
Abstract
The eukaryotic genome can be roughly divided into euchromatin and heterochromatin domains that are structurally and functionally distinct. Heterochromatin is characterized by its high compaction that impedes DNA transactions such as gene transcription, replication, or recombination. Beyond its role in regulating DNA accessibility, heterochromatin plays essential roles in nuclear architecture, chromosome segregation, and genome stability. The formation of heterochromatin involves special histone modifications and the recruitment and spreading of silencing complexes that impact the higher-order structures of chromatin; however, its molecular nature varies between different chromosomal regions and between species. Although heterochromatin has been extensively characterized, its formation and maintenance throughout the cell cycle are not yet fully understood. The biggest challenge for the faithful transmission of chromatin domains is the destabilization of chromatin structures followed by their reassembly on a novel DNA template during genomic replication. This destabilizing event also provides a window of opportunity for the de novo establishment of heterochromatin. In recent years, it has become clear that different types of obstacles such as tight protein-DNA complexes, highly transcribed genes, and secondary DNA structures could impede the normal progression of the replisome and thus have the potential to endanger the integrity of the genome. Multiple studies carried out in different model organisms have demonstrated the capacity of such replisome impediments to favor the formation of heterochromatin. Our review summarizes these reports and discusses the potential role of replication stress in the formation and maintenance of heterochromatin and the role that silencing proteins could play at sites where the integrity of the genome is compromised.
Similar content being viewed by others

In eukaryotic cells, the genetic information is stored as a complex of DNA and proteins called chromatin. Based on cytological observations, the German botanist Emil Heitz distinguished the following two types of chromatin: euchromatin, corresponding to regions of mitotic chromosome that decondensed during interphase, and heterochromatin, corresponding to regions that remained condensed throughout the whole cell cycle. Heitz proposed that heterochromatin reflects a functionally inactive state of the genome. Heterochromatin was subsequently found to be associated with gene repression and specific patterns of post-translational histone modifications or chromatin marks that recruit general repressors of transcription. Apart from their role in transcriptional repression, heterochromatic regions are also an important determinant of the spatial organization of the genome. They tend to cluster together and localize close to the nuclear periphery or around nucleoli, forming functional nuclear subcompartments, which sequester the pool of silencing proteins (Padeken and Heun 2014; Meister and Taddei 2013; Saksouk et al. 2015).
Heterochromatin can be generally divided into two major types—constitutive and facultative—that display different molecular signatures. Constitutive heterochromatin forms at telomeres, centromeres, and repetitive elements, where it plays a major role in genome stability (Dillon 2004; Saksouk et al. 2015). Facultative heterochromatin is developmentally regulated and acts as a key regulator of cellular differentiation and morphogenesis (Trojer and Reinberg 2007).
How heterochromatin domains are formed at specific loci and maintained throughout cellular division is still an open question, but DNA replication appears to be an important player in this process.
The replication process is per se a disrupting event that challenges established patterns of chromatin marks but at the same time provides a window of opportunity to change chromatin status. Cells have thus evolved specific mechanisms to coordinate DNA replication with the re-assembly of a similar chromatin environment on the two daughter strands (Alabert and Groth 2012).
Direct molecular links between replication and heterochromatin factors have been reported in many different species. One striking example is the conserved role of the origin recognition complex (ORC) in heterochromatin formation that was first observed in budding yeast, where the ORC subunit Orc1 interacts directly with the silencing factor Sir1 (Bell et al. 1993). Furthermore, Sir3, which is the structural component of silent chromatin in this species, is a paralog of Orc1 that arose from the whole genome duplication event in the Saccharomycetaceae order (Hickman et al. 2011). It is likely that Sir3 acquired its silencing function after gene duplication, as the Orc1 protein fulfills the role of Sir3 in Kluyveromyces lactis, a species that diverged from Saccharomyces prior to Orc1 duplication (Hickman and Rusche 2010). Interestingly, although heterochromatin components are not conserved between budding yeast and metazoans, ORC has been shown to interact with heterochromatin proteins such as heterochromatin protein 1 (HP1) in metazoans, indicating the conserved cross-talk between DNA replication and chromatin state.
Although ORC is present at all replication origins, heterochromatin is formed and confined to specific domains, suggesting that the interaction between ORC and heterochromatic factors is restricted to specific regions or that it leads to heterochromatin formation only under specific circumstances.
What is the hallmark of heterochromatin domains that distinguishes them from euchromatin regions and triggers the formation of heterochromatic structures? One possibility that we will discuss in this review is that these regions are difficult to replicate and that this feature could favor heterochromatin formation. Indeed, it is noteworthy that sequences embedded into heterochromatin are often inherently difficult to replicate and are thus a potential source of replication stress (Ivessa et al. 2003; Miller et al. 2006; Sfeir et al. 2009; Zaratiegui et al. 2011b). Replication stress is defined as slowing or stalling in replication fork progression and arises from many different sources including repetitive sequences, tight DNA-protein complexes, DNA secondary structures, DNA–RNA hybrids (R-loops), collisions between replication and transcription machineries, DNA lesions, or misincorporation of ribonucleotides (Aguilera and Garcia-Muse 2012; Bochman et al. 2012; Tourriere and Pasero 2007). Although replication stress can occur in both euchromatin and heterochromatin, several lines of evidence indicate that replication stress could be an important, although not unique, determinant in the formation and maintenance of heterochromatin states.
In this review, we will try to summarize the current knowledge on the mechanisms that link replication stress with heterochromatin formation and maintenance and discuss the potential function of this link.
Replicating heterochromatin
As mentioned above, heterochromatin comes in different flavors; here, we will briefly discuss mechanisms involved in pericentric heterochromatin replication that can serve as a paradigm for heterochromatin duplication. In mammalian cells, heterochromatin is formed in pericentric regions via the recruitment of HP1, which exists in the following three isoforms: α, β, and γ (Maison and Almouzni 2004), which have distinct functions in heterochromatin formation depending on the stage of differentiation (Aucott et al. 2008; Caillier et al. 2010; Sridharan et al. 2013). HP1 association with chromatin relies on the activity of the Suv39h1 methyltransferase, which creates a docking site for HP1 by trimethylating H3 lysine 9 (K9) (Maison and Almouzni 2004). HP1 also physically interacts with Suv39h1, providing a feed-forward loop to spread heterochromatin to neighboring nucleosomes.
During DNA replication, nucleosomes disassemble ahead of the fork and parental H3–H4 histones are handled by the histone chaperone anti-silencing function 1 (ASF1), which associates with the replicative DNA helicase (Alabert and Groth 2012). ASF1 is thought to coordinate the recycling of parental histones and de novo deposition of newly synthesized histones on the daughter strands. Chromatin assembly is coupled to DNA synthesis by the interaction between the sliding clamp proliferating cell nuclear antigen (PCNA) and the chromatin assembly factor 1 (CAF1) (Shibahara and Stillman 1999). Additionally, CAF1 interacts with HP1α and the histone lysine N-methyltransferase SetDB1, which monomethylates H3.1 (Loyola et al. 2009). Thus, in addition to its role in depositing H3 onto newly replicated DNA, CAF1 helps to target SetDB1 and HP1α to sites of heterochromatin formation, promoting both the monomethylation of H3.1K9 and the loading of HP1α in these regions. H3K9me1 is subsequently recognized by the methyltransferase Suv39h1, which trimethylates H3K9, thus allowing HP1 binding. Furthermore, recycled parental H3K9me3 also have the potential to recruit HP1 (Rivera et al. 2014). Duplication of HP1-associated chromatin thus appears to be a robust process involving parallel pathways. Similar mechanisms seem to be at play in the transmission of other heterochromatin-associated histone marks such as H4K20me (Rivera et al. 2014). In addition, HP1α is targeted as a sumoylated form to pericentric heterochromatin via its association with non-coding transcripts corresponding to major satellite repeats, independently of the methyltransferase activity of Suv39h1 (Maison et al. 2011). Whether this pathway requires replication is still an open question.
