
Abstract
Quantitative multistage carcinogenesis models are used in radiobiology to estimate cancer risks and latency periods (time from exposure to clinical cancer). Steps such as initiation, promotion and transformation have been modeled in detail. However, progression, a later step during which malignant cells can develop into clinical symptomatic cancer, has often been approximated simply as a fixed lag time. This approach discounts important stochastic mechanisms in progression and evidence on the high prevalence of dormant tumors. Modeling progression more accurately is therefore important for risk assessment. Unlike models of earlier steps, progression models can readily utilize not only experimental and epidemiological data but also clinical data such as the results of modern screening and imaging. Here, a stochastic progression model is presented. We describe, with minimal parameterization: the initial growth or extinction of a malignant clone after formation of a malignant cell; the likely dormancy caused, for example, by nutrient and oxygen deprivation; and possible escape from dormancy resulting in a clinical cancer. It is shown, using cohort simulations with parameters appropriate for lung adenocarcinomas, that incorporating such processes can dramatically lengthen predicted latency periods. Such long latency periods together with data on timing of radiation-induced cancers suggest that radiation may influence progression itself.
Similar content being viewed by others


Introduction
The importance of tumor progression
Tumor progression is a late step in carcinogenesis, during which previously transformed, neoplastic cells may proliferate, become more malignant and evolve into cancer that presents the first clinical symptoms. In radiation risk-estimation, early steps such as initiation, promotion and transformation have been extensively investigated (Luebeck and Hazelton 2002; Moolgavkar and Luebeck 2003; Sachs et al. 2005; Heidenreich et al. 2007; Little et al. 2008). Progression, however, has as yet received less attention. Indeed, current quantitative radiobiology models often approximate progression as merely a fixed time lag from the appearance of the first malignant cell until clinical cancer incidence.
This deterministic fixed lag time approximation overlooks important mechanisms underlying progression, which may significantly affect risk assessment. After transformation has produced a malignant cell, this lesion needs to progress through various phases before reaching a clinical stage. First, it needs to escape extinction in a stochastic birth–death proliferation process. If the lesion does start to expand, immune suppression or lack of neo-vascularization may arrest or drastically slow its clone’s growth, leading to a dormant microscopic tumor (Aguirre-Ghiso 2007; Kim et al. 2007; Koebel et al. 2007). A subsequent switch from the dormancy phase to aggressive proliferation may take years or even be postponed indefinitely (Naumov et al. 2008). Hence, progression duration may be a major part of the latency period between radiation and clinical cancer.
The effects of stochastic proliferation of malignant lesions has been addressed and discussed in various ways but quantitative analyses have been limited to theoretical formalisms or to modeling rodent experiments (Dewanji et al. 1991; Yang and Chen 1991; Luebeck and Moolgavkar 1994; Tan and Chen 1998; Smith and Portier 2000). Recently, we analyzed the effect of stochastic extinction on progression distributions and hazard functions for simulated cohorts based on atomic bomb survivor data (Fakir et al. 2009). All these studies analyzed only a single aspect of progression, early stochastic proliferation of malignant cells, leading to extinction or clonal growth. We suggest that more investigations of progression, especially of tumor dormancy, are needed for the following reasons
-
1.
There is strong evidence that microscopic tumors are commonly present in adults in the form of dormant lesions (Black and Welch 1993). In such cases, progression is presumably delayed or arrested by nutrient or oxygen deprivation, signals from the microenvironment and interactions with associated stromal, immune system, or endothelial cells (Aguirre-Ghiso 2007).
-
2.
Progression is the one relevant process where extensive human data are directly available. Indeed, advances in molecular technologies are continuously providing more insights into the pathology of the tumors and the process of their development in the asymptomatic state or after clinical detection (Bunn 2002; Liu et al. 2008). In addition, continuing screening programs and measurements of sojourn time, the interval during which the disease does not produce symptoms leading to diagnosis but is already detectable by screening tests, are providing more precise and quantitative data (Chien and Chen 2008; Chien et al. 2008).
-
3.
Reactivation of dormant lesions occurs after other perturbations, e.g. surgery (Veronesi et al. 1995; Udagawa 2008) and trauma (El Saghir et al. 2005; Naumov et al. 2009). Recurrent cancer following treatment is reviewed in (Naumov et al. 2008). Radiation as well can act directly on progression for example by altering angiogenesis or immune responses [reviewed, e.g. in (Cunha et al. 2003; Folkman and Kalluri 2004; Ohuchida et al. 2004)]. Clinical cancer presentation for radiation-induced reactivation of a pre-existing dormant neoplasm is likely to occur earlier than for radiation initiation, promotion, or transformation. Hence, radiation perturbations of progression may be especially important both in risk estimation and for prevention or amelioration measures.
