
Abstract
TMEM106B is a risk modifier of multiple neurological conditions, where a single coding variant and multiple non-coding SNPs influence the balance between susceptibility and resilience. Two key questions that emerge from past work are whether the lone T185S coding variant contributes to protection, and if the presence of TMEM106B is helpful or harmful in the context of disease. Here, we address both questions while expanding the scope of TMEM106B study from TDP-43 to models of tauopathy. We generated knockout mice with constitutive deletion of TMEM106B, alongside knock-in mice encoding the T186S knock-in mutation (equivalent to the human T185S variant), and crossed both with a P301S transgenic tau model to study how these manipulations impacted disease phenotypes. We found that TMEM106B deletion accelerated cognitive decline, hind limb paralysis, tau pathology, and neurodegeneration. TMEM106B deletion also increased transcriptional correlation with human AD and the functional pathways enriched in KO:tau mice aligned with those of AD. In contrast, the coding variant protected against tau-associated cognitive decline, synaptic impairment, neurodegeneration, and paralysis without affecting tau pathology. Our findings reveal that TMEM106B is a critical safeguard against tau aggregation, and that loss of this protein has a profound effect on sequelae of tauopathy. Our study further demonstrates that the coding variant is functionally relevant and contributes to neuroprotection downstream of tau pathology to preserve cognitive function.
Similar content being viewed by others

Introduction
TMEM106B was initially discovered as a risk modifier for frontotemporal lobar degeneration (FTLD) with TDP-43 inclusions [41], and has since been linked to elevated risk for multiple other neurodegenerative diseases, including Alzheimer’s disease (AD), chronic traumatic encephalopathy, and cognitive decline in ALS and Parkinson's disease [11]. Despite its broad influence, the precise function of TMEM106B in the healthy brain and how its dysfunction contributes to disease remain elusive.
Research in cell lines localized TMEM106B to the endolysosome, where its overexpression disrupts lysosomal pH, size, and trafficking [11, 19, 31]. Consistent with this deleterious effect, multiple studies have reported higher TMEM106B expression in FTLD cases than controls, leading to the hypothesis that protein reduction might protect against disease [7, 11, 17, 41]. Several additional lines of evidence support this notion. Single nucleotide polymorphisms (SNPs) associated with the risk allele alter chromatin architecture to promote transcription, and artificially increasing TMEM106B levels exacerbates cytotoxicity [16]. Furthermore, TMEM106B was recently found to accumulate in abnormal intracellular fibrils with age and disease [6, 18, 19, 33, 37].
Countering this support for TMEM reduction in forestalling disease, three recent studies investigating TMEM106B deletion in FTLD models reached the opposite conclusion. While TMEM106B deletion by itself had relatively mild consequences on overall fitness [12, 14, 24, 34, 38, 54], its loss profoundly worsened disease phenotypes in GRN deletion mice [13, 46, 53]. These findings suggest that TMEM106B may be indispensable for lysosomal function in the context of disease. They also indicate that absolute protein level may not be the only feature governing the association of TMEM106 sequence with disease risk or resilience.
TMEM106B SNPs are typically inherited together as a single linkage disequilibrium block, where multiple non-coding SNPs associated with disease risk co-segregate with the common coding variant encoding serine at position 185 [16, 41, 43]. Co-inheritance of both coding and non-coding SNPs has posed a challenge in discerning the causal variant. Previous work highlighted a variant in the 3’ UTR as a likely culprit [16], however, this SNP co-segregates with a single coding variant that replaces serine with threonine at residue 185. Although the substitution is biochemically conservative, past work in transfected cells suggested that serine at this site may indeed affect TMEM106B activity [20, 30]. One study suggested that the serine variant reduced TMEM106B half-life, while the other showed that it preserved autophagic flux relative to the threonine variant. To date, only one study has examined the impact of the coding variant in vivo, reporting that the T186S variant had little effect on pathology or microgliosis in a GRN deletion model of FTLD [5]. However, no study to date isolated the contribution of this variant to cognitive function and neural preservation that are fundamental to disease resilience. In this study, we extend our investigation from FTLD-TDP to tauopathy to ascertain whether the lone T185S coding variant contributes to protection, and if the presence of TMEM106B is helpful or harmful in the context of disease. We use a well-studied MAPT P301S model to examine how genetic manipulation of TMEM106B affects cognitive decline, CNS degeneration, and transcriptional signatures of dementia. Our findings not only corroborate a protective role for TMEM106B but also provide the first evidence that the T186S coding variant is functionally relevant and confers neuroprotection in vivo.