Heterochromatin protein at DNA damage sites
In response to DNA damage, chromatin undergoes global decondensation. This process has been proposed to facilitate genome surveillance by enhancing the access of DNA damage response (DDR) proteins to sites of damage (Adam et al. 2015). Paradoxically, heterochromatin factors such as KAP1, HP1, and the H3K9 methyltransferase Suv39h1 were shown to accumulate rapidly and transiently at DNA damage sites (Ayrapetov et al. 2014; Lemaitre and Soutoglou 2014). This occurs in both euchromatic and heterochromatic regions, suggesting a general role for this response in DNA repair. Indeed, depletion of two isoforms of HP1 (α and β) impairs homologous recombination (HR) efficiency, whereas depletion of HPγ gamma had the opposite effect, through mechanisms that are still under debate (Lemaitre and Soutoglou 2014; Soria and Almouzni 2013). A recent study showed that the rapid loading of HP1/KAP1/Suv39h1 leads to the trimethylation of H3K9 over tens of kilobase flanking double-strand breaks (DSBs). Although this H3K9me3 is often associated with heterochromatin, the modification of this large region is thought to help recruit and activate the Tip60 acetyltransferase, which recognizes H3K9me3. Tip60 promotes nucleosome turnover and activation of the ataxia telangiectasia mutated (ATM) checkpoint kinase, which acts as a negative feedback by removing the HP1/KAP1/suv39 complex from chromatin (Ayrapetov et al. 2014).
Beside its potential direct impact on HR, heterochromatin-associated proteins might also help coordinate transcription and repair. Indeed, polycomb proteins that are involved in gene silencing at facultative heterochromatin are also recruited to DSB sites, where they are thought to switch off transcription to facilitate DSB repair (Kakarougkas et al. 2014; Ui et al. 2015; Vissers et al. 2012).
HP1 recruitment at sites of DNA damage is again dependent on its interaction with CAF1 (Baldeyron et al. 2011; Loyola et al. 2009), which is recruited to DNA synthesis repair sites by PCNA (Moggs et al. 2000). CAF1 is conserved in budding yeast, where it also contributes to the maintenance of silent chromatin (Enomoto and Berman 1998) and genome stability (Kaufman et al. 1997). Other links between DNA repair and heterochromatin have been reported in budding yeast. Similar to HP1, its functional homologue in budding yeast (Sir3) has been found to localize at sites of DNA damage (Martin et al. 1999; Mills et al. 1999), although the function of this recruitment remains elusive. Furthermore, the Ku protein plays essential roles in protecting DSB against resection and in promoting subtelomeric gene repression (Boulton and Jackson 1996, 1998). More recently, artificially tethering the ATM checkpoint kinase Tel1 or the DNA repair protein Mre11 to a defective silencer was shown to promote silencing at this locus. Tel1 has been shown to interact with Sir2 in the two-hybrid system, as has Mre11 with both Sir3 and Sir4 (Kirkland et al. 2015). Hence, Tel1 and Mre11 may trigger silencing through the direct recruitment of the SIR complex.
Thus, both DDR and replication are linked to the recruitment of silencing proteins in different species, sometimes ultimately leading to chromatin opening. Silencing proteins may also contribute to genome stability, although this has not always been clearly demonstrated.
Replication stress and senescence-associated heterochromatin foci in mammalian cells
Cellular senescence is a well-studied example of a process during which chromatin undergoes a massive reorganization. Cellular senescence is an irreversible proliferation arrest, thought to contribute to tumor suppression, wound healing, development, and aging (Rai and Adams 2013). Entry into senescence can be triggered by different sources of stress, including telomere shortening (replicative senescence) and oncogene activation (oncogene-induced senescence (OIS)) (Rai and Adams 2013; Serrano et al. 1997). OIS is accompanied by the accumulation and compaction of chromatin into large subnuclear heterochromatic domains, termed senescence-associated heterochromatin foci (SAHFs) (Narita et al. 2003; Zhang et al. 2005). These structures harbor heterochromatic markers, such as H3K9me3, the histone variant macroH2A, high-mobility group A (HMGA), and the three HP1 isoforms (Corpet and Stucki 2014; Narita et al. 2003, 2006; Zhang et al. 2005).
Importantly, SAHF formation is not a universal feature of senescence as it is less prevalent in replicative senescence and is highly variable between cell types. Thus, SAHF formation appears to be influenced by the origin of the stress that triggers senescence (Kosar et al. 2011). Senescence induced by oncogenes arises from an acute stress at stalled replication forks, leading to irreparable DNA damage (Bartkova et al. 2006; Di Micco et al. 2006, 2011). However, DNA breaks per se do not seem to be the source of SAHF formation, as the activation of the S phase checkpoint kinase ataxia telangiectasia and Rad3-related (ATR) protein in the absence of DNA damage is sufficient to cause cellular senescence and appearance of SAHF (Toledo et al. 2008). Interestingly, oncogene-induced SAHF formation depends on DNA replication and ATR (Di Micco et al. 2011). Furthermore, topological and chromatin regulators involved in the DNA replication process, such as topoisomerase 1 and ASF1, are also essential for SAHF formation. Together, these data support a causal link between oncogene-altered DNA replication and SAHF formation (Humbert et al. 2009; Zhang et al. 2005, 2007).
It is still unclear to what extent SAHF plays a role in the tumor suppressive function of senescence in vivo. Heterochromatinization of proliferation genes through SAHF has been proposed to directly contribute to proliferation arrest. Indeed, disruption of the Suv39h1 H3K9 methyltransferase dramatically accelerates Ras-induced T cell lymphomagenesis in a mouse model (Braig et al. 2005). However, SAHF formation is not sufficient to drive proliferation arrest as SAHF persists in p53 and ATM mutants, in which the cell cycle arrest has been relieved (Di Micco et al. 2011; Rai and Adams 2013). In this case, it was proposed that SAHF protects cells expressing oncogenes from undergoing apoptosis by dampening the DNA damage response (Di Micco et al. 2011). In support of this hypothesis, heterochromatin has been reported to modulate DDR activation in different model systems (see above and Murga et al. 2007). Indeed, loss of HP1γ or the Suv39 methyltransferase led to an amplification of the DDR, eventually resulting in cell apoptosis (Di Micco et al. 2011). As several cancer cell lines display elevated levels of H3K9me3 and HP1, it has been proposed that the inactivation of tumor suppressors, such as p53, hijacks SAHFs to maintain DDR at sublethal levels, thus allowing proliferation. In summary, on the one hand, SAHF might contribute to tumor suppression via repression of proliferation-promoting genes such as cyclin A, while on the other hand, SAHF may dampen the DNA damage response, suppress apoptosis, and promote viability. As a consequence, altering heterochromatin might have different outcomes depending on the stage of oncogenic progression and on the history that led to transformation.