The two-stage clonal expansion carcinogenesis model
The most commonly used biologically based approach to quantitative cancer modeling and radiation risk estimation is the two-stage clonal expansion (TSCE) model (Moolgavkar and Venzon 1979; Luebeck and Hazelton 2002). Here, we analyze radiation-influenced carcinogenesis with the TSCE model augmented by a more detailed treatment of progression.
The TSCE model assumes that cancer results from normal stem cells after two sequential stages in a process comprising four steps (Fig. 1a). The steps can be described as follows. Initiation: a step in which normal stem cells acquire alterations such as mutations and thereby become stage 1 cells, referred to as pre-malignant cells. Promotion: a second step where pre-malignant cells proliferate with a slight growth advantage over the surrounding normal stem cells. Transformation: a step in which a pre-malignant cell acquires an additional alteration and becomes a stage 2 (i.e. malignant) cell. Progression: a final step leading from malignant cells to clinical cancer. Promotion and progression may last years or decades, while one initiation or transformation event is comparatively very rapid. In the standard TSCE model, progression is often approximated by a fixed relatively short lag time (5–10 years) (Luebeck et al. 1999; Moolgavkar and Luebeck 2003; Jacob et al. 2007). Here, we assume instead that progression can be affected by complex processes involving stochastic cell dynamics, tissue level systems effects, and intercellular interactions. Even when more than two stages are assumed, multi-stage models for radiogenic carcinogenesis usually assume the last step is automatic. A formalism similar to our progression model in principle could, and in our opinion probably should, be used to replace this last step.

Carcinogenesis steps a the standard two-stage clonal expansion (TSCE) model for overall carcinogenesis. Initiation rapid alteration that produces a pre-malignant cell from the pool of normal stem cells; promotion stochastic proliferation of the pre-malignant cells; transformation a second rapid alteration which generates a malignant cell from the pool of pre-malignant cells; progression occurs during the time from the first malignant cell to clinical cancer. b More realistic model of the progression step. After transformation, a lesion may need to progress through bottlenecks, including stochastic extinction and/or dormancy, in order to generate a clinical cancer. If more than one malignant cell is formed by transformation, the different clones evolve independently of each other. For example, the second malignant cell could lead to the first clinical cancer if the first malignant clone becomes extinct, remains dormant indefinitely, or happens to grow slowly
Progression: early proliferation of malignant cells
Primary malignant lesions are subject to a stochastic birth–death process and hence may become extinct soon after their formation, in which case clinical cancer can only arise from malignant cells formed later. In a simulation of lung cancer in atomic bomb survivors, we have previously shown that progression probability distributions for the lag time from first malignant cell to first invasive tumor can extend to more than 50 years (Fakir et al. 2009). While modeling stochastic growth of malignant cells considerably improves the understanding of tumor progression, it may not entirely explain large tumor latency periods and discrepancies between observed microscopic tumors and clinically observed cancers.
Progression: dormancy and invasiveness
Non-extinct malignant clones or microscopic neoplastic lesions may persist in an asymptomatic state for years or decades (Aguirre-Ghiso 2007). This behavior has been shown for primary malignant tumors, for micrometastases and for residual disease, after treatment (Udagawa 2008). A corroboration of these phenomena is the observation that the frequency of microscopic neoplastic lesions in adults is much larger than the frequency of invasive cancers (Black and Welch 1993; Folkman and Kalluri 2004).
These large delays are generally attributed mainly to tumor dormancy. The specific features of dormancy may vary; they depend on the phase in tumor growth and the underlying mechanisms. In fact, dormancy can be related to single dormant cells subject to cell cycle arrest or to microscopic dormant tumors that are in a state of balanced apoptosis and proliferation resulting in no net increase in tumor mass (Aguirre-Ghiso 2007; Naumov et al. 2008; Udagawa 2008). In the second case, dormancy is usually due to immune suppression or the lack of neovascularization (Naumov et al. 2008). In the following, we refer to dormancy as the phase in the progression step where the growth of microscopic malignant lesions is inhibited. It can be considered as a dynamic equilibrium from which tumors may re-emerge when that equilibrium is disrupted. If the tumor escapes dormancy, it resumes growing at a net per-cell growth rate comparable with the pre-dormancy rate (Naumov et al. 2006b).
