00415-020-09705-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-020-09705-7