Replication roadblocks and heterochromatin formation
Tight DNA-protein complexes
Budding yeast does not show condensed chromatin during interphase and lacks most of the molecular markers that typify heterochromatin in most other eukaryotes, such as H3K9me3 and HP1. Instead, repressed chromatin is generated specifically at telomeres and cryptic mating-type loci (HMs) by the recruitment of a complex of silent information regulators, namely, Sir2, Sir3, and Sir4 (Kueng et al. 2013; Taddei and Gasser 2012). The complex has the ability to spread along the chromatin fiber from nucleation sites due to the enzymatic activity of Sir2 that deacetylates histone H4 tails from neighboring nucleosomes and to the affinity of Sir3 for deacetylated histones (Hecht et al. 1995; Imai et al. 2000; Rusche et al. 2003; Smith et al. 2000; Tanner et al. 2000). At telomeres, heterochromatin formation is nucleated by Rap1, which binds telomeric TG1–3 repeats and interacts with both Sir3 and Sir4 (Konig and Rhodes 1997; Moretti et al. 1994; Oppikofer et al. 2013). At HMs, nucleation is dependent on the presence of “silencer” elements. These elements contain combinations of binding sites for a set of factors Rap1, Abf1, and Sum1 and a member of the origin recognition complex (Orc1) (Kueng et al. 2013; Taddei and Gasser 2012). Intriguingly, Rap1 and Abf1 are two of the most common transcription factors in the yeast genome, and Orc1, as already discussed, is a member of the essential ORC complex, important for the firing of origins of replication (Bell and Stillman 1992; Shore 1994; Yarragudi et al. 2007). How the juxtaposition of these factors leads to the nucleation of silencing is still unknown. The current model proposes that the juxtaposition of factors with a low affinity for the SIR complex could generate a high-affinity binding site. However, although Rap1 interacts directly with Sir3 and Sir4 and Orc1 with Sir1, which in turn recruits Sir4, there is no clear evidence so far supporting a direct interaction between Abf1 and the SIR complex.
Interestingly, replication has been proposed to be required to establish silencing in Saccharomyces cerevisiae (Miller and Nasmyth 1984). However, this notion has been challenged by the observation that silencing can be established upon targeting of Sir1 to a non-replicative extrachromosomal cassette containing a crippled HMR locus (Kirchmaier and Rine 2001; Li et al. 2001). In this artificial context, silencing establishment retained cell cycle dependence, arguing that the passage of the replication fork is not required for establishment of yeast silent chromatin, although this does not exclude that it may contribute under some conditions. Interestingly, heterochromatin formation also shows cell cycle specificity in mammalian cells as microinjection of non-replicating plasmids in late S phase is transcriptionally repressed and associated with non-acetylated histones, whereas plasmid microinjected in early S phase is hyperacetylated and transcriptionally active (Zhang et al. 2002).
Although replication is not always necessary to establish silencing on non-chromosomal DNA, several observations indicate that fork pausing can contribute to heterochromatin establishment in budding yeast. First, it is noteworthy that natural sites of silencing (telomeres, HM, and ribosomal DNA (rDNA)) are all sites of transient replication fork pausing. Indeed, Rap1, ORC, Abf1, and Fob1 bind directly to the DNA and their tight interaction with their DNA-binding sequences acts as a replication roadblock (Brewer and Fangman 1988; Ivessa et al. 2003; Makovets et al. 2004). Interestingly, ORC binds more tightly at the HMR-E silencer element than at a very efficient replication origin found in euchromatin. Furthermore, this high-affinity binding site is required for ORC-dependent silencing at HMR (Palacios DeBeer et al. 2003). Second, we showed that natural or artificial pause sites can favor silencing establishment at ectopic loci (Dubarry et al. 2011). Importantly, this effect is increased in the absence of the DNA helicase Rrm3, which is known to facilitate replication at non-histone protein-DNA complexes. These data support a model where prolonged replication pausing increases the probability to form heterochromatin.
At endogenous loci, massive and stable SIR recruitment occurs only at sites harboring multiple DNA-protein complexes that have affinity for components of the SIR complex (i.e., Rap1 and Orc1). It is thus possible that replication fork pausing imposed by these complexes contributes to SIR recruitment at these sites, where they are then maintained by the affinities of Rap1 and Orc1 for Sir3, Sir4, and Sir1. Consistent with this hypothesis, SIR-dependent silencing of cryptic mating-type loci in K. lactis requires a different set of DNA-binding proteins for silencing establishment (Barsoum et al. 2010; Sjostrand et al. 2002). Further supporting this model is a recent report that a direct interaction between Sir3 and the ATPase subunit of the nucleosome remodeler Swi2 is key for SWI/SNF to promote resistance to replication stress in vivo and for the establishment of heterochromatin at telomeres (Manning and Peterson 2014).
In Schizosaccharomyces pombe, whose heterochromatin is more related to that of metazoans, mutations affecting the pool of dNTPS, thus slowing the replication fork and contributing to replication stress—favor the spreading of silent chromatin across the heterochromatin barriers of the silent mating-type locus mat2/3 (Singh and Klar 2008). Intriguingly, this extended heterochromatin depends on the binding of the transcription factor Atf1 that recruits histone deacetylases at this locus. Thus, the paradoxical role of transcription factors in heterochromatin formation in relation with replication stress appears as a recurrent theme.
In mouse and human genomes, the repetitive sequences associated with Suv39 H3K9me3-dependent heterochromatin contain transcription factor binding sites (Bulut-Karslioglu et al. 2012). Importantly, depletion of the two homeotic transcription factors Pax3 and Pax9 that bind mouse satellite repeats results in a derepression of these sequences and loss of heterochromatic marks. This led to a proposal that the reiterate arrangement of transcription factor binding sites in repetitive sequence is a general mechanism for heterochromatin formation (Bulut-Karslioglu et al. 2012). It will be interesting to test whether these factors represent obstacles for the replisome in this context.
What could be the function of heterochromatin formation at tight DNA-protein complexes sites? In S. cerevisiae, the histone deacetylase Sir2 represses PolII transcription and reduces recombination that would otherwise arise from the tight binding of Fob1 at rDNA repeats (Kobayashi 2011).
A similar function has been proposed in S. pombe for the CENP-B heterochromatin proteins that stabilize replication roadblocks imposed by the DNA-binding protein Sap1 at long terminal repeat retrotransposons (Zaratiegui et al. 2011b). Outside repetitive sequences, repressing transcription close to replication stress sites could contribute to genome integrity by preventing collision between the replication and transcription machineries.
DNA secondary structures
DNA secondary structures such as hairpin or G-quadruplex (G4) pose specific threats to the progression of the replication machinery (Leon-Ortiz et al. 2014). G4 are four-strand DNA structures held together by guanine, which can be unwound by several conserved helicases in vitro (WRN, BLM, FANCJ, and PIF1). Telomere sequences are predicted to form G4 and were shown to pose a challenge for the replication machinery in the absence of the telomere binding factors Taz1 in fission yeast and telomeric repeat factor 1 (TRF1) in human cells (Miller et al. 2006; Sfeir et al. 2009). TRF1 possibly alleviates this replication problem by recruiting the BLM helicase (Sfeir et al. 2009). In human cancer cells, the G4 stabilizing molecule pyridostatin targets nuclear sites overlapping with hPif1 (Rodriguez et al. 2012). Furthermore, this drug promotes growth arrest via inducing replication- and transcription-dependent DNA damage at telomeric and non-telomeric sequences with a propensity to form G4, arguing that these structures do form in vivo.
Furthermore, G4 structures were shown to cause epigenetic instability in the absence of G4 processing enzymes in metazoan and yeast (Sarkies et al. 2012). In S. cerevisiae, inserting a G4 motif 40 kb away from telomere VL in Pif1-defective cells induces SIR-dependent epigenetic instability on reporter genes located on the telomere proximal flanking region (Paeschke et al. 2013). Along the same lines, DT40 chicken cells defective for FANCJ show increased chromatin compaction in active regions of the genome (Schwab et al. 2013). Furthermore, loss of gene expression and active chromatin marks occur in the vicinity of G4 structure in the absence of the translesion polymerase Rev1 (Schiavone et al. 2014).