Methods
In order to account for accidental extinction of malignant clones and tumor dormancy, we augmented the TSCE model (Fig. 1a) with a progression model involving a sequence of three successive phases (Fig. 1b): 1, early stochastic proliferation of malignant cells that leads either to clonal extinction or to dormancy; 2, a stochastic dormancy duration that depends on tumor type and site and can, in some cases, continue indefinitely; 3, invasiveness during which the tumor resumes growing and causes the first symptoms. Parameter choices were taken to be those appropriate for lung adenocarcinomas. We next describe methods for analyzing these three phases of progression, then implementation of the augmented model, then the parameter choices.
Progression phase 1: initial birth–death process
Non-extinct malignant lesions grow first exponentially and homogeneously, being mainly composed of proliferative cells. Then because of limited resources (number of capillaries in the tissue and oxygen diffusion within the tumor), some cells may become hypoxic, anoxic, or necrotic as the volume of the tumor increases (Adam and Maggelakis 1990). The tumor then shows a certain degree of heterogeneity in its composition, its growth gradually slows and its volume converges to a limiting value, usually called carrying capacity. The growth of tumors is thus better described by S-shaped curves of non-homogeneous systems (see e.g. Gyllenberg and Webb 1990; Michelson and Leith 1997; Sachs et al. 2001; Albano and Giorno 2006). This limited growth may, actually, occur at different levels of tumor proliferation controlled by a sequential increase of the carrying capacity. Indeed, Speer et al. (1984) showed that stepwise growth offers a better representation of in vivo data for tumor growth (Speer et al. 1984). In our study, microscopic dormancy is assumed to be a first pause in tumor proliferation. Growth profiles at the macroscopic scale (after clinical presentation) are not of direct interest here.
In our study, tumor growth in phase 1 of progression is described by a stochastic logistic model with birth rate b(t) and death rate d(t) given by
where n(t) is a random function representing the number of cells in a malignant clone; n(t) jumps among various non-negative integers as the clone grows or shrinks; b 0 and d 0 , with b 0 > d 0 , are positive constants representing the maximum birth and death rates, respectively. The positive constant C in Eq. 1 is the carrying capacity. The carrying capacity corresponds to the number of cells in a dormant tumor. In a deterministic formalism n(t) would approach but never reach C; in the stochastic logistic process n(t) eventually reaches either values ≥C (dormant tumor) or 0 (extinction) (Tan 2002).
Progression phase 2: dormancy
Once the number of cells reaches or exceeds the carrying capacity C, the tumor is here considered dormant and modeled as a single unit. Dormant tumors are taken to progress according to a Markov process to permanent suppression or to invasiveness (escape from growth inhibition). The time of dormancy can then be approximated as a stochastic variable which follows an exponential distribution that depends on one parameter, the waiting time. The waiting time is determined by angiogenesis, immune system interactions, the tumor location, and other factors.
Progression phase 3: invasion
The growth of the invasive state is taken to be deterministic, an approximation valid to high accuracy in view of the large number of cells at this stage. The growth time from a dormant lesion to a clinically observable tumor is approximated using a simple exponential growth model.
Implementation of the augmented model
The TSCE model is implemented by Monte-Carlo instead of analytic methods, in order to fit smoothly with the Monte-Carlo progression calculations. The Monte Carlo program, based on earlier work (Fakir et al. 2009), simulates the full stochastic carcinogenesis process, including progression analyzed with our stochastic progression model described previously. Radiation is represented by an additive instantaneous dose-linear initiation term. Thus, the initiation rate is expressed as \( \alpha_{I} (t) = \alpha_{IB} + \alpha_{IR} D\delta (t - e) \), where α IB is an adjustable background initiation rate constant, α IR is an adjustable constant representing the initiation probability per unit radiation dose, D is the radiation dose, e is age at exposure, and δ(t − e)is the Dirac delta function. We did not explicitly consider more complex models where radiation acts also on promotion, transformation or progression itself because the model with radiation only perturbing initiation already gives considerable insight into the later progression mechanisms of interest.