Methods
Mice
TMEM106B deletion
Cryopreserved sperm for the line C57BL/6N-Tmem106btm2a(KOMP)Wtsi was purchased from the KnockOut Mouse Project (KOMP) Repository at University of California Davis. This line was created from ES cell clone EPD0047_1_E02, generated by the Wellcome Trust Sanger Institute which was used to generate founder mice by the KOMP Repository (www.komp.org) and the Mouse Biology Program (www.mousebiology.org) at the University of California Davis. The recovered sperm were used for in vitro fertilization of C57BL/6J oocytes by the BCM Genetically Engineered Mouse Core. 75 pups were recovered from 8 implanted females, of which 36 were heterozygous for the targeted allele. The resulting TMEM106B tm2a line was subsequently maintained by backcross to C57BL/6J. Tm2a mice were intercrossed with mice expressing Flp recombinase under control of the β-actin promoter which had been backcrossed to C57BL/6J for 8 generations (kind gift of Russell Ray, BCM, available from Jax as strain #3800 on a mixed background [36]) to generate the conditional deletion tm2c line, which was again maintained by mating with C57BL/6J. Offspring from the tm2c line were mated with mice expressing Cre recombinase under control of the β-actin promoter which had been backcrossed to C57BL/6J for 3 generations (kind gift of Russell Ray, BCM, available from Jax as strain #19,099 on a mixed background [23]) to generate the final tm2d constitutive knockout allele lacking the targeted exon 4 and the floxed β-galactosidase/neomycin resistance cassette. The line was subsequently outcrossed with C3HeJ before being intercrossed with PS19 animals on a C3B6 background. Mice were genotyped by PCR using the following primers: Primer 58 (intron 3 upstream of first FRT site): TTC TCT CCA TGT GCT GCA TTA T, Primer 59 (just before exon 4 but inside the loxP sites; deleted by Cre): GAA TAC TTC TCT CCT TAG CCC TTT AG, Primer 60 (part of loxP site downstream of exon 4): GCG AGC TCA GAC CAT AAC TT. Animals homozygous for TMEM106Btm2d exhibit a band at 304 bp. Wild-type animals exhibit a band at 496 bp, and heterozygous TMEM106Btm2d have bands at 304 bp and 496 bp. TMEM106Btm2d mice will be available through the MMRRC as stock # 71637.
TMEM106B T186S knock-in
The TMEM106B locus was targeted by CRISPR/Cas9 to change threonine 186 to serine within the endogenous mouse TMEM106B locus (homologous to human T185S). 200 C57BL/6J embryos were injected with 100 ng/µL Cas9 mRNA, 20 ng/µL guide RNA, and 100 ng/µL single stranded insert oligonucleotide by the BCM Genetically Engineered Mouse Core. The guide RNA sequence was TTACCTGCTTCATATCAAGT targeting a PAM site at exon 6 of the TMEM106B locus. A single-stranded oligonucleotide was used as the donor template (AATAACTATTATTCTGTTGAAGTTGAAAACATCACTGCTCAAGTCCAGTTTTCAAAAACCGTGATTGGAAAGGCTCGTTTAAACAACATTTCGAACATTGGCCCATTAGATATGAAGCAGGTAAGCCTATTCAGGAGTTGAGGGG) to convert TMEM106B Thr 186 into Ser (ACT > TCG) with two additional silent mutations: I185I (ATA > ATT) was added to introduce a novel BstBI site, and L191L (CTT > TTA) was added to block re-cutting of the correctly edited genome. 209 injected embryos were transferred to pseudopregnant females, yielding 59 animals of which 35 were positive for the T186S allele. Three positive founders were expanded by mating with C57BL/6J wild-type animals. Offspring were screened by Sanger sequencing across the targeted region and by high resolution melting analysis for the top 4 predicted off-target sites; all sites except the intended target came back as wild-type. No differences in brain TMEM106B expression level were observed between the three knock-in lines and so further studies focused on a single line identified as 5825. Sanger sequencing was repeated on tail DNA from 5825 offspring covering an expanded area surrounding the targeting site to include exon 6 plus approximately 50 intronic base pairs. The line was subsequently outcrossed with C3B6 before being intercrossed with PS19 animals on a C3B6 background. All offspring for study were generated from mating TMEM106B T186S heterozygous mice with PS19; TMEM106B T186S heterozygous mates. Animals were genotyped by PCR using the following primers: Primer 46: GTG TGT ATT TCC TGT CTC TGT T; Primer 47: GGG AGA GGA TGA GGG ATT T. All animals should exhibit a band at 407 bp before BstBI restriction digest at 65 °C. T186S animals exhibit a band at 97 bp and 208 bp; WT mice exhibit a band only at 407 bp, and heterozygous mice have 3 bands at 97, 208, and 407 bp. TMEM106B T186S mice will be available through the Jackson Laboratory as stock #39,047.
MAPT P301S transgenic line PS19
B6;C3-Tg(Prnp-MAPT*P301S)PS19Vle/J mice (hereafter referred to as tau or PS19) were obtained from Jackson Laboratory (stock #8169) and maintained on a B6;C3 background. These mice express the 4R1N form of human MAPT encoding the P301S mutation under control of the mouse prion protein promoter as described by Yoshiyama et al. [48]. All animal experiments were reviewed and approved by the Baylor College of Medicine Institutional Care and Use Committee.