Trinucleotide repeats are prone to genomic instability, and their expansion is a recurrent cause of several human diseases including Friedreich’s ataxia (GAA/TTC) and fragile X syndrome (CGG). In both cases, trinucleotide expansion in a non-coding region leads to the silencing of the surrounding locus. Studies in transgenic mice indicate that GAA-triplet expansions stimulate heterochromatinization of a neighboring gene (Saveliev et al. 2003), a process that can be reduced by inhibiting the Sir2 family of deacetylases (Chan et al. 2013). Mechanisms leading to heterochromatin formation at expanded trinucleotide repeats are not well understood. Given that trinucleotide repeats are prone to form hairpin secondary structures and interfere with replication, replication stress could trigger heteterochromatin formation at these sites (Mirkin 2007).
Heterochromatin at replication/transcription collisions
Another source of replication stress arises from transcription—replication collisions (Azvolinsky et al. 2009). Such collisions seem to play a central role in the maintenance of pericentromeric heterochromatin in fission yeast. Genetic and biochemical evidence supports a model in which collisions between the replisome and the RNA polymerase II transcription complex generate stalled replication forks at these sites. It has been shown that, during replication, co-transcriptional RNA interference (RNAi) mechanisms release RNA polymerase II (Pol II) and avoid conflicts with the replication machinery (Li et al. 2011; Zaratiegui et al. 2011a). This is accompanied by the stable recruitment of the major silencing complex in fission yeast-cryptic loci regulator complex (CLRC) and the faithful propagation of heterochromatic states. The CLRC complex bridges these processes by interacting with subunits of the RNA-induced transcriptional silencing (RITS) complex as well as Cdc20 and Mms19, a subunit of the leading strand polymerase polε and a regulatory subunit of the Pol II transcription factor TFIIH, respectively (Bayne et al. 2010; Li et al. 2011; Motamedi et al. 2008; Svejstrup 2010). Zaratiegui and colleagues proposed that CLRC recruits the RNAi machinery at sites where the transcriptional and replication machineries clash. The RNAi machinery is able to remove Pol II and allow the resumption of DNA synthesis and further spreading of heterochromatin along the chromatin fiber (Kloc et al. 2008; Zaratiegui et al. 2011a). Whether this mechanism is conserved in other eukaryotes is not clear.
Mechanisms linking replication stress with heterochromatin formation
If replication stress contributes to heterochromatin formation, one can wonder what prevents heterochromatin formation at coding euchromatic regions where replication stress is likely to occur. One possibility is that counteracting activities associated with transcription destabilize heterochromatin or prevent its formation. Interestingly, the William syndrome transcription factor (WSTF) was shown to interact with PCNA to target the chromatin remodeler Snf2h to replication forks, thus preventing heterochromatin formation at ectopic sites (Culver-Cochran and Chadwick 2013; Poot et al. 2004). Whether these sites correspond to sites of replication stress is not known.
Although the mechanism(s) linking replication stress with heterochromatin formation remains largely unknown, several conserved factors arise as good candidates to link these two processes. First, cohesin is a conserved and essential multiprotein complex that holds together newly replicated chromatids and plays a crucial role in the maintenance of genomic stability (Jeppsson et al. 2014). Cohesins were shown to accumulate at replication sites when DNA synthesis is impeded and are critical for the recovery of stalled forks in budding yeast (Tittel-Elmer et al. 2012) and possibly in human cells (Gatei et al. 2014). Importantly, both Sir2 in budding yeast and HP1/Swi6 in metazoan and fission yeast interact directly with cohesins (Nonaka et al. 2002; Wu et al. 2011), again supporting a model in which replication stress sites could stimulate the establishment of heterochromatin.
Replication stress, like DNA damage, triggers phosphorylation of serine 129 of histone H2A in yeast (γH2A) or the histone variant H2A.X in mammalian cells (Downs et al. 2000; Foster and Downs 2005; Rogakou et al. 1998). This modification aids chromatin remodeling and the recruitment of repair factors during DNA repair, but its role at replication-stalled sites is unclear. This modification is enriched at heterochromatic domains in both budding and fission yeast, and this depends, respectively, on the SIR complex and the histone methyltransferase Clr4 (Kim et al. 2007; Kitada et al. 2011; Rozenzhak et al. 2010; Szilard et al. 2010). Although enrichment of this mark could be a passive consequence of the low turnover of nucleosomes in heterochromatic regions, it has the potential to recruit specific factors.
In S. pombe, one of the factors that recognizes γH2A is the Brct containing domain protein Brc1 (Williams et al. 2010) that is recruited to pericentric heterochromatin during S phase. Interestingly, Brc1 mutants show defects in centromeric silencing and increased chromosome missegregation in the presence of a microtubule-destabilizing agent (Lee et al. 2013; Lee and Russell 2013). Genetic evidences suggest that Brc1 stabilizes the replisome in these regions, thus avoiding replication restart through the recombination machinery, which can lead to loss of genetic and epigenetic information. Again, although budding yeast heterochromatin has diverged from one of the S. pombe, the functional link between scaffolding repair proteins and heterochromatin appears to be conserved. Indeed, Brc1 resembles Rtt107/Esc4 in budding yeast, both structurally and functionally (Zappulla et al. 2006). Esc4 also interacts with γH2A and was recently shown to dampen checkpoint activation at replication-induced lesions (Ohouo et al. 2013). Intriguingly, Esc4 also interacts with the silencing factor Sir3 and mediates Sir3-dependent establishment of heterochromatin when targeted (Ohouo et al. 2010; Roberts et al. 2006; Rouse 2004; Zappulla et al. 2006). Whether this interaction plays a role in dampening checkpoint activation has not yet been tested. However, silent chromatin was shown to suppress the checkpoint response upon induction of massive replication fork blocks in the rDNA of S. cerevisiae, indicating that this mechanism could be conserved in budding yeast (Bentsen et al. 2013). Esc2 provides another potential molecular link between replication stress and silent chromatin. Like Esc4, Esc2 was first identified for its ability to establish silencing when targeted to a modified silencer, possibly through its ability to recruit Sir2 (Dhillon and Kamakaka 2000; Cuperus and Shore 2002). Esc2 is a protein conserved from yeast to human containing two SUMO-like domains (Yu et al. 2010) that play a role in the resolution of replication coupled recombination intermediates in conjunction with Smc5-Smc6 and Mms21 (Albuquerque et al. 2013; Choi et al. 2010; Mankouri et al. 2009; Mimura et al. 2010; Sollier et al. 2009).
More indirect mechanisms could also initiate heterochromatin formation at sites of replication stress. These include defects in recycling of parental histones and unscheduled histone incorporation that could lead to loss or gain of specific histone marks or histone modifications and remodeling associated with post-replicative repair events (Alabert and Groth 2012).
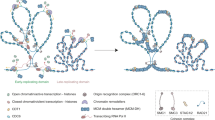
As mentioned above, some factors involved in DSB repair are also linked to gene silencing. It is tempting to speculate that DNA insults, irrespective of whether they occur during S phase or outside of it, could serve as an initial signal for the recruitment of silencing proteins with the capacity to induce the formation of heterochromatin structures. This local recruitment of silencing factors would likely be transient as shown for the recruitment of heterochromatin factors at euchromatic DNA damage sites (see above) and would thus not systematically lead to heterochromatin formation at most loci. However, one can speculate that the density of these events and/or the lack of antagonistic activities (i.e., histone remodeler of modifier) could lead to heterochromatin formation and maintenance at repetitive sequences. In these regions, heterochromatin formation would in turn avoid further stress by repressing transcription (avoiding replication/transcription collision), stabilizing the replication fork, and/or limiting checkpoint activation (Fig. 1).