The Monte Carlo progression calculation proceeds in small steps of size Δt. For phase 1 of progression, the numbers of births and deaths during Δt are generated from a multinomial distribution with probabilities b(t)Δt and d(t)Δt, respectively. If a tumor then reaches dormancy, it is added to the pool of dormant tumors. At each time step Δt, a multinomial distribution is used to select the numbers of permanently suppressed and reactivated dormant tumors with probabilities αΔt and βΔt, respectively. Finally, if a dormant tumor is reactivated, its subsequent growth is modeled simply as exponential growth up to a macroscopic or clinically observable size. Fig. 2 displays the transition parameters and growth rates of the progression step.

Progression parameters. The figure shows the same boxes as in Fig. 1b, specifying the clone states that are used in our model to analyze progression processes. Labels on the arrows specify the parameters relevant to transitions between these states. The parameters are fixed by experimental (not epidemiological) data and are described in more detail in the Methods section. The transition from a malignant cell to a harmless clone (shown by the downward arrow; it is actually an extinct clone in this case) is governed mainly by the maximum death rate d0 and birth rate b 0, with carrying capacity C playing almost no role. In the transition from dormant tumor to clinical cancer, only the difference SGR ≡ b 0 − d 0 is relevant, not b0 and d0 seperately. In our calculations here, α was taken as 0
Parameter values
The simulated cohort contains 105 individuals. Based on measurements of Mercer et al. (1994), we assume that the number of stem cells (basal and secretory) in the lung is 4 × 109. Their birth rate is assumed equal to 12 per year (NRC 1999). A tumor may give rise to the first symptoms and be first diagnosed at volume of ~1 ccm that contains ~109 cells (Friberg and Mattson 1997). It is assumed here that lung cancer behaves totally as lung adenocarcinoma. Growth kinetics and volume doubling time have been measured in vivo for early stage lung adenocarcinoma using low-dose spiral computed tomography by Wang and colleagues (Wang et al. 2000). However, Mehrara et al. (2007) have shown that the specific growth rate (SGR) is a more suitable quantity for the description of tumor growth kinetics. The SGR represents the volume growth rate of the tumor in an exponential growth model. The corresponding SGR is equal to 2.37 per year (Mehrara et al. 2007). This growth rate implies that exponential deterministic growth of a malignant cell would produce a clinical cancer in about 8.7 years. It has been suggested that tumor growth is due to a reduction in cell loss, while birth rates remain similar in normal tissues and tumors (Rew and Wilson 2000). Accordingly, it is assumed here that tumor growth is due to a reduction in cell death rates, while birth rates remain similar to the birth rates in normal tissues. Therefore, the death rate d 0 for lung adenocarcinomas is 9.63 per year. A dormant tumor is generally assumed to have a diameter of ~1 mm, and the number of cells equal to approximately 106 (Naumov et al. 2006a). Despite a substantial literature about tumor dormancy, very few papers present quantitative data about dormancy in human cancer. For simplicity, we here set the permanent suppression rate, α, to 0. The rate of invasiveness, β, is set equal to 0.26 per year based on a mean sojourn time (MST) for lung cancer equal to 3.9 years (95% CI: 3.42–3.99) (Chien and Chen 2008). The MST represents the average duration of the preclinical screen detectable phase. The data on which the calculation of the MST is based consist of a large screening study of asymptomatic individuals at risk for lung cancer using low-dose computed tomography (Henschke et al. 2006). Following our earlier discussion, we assume that tumors grow with the same rate before and after dormancy. The time between the reactivation of dormant tumors and clinically observable tumors is then 2.9 years based on a deterministic exponential growth with a SGR equal to 2.37 per year. Progression is thus characterized in our study by independently measured tumor growth kinetics and involves no parameters that can be adjusted to fit epidemiological data. In contrast, in more standard versions of the TSCE model only one progression parameter, the lag time, is used but this parameter is adjusted to the epidemiological data.
The remaining parameters are those of the standard TSCE model and were determined in order to have the overall model reproduce lung cancer data for atomic bomb survivors based on the most recent life span study (LSS) 1958–1998 (Preston et al. 2007). The purpose of using the LSS data was not to give optimal fits to the atomic bomb survivor data but rather to obtain a set of reasonable parameters with which to analyze the properties of the progression step in an overall, combined model. Detailed descriptions of the cohort simulation procedure and the analysis methods are given in (Fakir et al. 2009). The adjusted TSCE parameter values are the spontaneous initiation rate α IB = 9.5 × 10−11 year−1, the radiation initiation rate α IR = 8 × 10−8 G year−1, the transformation rate α T = 3.9 × 10−6 year−1, and birth and death rates of pre-malignant cells, b P and d P , which are 12 and 11.85 year−1, respectively. In our case, all these parameters are in principle separately identifiable.