During S phase, the replisome encounters a number of impediments, which could interfere with its progression and have the potential to endanger the stability of the genome. When faced with such obstacles, in many cases, the cell triggers the S phase checkpoint and stabilizes the replication fork. In the event of replication fork breakdown or “collapse,” replication can restart by recruiting an alternative pathway requiring homology-directed repair (HDR), which could eventually result in loss of genetic and/or epigenetic information. If stalled forks fail to restart, persistent checkpoint activation can lead to apoptosis. As discussed in the text, silencing factors are also recruited to sites of replication stress. Possible roles of heterochromatin proteins at these loci could be in preserving the stability of the replisome or in modulating the cellular response to replication stress via largely unknown mechanism(s). Such events could also serve as an initial signal and potentially trigger the formation of heterochromatin
Concluding remarks
Many studies from different organisms have identified a crosstalk between replication stress and heterochromatin formation. It is striking that similar processes seem to be at play despite the un-conserved molecular nature of heterochromatin. This raises the question of the potential role(s) of this crosstalk. The following two recurrent themes emerge from the above-discussed studies (Fig. 1): heterochromatin dampening the checkpoint activation at replication stress sites and heterochromatin stabilizing the replisome by avoiding collision between the replication and transcription machineries or by avoiding unscheduled recombination events. On the other hand, the numerous links between replication stress and heterochromatin strongly indicate that replication stress could be an evolutionary conserved auxiliary mechanism for the establishment of heterochromatin and silencing domains. Further studies are needed to unravel the candidates and cascade of events that govern this complex crosstalk.
References
Adam S, Dabin J, Polo SE (2015) Chromatin plasticity in response to DNA damage: the shape of things to come. DNA Repair (Amst) 32:120–126
Aguilera A, Garcia-Muse T (2012) R loops: from transcription byproducts to threats to genome stability. Mol Cell 46:115–124
Alabert C, Groth A (2012) Chromatin replication and epigenome maintenance. Nat Rev Mol Cell Biol 13:153–167
Albuquerque CP, Wang G, Lee NS, Kolodner RD, Putnam CD, Zhou H (2013) Distinct SUMO ligases cooperate with Esc2 and Slx5 to suppress duplication-mediated genome rearrangements. PLoS Genet 9:e1003670
Aucott R, Bullwinkel J, Yu Y, Shi W, Billur M, Brown JP, Menzel U, Kioussis D, Wang G, Reisert I, Weimer J, Pandita RK, Sharma GG, Pandita TK, Fundele R, Singh PB (2008) HP1-beta is required for development of the cerebral neocortex and neuromuscular junctions. J Cell Biol 183:597–606
Ayrapetov MK, Gursoy-Yuzugullu O, Xu C, Xu Y, Price BD (2014) DNA double-strand breaks promote methylation of histone H3 on lysine 9 and transient formation of repressive chromatin. Proc Natl Acad Sci U S A 111:9169–9174
Azvolinsky A, Giresi PG, Lieb JD, Zakian VA (2009) Highly transcribed RNA polymerase II genes are impediments to replication fork progression in Saccharomyces cerevisiae. Mol Cell 34:722–734
Baldeyron C, Soria G, Roche D, Cook AJ, Almouzni G (2011) HP1alpha recruitment to DNA damage by p150CAF-1 promotes homologous recombination repair. J Cell Biol 193:81–95
Barsoum E, Sjostrand JO, Astrom SU (2010) Ume6 is required for the MATa/MATalpha cellular identity and transcriptional silencing in Kluyveromyces lactis. Genetics 184:999–1011
Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, Vassiliou LV, Kolettas E, Niforou K, Zoumpourlis VC, Takaoka M, Nakagawa H, Tort F, Fugger K, Johansson F, Sehested M, Andersen CL, Dyrskjot L, Orntoft T, Lukas J, Kittas C, Helleday T, Halazonetis TD, Bartek J, Gorgoulis VG (2006) Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 444:633–637
Bayne EH, White SA, Kagansky A, Bijos DA, Sanchez-Pulido L, Hoe KL, Kim DU, Park HO, Ponting CP, Rappsilber J, Allshire RC (2010) Stc1: a critical link between RNAi and chromatin modification required for heterochromatin integrity. Cell 140:666–677
Bell SP, Stillman B (1992) ATP-dependent recognition of eukaryotic origins of DNA replication by a multiprotein complex. Nature 357:128–134
Bell SP, Kobayashi R, Stillman B (1993) Yeast origin recognition complex functions in transcription silencing and DNA replication. Science 262:1844–1849
Bentsen IB, Nielsen I, Lisby M, Nielsen HB, Gupta SS, Mundbjerg K, Andersen AH, Bjergbaek L (2013) MRX protects fork integrity at protein-DNA barriers, and its absence causes checkpoint activation dependent on chromatin context. Nucleic Acids Res 41:3173–3189
Bochman ML, Paeschke K, Zakian VA (2012) DNA structures: stability and function of G-quadruplex structures. Nat Rev Genet 13:770–780
Boulton SJ, Jackson SP (1996) Saccharomyces-cerevisiae ku70 potentiates illegitimate DNA double-strand break repair and serves as a barrier to error-prone DNA-repair pathways. EMBO J 15:5093–5103
Boulton SJ, Jackson SP (1998) Components of the Ku-dependent non-homologous end-joining pathway are involved in telomeric length maintenance and telomeric silencing. EMBO J 17:1819–1828
Braig M, Lee S, Loddenkemper C, Rudolph C, Peters AH, Schlegelberger B, Stein H, Dorken B, Jenuwein T, Schmitt CA (2005) Oncogene-induced senescence as an initial barrier in lymphoma development. Nature 436:660–665
Brewer BJ, Fangman WL (1988) A replication fork barrier at the 3′ end of yeast ribosomal RNA genes. Cell 55:637–643
Bulut-Karslioglu A, Perrera V, Scaranaro M, de la Rosa-Velazquez IA, van de Nobelen S, Shukeir N, Popow J, Gerle B, Opravil S, Pagani M, Meidhof S, Brabletz T, Manke T, Lachner M, Jenuwein T (2012) A transcription factor-based mechanism for mouse heterochromatin formation. Nat Struct Mol Biol 19:1023–1030
Caillier M, Thenot S, Tribollet V, Birot AM, Samarut J, Mey A (2010) Role of the epigenetic regulator HP1gamma in the control of embryonic stem cell properties. PLoS One 5:e15507
Chan PK, Torres R, Yandim C, Law PP, Khadayate S, Mauri M, Grosan C, Chapman-Rothe N, Giunti P, Pook M, Festenstein R (2013) Heterochromatinization induced by GAA-repeat hyperexpansion in Friedreich’s ataxia can be reduced upon HDAC inhibition by vitamin B3. Hum Mol Genet 22:2662–2675
Choi K, Szakal B, Chen YH, Branzei D, Zhao X (2010) The Smc5/6 complex and Esc2 influence multiple replication-associated recombination processes in Saccharomyces cerevisiae. Mol Biol Cell 21:2306–2314