Results
In the simulated cohort, only 51% of the individuals who developed at least one malignant cell presented clinical cancers. Figure 3 shows the incidence probabilities for both the first malignant cells and clinical cancers. At early and middle ages individuals are more likely to build up malignant clones that may or may not continue through all the progression phases. Later the persistent lesions show clinical cancers, while the probability of producing a malignant cell for the first time decreases. A clinical cancer incident at 70 years for instance may have resulted from a malignant cell that has been produced at an age of 20 years.

Probability density distributions of time of first occurrence of primary malignant cells and clinical tumors in a heterogeneous cohort mimicking the atomic bomb survivor cohort. Competing risks are not taken into account. Tumor growth parameters correspond to lung adenocarcinoma. The other parameters are adjusted in order to simulate clinically observed lung cancer incidence. Fluctuations are due to using Monte-Carlo methods
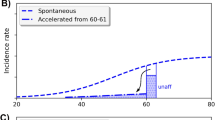
In order to compare our stochastic progression model to the deterministic approximation, we calculated the lag time distributions from the first malignant transformation to clinical cancer (Fig. 4). For stochastic-exponential growth without dormancy, the first peak represents the cases where the first malignant cell escaped stochastic extinction and progressed to the clinical (symptomatic) phase. For stochastic-logistic growth followed by dormancy, the first peak represents cases where the first malignant cells produced escaped stochastic extinction and had only short dormancy. The width of the first peak depends on the birth and death rates, and its location represents the mean time needed for proliferation (for exponential growth) or proliferation plus escaping dormancy plus post-dormancy exponential growth (for logistic growth). The broader, second peak represents cases where the first malignant clone becomes extinct and clinical cancers are generated from a malignant cell produced later. Dormancy shifts the distribution by about 6 years and considerably flattens it. The difference in distributions between the solely exponential growth and the growth with dormancy is due to three components: the logistic growth model, the dormancy period and the deterministic re-growth period after “awakening” from dormancy. The logistic shape has only a minor effect on the latency period. The six-year shift is mainly due to the dormancy period (mean 3.9 years) and the deterministic re-growth (2.9 years). As discussed in our earlier work (Fakir et al. 2009), variations in the rates for processes that precede progression (i.e. initiation, promotion and transformation rates) may indirectly but significantly affect our estimated lag time distributions due to the way in which we fit the epidemiological data. Figure 4 shows the substantial effect of varying a promotion parameter, the death rate d P of pre-malignant cells. In contrast, the lag time distributions don’t show any significant dependence on the background initiation rate (results not shown here).

Stochastic progression lag time distributions (first malignant cell to first clinical tumor) for two values of the pre-malignant cell death rate d p . For comparison, the figure also shows the lag time distribution when stochastic exponential malignant cell proliferation without dormancy is assumed and shows the delta-function distribution for a 10-year deterministic lag time model. Tumor growth parameters during progression correspond to lung adenocarcinoma; for other kinds of lung cancer (results shown in [Fakir et al. (2009) but not in the present paper] the first peak of the curve assuming no dormancy is considerably further to the left. The parameters for earlier steps (initiation, promotion, and transformation) are adjusted in order to simulate lung cancer incidence in atomic bomb survivors
For specific exposure conditions and parameter values, it is of interest to calculate the frequency distributions of tumors for an individual at a given age. Such distributions may be useful for studying potential effects of environmental factors on progression, and especially on malignant microscopic disease and its evolution into symptomatic cancer. As an illustration, Fig. 5 shows the frequency distributions of malignant transformations and clinical symptomatic tumors for age 90 based on the present parameters. In our example, it is found that by age 90 an individual who has never developed a clinical cancer has on average developed 48 separate malignant transformations but would present only 1.2 dormant tumors.

Predicted distribution of the number of malignant transformations and of clinical tumors that occur in the lifetime of a non-irradiated individual of age 90 years assuming the parameters computed here and no competing risks
Conclusion
In many calculations of radiation risks using the TSCE model, the production of the first malignant cell inevitably gives cancer after a fixed lag time. Some studies have addressed the issues of progression and the fixed lag time assumption. For instance, Hazelton et al. have analyzed progression in the TSCE model by replacing the fixed lag time by a gamma distribution that considerably improved risk estimation and predicted larger lag times (Hazelton et al. 2001; 2006). In our work, we aimed to give a biologically motivated quantitative description of the progression step, involving tumor proliferation mechanisms and observed phenomena. We aimed to help bridge the gap between radiation biology modeling based on experimental and epidemiological studies, and the clinical aspects of carcinogenesis: at present clinical observations tell us more about progression than about earlier carcinogenesis steps.