Corpet A, Stucki M (2014) Chromatin maintenance and dynamics in senescence: a spotlight on SAHF formation and the epigenome of senescent cells. Chromosoma 123:423–436
Culver-Cochran AE, Chadwick BP (2013) Loss of WSTF results in spontaneous fluctuations of heterochromatin formation and resolution, combined with substantial changes to gene expression. BMC Genomics 14:740
Cuperus G, Shore D (2002) Restoration of silencing in Saccharomyces cerevisiae by tethering of a novel Sir2-interacting protein, Esc8. Genetics 162:633–645
Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, Luise C, Schurra C, Garre M, Nuciforo PG, Bensimon A, Maestro R, Pelicci PG, d’Adda di Fagagna F (2006) Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 444:638–642
Di Micco R, Sulli G, Dobreva M, Liontos M, Botrugno OA, Gargiulo G, dal Zuffo R, Matti V, d’Ario G, Montani E, Mercurio C, Hahn WC, Gorgoulis V, Minucci S, d’Adda di Fagagna F (2011) Interplay between oncogene-induced DNA damage response and heterochromatin in senescence and cancer. Nat Cell Biol 13:292–302
Dhillon N, Kamakaka RT (2000) A histone variant, Htz1p, and a Sir1p-like protein, Esc2p, mediate silencing at HMR. Mol Cell 6:769–780
Dillon N (2004) Heterochromatin structure and function. Biol Cell 96:631–637
Downs JA, Lowndes NF, Jackson SP (2000) A role for Saccharomyces cerevisiae histone H2A in DNA repair. Nature 408:1001–1004
Dubarry M, Loiodice I, Chen CL, Thermes C, Taddei A (2011) Tight protein-DNA interactions favor gene silencing. Genes Dev 25:1365–1370
Enomoto S, Berman J (1998) Chromatin assembly factor I contributes to the maintenance, but not the re-establishment, of silencing at the yeast silent mating loci. Genes Dev 12:219–232
Foster ER, Downs JA (2005) Histone H2A phosphorylation in DNA double-strand break repair. FEBS J 272:3231–3240
Gatei M, Kijas AW, Biard D, Dork T, Lavin MF (2014) RAD50 phosphorylation promotes ATR downstream signaling and DNA restart following replication stress. Hum Mol Genet 23:4232–4248
Hecht A, Laroche T, Strahl-Bolsinger S, Gasser SM, Grunstein M (1995) Histone H3 and H4 N-termini interact with Sir3 and Sir4 proteins: a molecular model for the formation of heterochromatin in yeast. Cell 80:583–592
Hickman MA, Rusche LN (2010) Transcriptional silencing functions of the yeast protein Orc1/Sir3 subfunctionalized after gene duplication. Proc Natl Acad Sci U S A 107:19384–19389
Hickman MA, Froyd CA, Rusche LN (2011) Reinventing heterochromatin in budding yeasts: Sir2 and the origin recognition complex take center stage. Eukaryot Cell 10:1183–1192
Humbert N, Martien S, Augert A, Da Costa M, Mauen S, Abbadie C, de Launoit Y, Gil J, Bernard D (2009) A genetic screen identifies topoisomerase 1 as a regulator of senescence. Cancer Res 69:4101–4106
Imai S, Armstrong CM, Kaeberlein M, Guarente L (2000) Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 403:795–800
Ivessa AS, Lenzmeier BA, Bessler JB, Goudsouzian LK, Schnakenberg SL, Zakian VA (2003) The Saccharomyces cerevisiae helicase Rrm3p facilitates replication past nonhistone protein-DNA complexes. Mol Cell 12:1525–1536
Jeppsson K, Kanno T, Shirahige K, Sjogren C (2014) The maintenance of chromosome structure: positioning and functioning of SMC complexes. Nat Rev Mol Cell Biol 15:601–614
Kakarougkas A, Ismail A, Chambers AL, Riballo E, Herbert AD, Kunzel J, Lobrich M, Jeggo PA, Downs JA (2014) Requirement for PBAF in transcriptional repression and repair at DNA breaks in actively transcribed regions of chromatin. Mol Cell 55:723–732
Kaufman PD, Kobayashi R, Stillman B (1997) Ultraviolet radiation sensitivity and reduction of telomeric silencing in Saccharomyces cerevisiae cells lacking chromatin assembly factor I. Genes Dev 11:345–357
Kim JA, Kruhlak M, Dotiwala F, Nussenzweig A, Haber JE (2007) Heterochromatin is refractory to gamma-H2AX modification in yeast and mammals. J Cell Biol 178:209–218
Kirchmaier AL, Rine J (2001) DNA replication-independent silencing in S. cerevisiae. Science 291:646–650
Kirkland JG, Peterson MR, Still CD 2nd, Brueggeman L, Dhillon N, Kamakaka RT (2015) Heterochromatin formation via recruitment of DNA repair proteins. Mol Biol Cell 26:1395–1410
Kitada T, Schleker T, Sperling AS, Xie W, Gasser SM, Grunstein M (2011) GammaH2A is a component of yeast heterochromatin required for telomere elongation. Cell Cycle (Georgetown, Tex) 10:293–300
Kloc A, Zaratiegui M, Nora E, Martienssen R (2008) RNA interference guides histone modification during the S phase of chromosomal replication. Curr Biol 18:490–495
Kobayashi T (2011) How does genome instability affect lifespan?: roles of rDNA and telomeres. Genes Cells 16:617–624
Konig P, Rhodes D (1997) Recognition of telomeric DNA. Trends Biochem Sci 22:43–47
Kosar M, Bartkova J, Hubackova S, Hodny Z, Lukas J, Bartek J (2011) Senescence-associated heterochromatin foci are dispensable for cellular senescence, occur in a cell type- and insult-dependent manner and follow expression of p16(ink4a). Cell Cycle 10:457–468
Kueng S, Oppikofer M, Gasser SM (2013) SIR proteins and the assembly of silent chromatin in budding yeast. Annu Rev Genet 47:275–306
Lee SY, Russell P (2013) Brc1 links replication stress response and centromere function. Cell Cycle 12:1665–1671
Lee SY, Rozenzhak S, Russell P (2013) gammaH2A-binding protein Brc1 affects centromere function in fission yeast. Mol Cell Biol 33:1410–1416
Lemaitre C, Soutoglou E (2014) Double strand break (DSB) repair in heterochromatin and heterochromatin proteins in DSB repair. DNA Repair (Amst) 19:163–168
Leon-Ortiz AM, Svendsen J, Boulton SJ (2014) Metabolism of DNA secondary structures at the eukaryotic replication fork. DNA Repair (Amst) 19:152–162
Li YC, Cheng TH, Gartenberg MR (2001) Establishment of transcriptional silencing in the absence of DNA replication. Science 291:650–653
Li F, Martienssen R, Cande WZ (2011) Coordination of DNA replication and histone modification by the Rik1-Dos2 complex. Nature 475:244–248
Loyola A, Tagami H, Bonaldi T, Roche D, Quivy JP, Imhof A, Nakatani Y, Dent SY, Almouzni G (2009) The HP1alpha-CAF1-SetDB1-containing complex provides H3K9me1 for Suv39-mediated K9me3 in pericentric heterochromatin. EMBO Rep 10:769–775
Maison C, Almouzni G (2004) HP1 and the dynamics of heterochromatin maintenance. Nat Rev Mol Cell Biol 5:296–304
Maison C, Bailly D, Roche D, Montes de Oca R, Probst AV, Vassias I, Dingli F, Lombard B, Loew D, Quivy JP, Almouzni G (2011) SUMOylation promotes de novo targeting of HP1alpha to pericentric heterochromatin. Nat Genet 43:220–227