Here, we argued that the progression step and the ability of malignant cells to proliferate, escape extinction, and escape dormancy are at least as important as the earlier steps. Progression may constitute a much larger portion of the evolution time of cancer than is often assumed in radiobiological models. In agreement with observations, our model predicts that an individual may contain a significant number of dormant tumors.
Other calculations (not shown here), e.g. for faster growing tumors, give somewhat smaller progression times (Fakir et al. 2009). However, in view of the long progression times estimated here, the data on timing of extra cancers in the atomic bomb survivors and on radiotherapy patients (Little et al. 1999; Preston et al. 2007) indicate that radiation probably acts directly on promotion, on transformation, and even on progression itself. If radiation action was confined to initiation in Fig. 1a, there would not be enough time for the extra radiogenic lesions to go through the subsequent phases prior to observed extra clinical cancers. Instead, clinical cancers that appear within ~10 years of the irradiation may result from reactivation of dormant tumors, other progression acceleration, or malignant transformation of already present pre-malignant cells that then undergo a comparatively rapid progression (the first peak in Fig. 4). If confirmed, radiation action directly on progression would have major implications for radiation risk estimation and protective measures.
Incorporating progression details complicates the analysis of cancer development (Enderling et al. 2009) and introduces additional parameters. Hence, interdisciplinary studies are needed to better comprehend the different aspects of the problem. Cancer screening studies can give valuable information about carcinogenesis. Studies of second cancers after fractionated radiotherapy are done under controlled exposure conditions and with good knowledge of tumor incidence in each patient. Autopsies can uncover microscopic tumors. Such clinical studies can supplement the usual analyses of radiogenic carcinogenesis, based on data from in vitro or animal experiments and epidemiological data (on atomic bomb survivors and occupationally or environmentally exposed cohorts). The clinical data are relevant to later steps of carcinogenesis, especially progression. Thus, epidemiological data, the possibility of radiation influencing progression, and the need to incorporate increasingly available clinical data all point to the importance of considering detailed progression scenarios in mathematical/computational carcinogenesis modeling.
References
Adam JA, Maggelakis SA (1990) Diffusion regulated growth characteristics of a spherical prevascular carcinoma. Bull Math Biol 52:549–582
Aguirre-Ghiso JA (2007) Models, mechanisms and clinical evidence for cancer dormancy. Nat Rev Cancer 7:834–846
Albano G, Giorno V (2006) A stochastic model in tumor growth. J Theor Biol 242:329–336
Black WC, Welch HG (1993) Advances in diagnostic imaging and overestimations of disease prevalence and the benefits of therapy. N Engl J Med 328:1237–1243
Bunn PA Jr (2002) Molecular biology and early diagnosis in lung cancer. Lung Cancer 38:S5–S8
Chien CR, Chen TH (2008) Mean sojourn time and effectiveness of mortality reduction for lung cancer screening with computed tomography. Int J Cancer 122:2594–2599
Chien CR, Lai MS, Chen TH (2008) Estimation of mean sojourn time for lung cancer by chest X-ray screening with a Bayesian approach. Lung Cancer 62:215–220
Cunha GR, Hayward SW, Wang YZ, Ricke WA (2003) Role of the stromal microenvironment in carcinogenesis of the prostate. Int J Cancer 107:1–10
Dewanji A, Moolgavkar SH, Luebeck EG (1991) Two-mutation model for carcinogenesis: joint analysis of premalignant and malignant lesions. Math Biosci 104:97–109
El Saghir NS, Elhajj II, Geara FB, Hourani MH (2005) Trauma-associated growth of suspected dormant micrometastasis. BMC Cancer 5:94
Enderling H, Anderson ARA, Chaplain MAJ, Beheshti A, Hlatky L, Hahnfeldt P (2009) Paradoxical dependencies of tumor dormancy and progression on basic cell kinetics. Cancer Res: 0008-5472.CAN-09-2115
Fakir H, Tan WY, Hlatky L, Hahnfeldt P, Sachs RK (2009) Stochastic population dynamic effects for lung cancer progression. Radiat Res 172:383–393
Folkman J, Kalluri R (2004) Cancer without disease. Nature 427:787
Friberg S, Mattson S (1997) On the growth rates of human malignant tumors: implications for medical decision making. J Surg Oncol 65:284–297
Gyllenberg M, Webb GF (1990) A nonlinear structured population model of tumor growth with quiescence. J Math Biol 28:671–694
Hazelton WD, Luebeck EG, Heidenreich WF, Moolgavkar SH (2001) Analysis of a historical cohort of Chinese tin miners with arsenic, radon, cigarette smoke, and pipe smoke exposures using the biologically based two-stage clonal expansion model. Radiat Res 156:78–94
Hazelton WD, Moolgavkar SH, Curtis SB, Zielinski JM, Ashmore JP, Krewski D (2006) Biologically based analysis of lung cancer incidence in a large Canadian occupational cohort with low-dose ionizing radiation exposure, and comparison with Japanese atomic bomb survivors. J Toxicol Environ Health A 69:1013–1038
Heidenreich WF, Cullings HM, Funamoto S, Paretzke HG (2007) Promoting action of radiation in the atomic bomb survivor carcinogenesis data? Radiat Res 168:750–756
Henschke CI, Yankelevitz DF, Libby DM, Pasmantier MW, Smith JP, Miettinen OS (2006) Survival of patients with stage I lung cancer detected on CT screening. N Engl J Med 355:1763–1771
Jacob P, Meckbach R, Sokolnikov M, Khokhryakov VV, Vasilenko E (2007) Lung cancer risk of Mayak workers: modelling of carcinogenesis and bystander effect. Radiat Environ Biophys 46:383–394
Kim R, Emi M, Tanabe K (2007) Cancer immunoediting from immune surveillance to immune escape. Immunology 121:1–14
Koebel CM, Vermi W, Swann JB, Zerafa N, Rodig SJ, Old LJ, Smyth MJ, Schreiber RD (2007) Adaptive immunity maintains occult cancer in an equilibrium state. Nature 450:903–907
Little MP, Muirhead CR, Haylock RG, Thomas JM (1999) Relative risks of radiation-associated cancer: comparison of second cancer in therapeutically irradiated populations with the Japanese atomic bomb survivors. Radiat Environ Biophys 38:267–283
Little MP, Heidenreich WF, Moolgavkar SH, Schöllnberger H, Thomas DC (2008) Systems biological and mechanistic modelling of radiation-induced cancer. Radiat Environ Biophys 47:39–47
Liu P, Sun J, Guan Y, Yue W, Xu LX, Li Y, Zhang G, Hwu Y, Je JH, Margaritondo G (2008) Morphological study of early-stage lung cancer using synchrotron radiation. J Synchrotron Radiat 15:36–42
Luebeck EG, Hazelton WD (2002) Multistage carcinogenesis and radiation. J Radiol Prot 22:A43–A49
Luebeck EG, Moolgavkar SH (1994) Simulating the process of malignant transformation. Math Biosci 123:127–146
Luebeck EG, Heidenreich WF, Hazelton WD, Paretzke HG, Moolgavkar SH (1999) Biologically based analysis of the data for the Colorado uranium miners cohort: age, dose and dose-rate effects. Radiat Res 152:339–351
Mehrara E, Forssell-Aronsson E, Ahlman H, Bernhardt P (2007) Specific growth rate versus doubling time for quantitative characterization of tumor growth rate. Cancer Res 67:3970–3975
Mercer RR, Russell ML, Roggli VL, Crapo JD (1994) Cell number and distribution in human and rat airways. Am J Respir Cell Mol Biol 10:613–624
Michelson S, Leith JT (1997) Positive feedback and angiogenesis in tumor growth control. Bull Math Biol 59:233–254
Moolgavkar SH, Luebeck EG (2003) Multistage carcinogenesis and the incidence of human cancer. Genes Chromosomes Cancer 38:302–306
Moolgavkar HS, Venzon JD (1979) Two-event models for carcinogenesis: incidence curves for childhood and adult tumors. Math Biosci 47:55–77
Naumov GN, Akslen LA, Folkman J (2006a) Role of angiogenesis in human tumor dormancy: animal models of the angiogenic switch. Cell Cycle 5:1779–1787
Naumov GN, Bender E, Zurakowski D, Kang SY, Sampson D, Flynn E, Watnick RS, Straume O, Akslen LA, Folkman J, Almog N (2006b) A model of human tumor dormancy: an angiogenic switch from the nonangiogenic phenotype. J Natl Cancer Inst 98:316–325