Makovets S, Herskowitz I, Blackburn EH (2004) Anatomy and dynamics of DNA replication fork movement in yeast telomeric regions. Mol Cell Biol 24:4019–4031
Mankouri HW, Ngo HP, Hickson ID (2009) Esc2 and Sgs1 act in functionally distinct branches of the homologous recombination repair pathway in Saccharomyces cerevisiae. Mol Biol Cell 20:1683–1694
Manning BJ, Peterson CL (2014) Direct interactions promote eviction of the Sir3 heterochromatin protein by the SWI/SNF chromatin remodeling enzyme. Proc Natl Acad Sci U S A 111:17827–17832
Martin SG, Laroche T, Suka N, Grunstein M, Gasser SM (1999) Relocalization of telomeric Ku and SIR proteins in response to DNA strand breaks in yeast. Cell 97:621–633
Meister P, Taddei A (2013) Building silent compartments at the nuclear periphery: a recurrent theme. Curr Opin Genet Dev 23:96–103
Miller AM, Nasmyth KA (1984) Role of DNA replication in the repression of silent mating type loci in yeast. Nature 312:247–251
Miller KM, Rog O, Cooper JP (2006) Semi-conservative DNA replication through telomeres requires Taz1. Nature 440:824–828
Mills KD, Sinclair DA, Guarente L (1999) MEC1-dependent redistribution of the Sir3 silencing protein from telomeres to DNA double-strand breaks. Cell 97:609–620
Mimura S, Yamaguchi T, Ishii S, Noro E, Katsura T, Obuse C, Kamura T (2010) Cul8/Rtt101 forms a variety of protein complexes that regulate DNA damage response and transcriptional silencing. J Biol Chem 285:9858–9867
Mirkin SM (2007) Expandable DNA repeats and human disease. Nature 447:932–940
Moggs JG, Grandi P, Quivy JP, Jonsson ZO, Hubscher U, Becker PB, Almouzni G (2000) A CAF-1-PCNA-mediated chromatin assembly pathway triggered by sensing DNA damage. Mol Cell Biol 20:1206–1218
Moretti P, Freeman K, Coodly L, Shore D (1994) Evidence that a complex of SIR proteins interacts with the silencer and telomere-binding protein RAP1. Genes Dev 8:2257–2269
Motamedi MR, Hong EJ, Li X, Gerber S, Denison C, Gygi S, Moazed D (2008) HP1 proteins form distinct complexes and mediate heterochromatic gene silencing by nonoverlapping mechanisms. Mol Cell 32:778–790
Murga M, Jaco I, Fan Y, Soria R, Martinez-Pastor B, Cuadrado M, Yang SM, Blasco MA, Skoultchi AI, Fernandez-Capetillo O (2007) Global chromatin compaction limits the strength of the DNA damage response. J Cell Biol 178:1101–1108
Narita M, Nunez S, Heard E, Narita M, Lin AW, Hearn SA, Spector DL, Hannon GJ, Lowe SW (2003) Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 113:703–716
Narita M, Narita M, Krizhanovsky V, Nunez S, Chicas A, Hearn SA, Myers MP, Lowe SW (2006) A novel role for high-mobility group a proteins in cellular senescence and heterochromatin formation. Cell 126:503–514
Nonaka N, Kitajima T, Yokobayashi S, Xiao G, Yamamoto M, Grewal SI, Watanabe Y (2002) Recruitment of cohesin to heterochromatic regions by Swi6/HP1 in fission yeast. Nat Cell Biol 4:89–93
Ohouo PY, Bastos de Oliveira FM, Almeida BS, Smolka MB (2010) DNA damage signaling recruits the Rtt107-Slx4 scaffolds via Dpb11 to mediate replication stress response. Mol Cell 39:300–306
Ohouo PY, Bastos de Oliveira FM, Liu Y, Ma CJ, Smolka MB (2013) DNA-repair scaffolds dampen checkpoint signalling by counteracting the adaptor Rad9. Nature 493:120–124
Oppikofer M, Kueng S, Gasser SM (2013) SIR-nucleosome interactions: structure-function relationships in yeast silent chromatin. Gene 527:10–25
Paeschke K, Bochman ML, Garcia PD, Cejka P, Friedman KL, Kowalczykowski SC, Zakian VA (2013) Pif1 family helicases suppress genome instability at G-quadruplex motifs. Nature 497:458–462
Palacios DeBeer MA, Muller U, Fox CA (2003) Differential DNA affinity specifies roles for the origin recognition complex in budding yeast heterochromatin. Genes Dev 17:1817–1822
Padeken J, Heun P (2014) Nucleolus and nuclear periphery: velcro for heterochromatin. Curr Opin Cell Biol 28:54–60
Poot RA, Bozhenok L, van den Berg DL, Steffensen S, Ferreira F, Grimaldi M, Gilbert N, Ferreira J, Varga-Weisz PD (2004) The Williams syndrome transcription factor interacts with PCNA to target chromatin remodelling by ISWI to replication foci. Nat Cell Biol 6:1236–1244
Rai TS, Adams PD (2013) Lessons from senescence: chromatin maintenance in non-proliferating cells. Biochim Biophys Acta 1819:322–331
Rivera C, Gurard-Levin ZA, Almouzni G, Loyola A (2014) Histone lysine methylation and chromatin replication. Biochim Biophys Acta 1839:1433–1439
Roberts TM, Kobor MS, Bastin-Shanower SA, Ii M, Horte SA, Gin JW, Emili A, Rine J, Brill SJ, Brown GW (2006) Slx4 regulates DNA damage checkpoint-dependent phosphorylation of the BRCT domain protein Rtt107/Esc4. Mol Biol Cell 17:539–548
Rodriguez R, Miller KM, Forment JV, Bradshaw CR, Nikan M, Britton S, Oelschlaegel T, Xhemalce B, Balasubramanian S, Jackson SP (2012) Small-molecule-induced DNA damage identifies alternative DNA structures in human genes. Nat Chem Biol 8:301–310
Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM (1998) DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem 273:5858–5868
Rouse J (2004) Esc4p, a new target of Mec1p (ATR), promotes resumption of DNA synthesis after DNA damage. EMBO J 23:1188–1197
Rozenzhak S, Mejia-Ramirez E, Williams JS, Schaffer L, Hammond JA, Head SR, Russell P (2010) Rad3 decorates critical chromosomal domains with gammaH2A to protect genome integrity during S-phase in fission yeast. PLoS Genet 6:e1001032
Rusche LN, Kirchmaier AL, Rine J (2003) The establishment, inheritance, and function of silenced chromatin in Saccharomyces cerevisiae. Annu Rev Biochem 72:481–516
Saksouk N, Simboeck E, Dejardin J (2015) Constitutive heterochromatin formation and transcription in mammals. Epigenetics Chromatin 8:3
Sarkies P, Murat P, Phillips LG, Patel KJ, Balasubramanian S, Sale JE (2012) FANCJ coordinates two pathways that maintain epigenetic stability at G-quadruplex DNA. Nucleic Acids Res 40:1485–1498
Saveliev A, Everett C, Sharpe T, Webster Z, Festenstein R (2003) DNA triplet repeats mediate heterochromatin-protein-1-sensitive variegated gene silencing. Nature 422:909–913
Schiavone D, Guilbaud G, Murat P, Papadopoulou C, Sarkies P, Prioleau MN, Balasubramanian S, Sale JE (2014) Determinants of G quadruplex-induced epigenetic instability in REV1-deficient cells. EMBO J 33:2507–2520
Schwab RA, Nieminuszczy J, Shin-ya K, Niedzwiedz W (2013) FANCJ couples replication past natural fork barriers with maintenance of chromatin structure. J Cell Biol 201:33–48
Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW (1997) Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 88:593–602