Naumov GN, Folkman J, Straume O, Akslen LA (2008) Tumor-vascular interactions and tumor dormancy. Apmis 116:569–585
Naumov GN, Folkman J, Straume O (2009) Tumor dormancy due to failure of angiogenesis: role of the microenvironment. Clin Exp Metastasis 26:51–60
NRC (1999) The health effects of exposure to indoor radon. Biological effects of ionizing radiation (BEIR) VI report. National Research Council, Washington, DC
Ohuchida K, Mizumoto K, Murakami M, Qian LW, Sato N, Nagai E, Matsumoto K, Nakamura T, Tanaka M (2004) Radiation to stromal fibroblasts increases invasiveness of pancreatic cancer cells through tumor-stromal interactions. Cancer Res 64:3215–3222
Preston DL, Ron E, Tokuoka S, Funamoto S, Nishi N, Soda M, Mabuchi K, Kodama K (2007) Solid cancer incidence in atomic bomb survivors: 1958–1998. Radiat Res 168:1–64
Rew DA, Wilson GD (2000) Cell production rates in human tissues and tumours and their significance. Part II: clinical data. Eur J Surg Oncol 26:405–417
Sachs RK, Hlatky LR, Hahnfeldt P (2001) Simple ODE models of tumor growth and anti-angiogenic or radiation treatment. Math Comput Model 33:1297–1305
Sachs RK, Chan M, Hlatky L, Hahnfeldt P (2005) Modeling intercellular interactions during carcinogenesis. Radiat Res 164:324–331
Smith MV, Portier CJ (2000) Incorporating observability thresholds of tumors into the two-stage carcinogenesis model. Math Biosci 163:75–89
Speer JF, Petrosky VE, Retsky MW, Wardwell RH (1984) A stochastic numerical model of breast cancer growth that simulates clinical data. Cancer Res 44:4124–4130
Tan WY (2002) Stochastic models with applications to genetics, cancers AIDS and other biomedical systems. World Scientific Publishing Company, New Jersey
Tan WY, Chen CW (1998) Stochastic modeling of carcinogenesis: some new insights. Mathl Comput Modeling 28:49–71
Udagawa T (2008) Tumor dormancy of primary and secondary cancers. Apmis 116:615–628
Veronesi U, Marubini E, Del Vecchio M, Manzari A, Andreola S, Greco M, Luini A, Merson M, Saccozzi R, Rilke F (1995) Local recurrences and distant metastases after conservative breast cancer treatments: partly independent events. J Natl Cancer Inst 87:19–27
Wang JC, Sone S, Feng L, Yang ZG, Takashima S, Maruyama Y, Hasegawa M, Kawakami S, Honda T, Yamanda T (2000) Rapidly growing small peripheral lung cancers detected by screening CT: correlation between radiological appearance and pathological features. Br J Radiol 73:930–937
Yang GL, Chen CW (1991) A stochastic two-stage carcinogenesis model: a new approach to computing the probability of observing tumor in animal bioassays. Math Biosci 104:247–258
Acknowledgments
This research was supported by NASA NSCOR04-0014-0017. We are grateful to Drs. W. Y. Tan, P. Hahnfeldt and L. Hlatky for many detailed discussions. This report makes use of data obtained from the Radiation Effects Research Foundation (RERF) in Hiroshima, Japan. RERF is a private foundation funded equally by the Japanese Ministry of Health, Labour, and Welfare and the U.S. Department of Energy through the U.S. National Academy of Sciences. The data include information obtained from the Hiroshima City, Hiroshima Prefecture, Nagasaki City, and Nagasaki Prefecture Tumor Registries and the Hiroshima and Nagasaki Tissue Registries. The conclusions in this report are those of the authors and do not necessarily reflect the scientific judgment of RERF or its funding agencies.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
This paper is based on a presentation given at the International Conference on Late Health Effects of Ionizing Radiation, 4–6 May 2009, Georgetown University, Washington DC, USA.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Fakir, H., Hofmann, W. & Sachs, R.K. Modeling progression in radiation-induced lung adenocarcinomas. Radiat Environ Biophys 49, 169–176 (2010). https://doi.org/10.1007/s00411-009-0264-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00411-009-0264-6