Sfeir A, Kosiyatrakul ST, Hockemeyer D, MacRae SL, Karlseder J, Schildkraut CL, de Lange T (2009) Mammalian telomeres resemble fragile sites and require TRF1 for efficient replication. Cell 138:90–103
Shibahara K, Stillman B (1999) Replication-dependent marking of DNA by PCNA facilitates CAF-1-coupled inheritance of chromatin. Cell 96:575–585
Shore D (1994) RAP1: a protean regulator in yeast. Trends Genet 10:408–412
Singh G, Klar AJ (2008) Mutations in deoxyribonucleotide biosynthesis pathway cause spreading of silencing across heterochromatic barriers at the mating-type region of the fission yeast. Yeast 25:117–128
Sjostrand JO, Kegel A, Astrom SU (2002) Functional diversity of silencers in budding yeasts. Eukaryot Cell 1:548–557
Smith JS, Brachmann CB, Celic I, Kenna MA, Muhammad S, Starai VJ, Avalos JL, Escalante-Semerena JC, Grubmeyer C, Wolberger C, Boeke JD (2000) A phylogenetically conserved NAD+-dependent protein deacetylase activity in the Sir2 protein family. Proc Natl Acad Sci U S A 97:6658–6663
Sollier J, Driscoll R, Castellucci F, Foiani M, Jackson SP, Branzei D (2009) The Saccharomyces cerevisiae Esc2 and Smc5-6 proteins promote sister chromatid junction-mediated intra-S repair. Mol Biol Cell 20:1671–1682
Soria G, Almouzni G (2013) Differential contribution of HP1 proteins to DNA end resection and homology-directed repair. Cell Cycle 12:422–429
Sridharan R, Gonzales-Cope M, Chronis C, Bonora G, McKee R, Huang C, Patel S, Lopez D, Mishra N, Pellegrini M, Carey M, Garcia BA, Plath K (2013) Proteomic and genomic approaches reveal critical functions of H3K9 methylation and heterochromatin protein-1gamma in reprogramming to pluripotency. Nat Cell Biol 15:872–882
Svejstrup JQ (2010) The interface between transcription and mechanisms maintaining genome integrity. Trends Biochem Sci 35:333–338
Szilard RK, Jacques PE, Laramee L, Cheng B, Galicia S, Bataille AR, Yeung M, Mendez M, Bergeron M, Robert F, Durocher D (2010) Systematic identification of fragile sites via genome-wide location analysis of gamma-H2AX. Nat Struct Mol Biol 17:299–305
Taddei A, Gasser SM (2012) Structure and function in the budding yeast nucleus. Genetics 192:107–129
Tanner KG, Landry J, Sternglanz R, Denu JM (2000) Silent information regulator 2 family of NAD-dependent histone/protein deacetylases generates a unique product, 1-O-acetyl-ADP-ribose. Proc Natl Acad Sci U S A 97:14178–14182
Tittel-Elmer M, Lengronne A, Davidson MB, Bacal J, Francois P, Hohl M, Petrini JH, Pasero P, Cobb JA (2012) Cohesin association to replication sites depends on rad50 and promotes fork restart. Mol Cell 48:98–108
Toledo LI, Murga M, Gutierrez-Martinez P, Soria R, Fernandez-Capetillo O (2008) ATR signaling can drive cells into senescence in the absence of DNA breaks. Genes Dev 22:297–302
Tourriere H, Pasero P (2007) Maintenance of fork integrity at damaged DNA and natural pause sites. DNA Repair (Amst) 6:900–913
Trojer P, Reinberg D (2007) Facultative heterochromatin: is there a distinctive molecular signature? Mol Cell 28:1–13
Ui A, Nagaura Y, Yasui A (2015) Transcriptional elongation factor ENL phosphorylated by ATM recruits polycomb and switches off transcription for DSB repair. Mol Cell 58:468–482
Vissers JH, van Lohuizen M, Citterio E (2012) The emerging role of polycomb repressors in the response to DNA damage. J Cell Sci 125:3939–3948
Williams JS, Williams RS, Dovey CL, Guenther G, Tainer JA, Russell P (2010) gammaH2A binds Brc1 to maintain genome integrity during S-phase. EMBO J 29:1136–1148
Wu CS, Chen YF, Gartenberg MR (2011) Targeted sister chromatid cohesion by Sir2. PLoS Genet 7:e1002000
Yarragudi A, Parfrey LW, Morse RH (2007) Genome-wide analysis of transcriptional dependence and probable target sites for Abf1 and Rap1 in Saccharomyces cerevisiae. Nucleic Acids Res 35:193–202
Yu Q, Kuzmiak H, Olsen L, Kulkarni A, Fink E, Zou Y, Bi X (2010) Saccharomyces cerevisiae Esc2p interacts with Sir2p through a small ubiquitin-like modifier (SUMO)-binding motif and regulates transcriptionally silent chromatin in a locus-dependent manner. J Biol Chem 285:7525–7536
Zappulla DC, Maharaj AS, Connelly JJ, Jockusch RA, Sternglanz R (2006) Rtt107/Esc4 binds silent chromatin and DNA repair proteins using different BRCT motifs. BMC Mol Biol 7:40
Zaratiegui M, Castel SE, Irvine DV, Kloc A, Ren J, Li F, de Castro E, Marin L, Chang AY, Goto D, Cande WZ, Antequera F, Arcangioli B, Martienssen RA (2011a) RNAi promotes heterochromatic silencing through replication-coupled release of RNA Pol II. Nature 479:135–138
Zaratiegui M, Vaughn MW, Irvine DV, Goto D, Watt S, Bahler J, Arcangioli B, Martienssen RA (2011b) CENP-B preserves genome integrity at replication forks paused by retrotransposon LTR. Nature 469:112–115
Zhang J, Xu F, Hashimshony T, Keshet I, Cedar H (2002) Establishment of transcriptional competence in early and late S phase. Nature 420:198–202
Zhang R, Poustovoitov MV, Ye X, Santos HA, Chen W, Daganzo SM, Erzberger JP, Serebriiskii IG, Canutescu AA, Dunbrack RL, Pehrson JR, Berger JM, Kaufman PD, Adams PD (2005) Formation of MacroH2A-containing senescence-associated heterochromatin foci and senescence driven by ASF1a and HIRA. Dev Cell 8:19–30
Zhang R, Chen W, Adams PD (2007) Molecular dissection of formation of senescence-associated heterochromatin foci. Mol Cell Biol 27:2343–2358
Acknowledgments
We thank Christèle Maison, Jean-Pierre Quivy, Xavier Tadeo, and Myriam Ruault for their critical reading of the manuscript and the members of the Taddei laboratory. Ivaylo Nikolov was supported by a doctoral fellowship from the Association pour la Recherche sur le Cancer, and the University Pierre et Marie Curie (UPMC) is the recipient of an Association pour la Recherche sur le Cancer fellowship 20100600373. Our group has received funding from the European Research Council under the European Community’s Seventh Framework Programme (FP7/2007-2013)/ERC grant agreement number 210508 and receives funding from the investissement d’avenir (Labex DEEP) Agence Nationale de la Recherche (ANR), the Association pour la Recherche sur le Cancer, and the Fondation pour la recherche médicale (FRM DEP20131128535)
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Angela Taddei declares that she has no conflict of interest. Ivaylo Nikolov declares that he has no conflict of interest.
This article does not contain any studies with human participants or animals performed by any of the authors.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Nikolov, I., Taddei, A. Linking replication stress with heterochromatin formation. Chromosoma 125, 523–533 (2016). https://doi.org/10.1007/s00412-015-0545-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00412-015-0545-6