Behavioral analysis
Animals began behavioral testing either at approximately 6 or 8 months of age. All animals were screened for paralysis prior to behavioral testing and eliminated from the study if present. Animals that developed hind limb weakness during study were removed from further testing, but were included in tasks completed prior to the onset of paralysis. The number of animals from the 8 mo testing groups that were removed for paralysis at some point between open field (OF) and radial arm water maze (RAWM) was: tau = 2, KO:tau = 3; tau = 4, KI:tau = 2. Additional mice from the 8 mo group could not be tested in RAWM due to the COVID-19 shutdown of our institute in March 2020, leaving group sizes for this task lower than for Morris water maze (MWM) or repeated reversal learning (RRL). Animals were included in analysis for all tasks they completed. Remaining animals underwent the complete behavioral battery at 8 mo: n = 18 WT, 18 KO, 21 tau, 20 KO:tau; n = 18 WT, 21 KI, 22 tau, 21 KI:tau, divided roughly evenly between sexes. The 6 mo cohort included 10 tau and 10 KO:tau animals (4F/6 M); all 6 mo animals completed the full behavioral battery. One KO:tau animal from the 8 mo cohort was subsequently removed from all analyses after it was discovered that this animal showed signs of visual impairment during visible platform testing.
Animals were handled for 5 min/d for 2 d prior to testing. Behavioral analysis began with open field (OF) on day 1, followed by straight swim on day 2, Morris water maze (MWM) on days 3–7 with final probe trial on day 10, followed by repeated reversal (RR) testing on days 10–12, visible platform water maze testing on day 13, and ending with RAWM on day 14 with final probe trial on day 17. Both male and female mice were used for testing in roughly equal numbers for all genotypes. All analyses were done using ANY-maze video tracking system (Version 4.99v, Stoelting Co.). Methods for OF, MWM, and RAWM are modified from Fowler et al. [15]. Animals were performed by a single experimenter blinded to genotype.
Open field
Testing was done in white acrylic open-top boxes (46 × 46 × 38 cm) in a room with indirect with fluorescent lighting to lower the brightness. Activity was recorded with an overhead camera for a single 30 min session.
Morris water maze
MWM testing was conducted in a circular tank measuring 58 cm high and 122 cm in diameter. The water level was 20 cm from the top of the tank and made opaque using nontoxic white paint. Water temperature was maintained between 19 to 21 °C. As described in Fowler et al., before the start of acquisition training, mice received 1 d of training in a straight swim channel to acclimate them to the water and check for motor deficits. Mice received four trials with a 15 min intertrial interval (ITI) in a channel constructed of white acrylic and measuring 107 × 56 × 14 cm, which was placed in the center of the pool. Visible cues were removed from the room during straight swim shaping trials. Mice were allowed 60 s to reach a submerged platform on the opposite side of the channel. Mice that failed to reach the platform were guided to the location by the experimenter. Mice were allowed to stay on the platform for 15 s before being removed from the water, dried, and returned to their home cage placed on a heating pad between trials.
Acquisition training in the MWM consisted of four trials per day with a 15–20 min ITI. A square platform (10 × 10 cm) covered in nylon mesh for traction was located 1 cm below the surface in the NE quadrant of the maze, half-way between the side and the center of the pool. Mice were placed in the maze facing the wall at each of four randomized, cardinal start locations and allowed 60 s to locate the hidden platform utilizing visible cues around the arena. As with straight swim, animals that failed to locate the platform in the allotted time were gently guided there by the experimenter. Mice were allowed to stay on the platform for 15 s before being returned to their home cage between trials. Mice underwent training for 5 consecutive days. On MWM days 2–5, a probe trial was done prior to the day’s 4 training trials, in which the platform was removed from the maze, and animals were placed in the tank for 45 s to navigate the maze. Training ended after 5 days. A final probe trial was conducted 3 d later to test long-term memory for the trained location.
Repeated reversals water maze
Following the final 3 d probe trial in MWM, animals underwent three days of repeated reversal testing. Animals were given 8 training trials per day with a 15–20 min ITI to a novel location: SW, NW, and SE quadrants of the maze. Once training was completed for the specified location, the day began with a probe trial testing recall for the prior platform location. Following the probe trial, the platform was returned to the pool to again test in a novel quadrant.
Visible platform water maze
Following the final 24 h probe trial for repeated reversal learning, all cues were removed from the walls, and the platform was returned to the pool but carrying a striped pole to mark its location. The platform was moved semi-randomly to test each quadrant of the pool twice thus totaling 8 cue trials with a 15–20 min ITI. Animals were placed into the pool from semi-random starting positions and allowed 60 s to arrive to the cued platform where they were allowed to rest for 15 s before being dried and placed back in the warmed home cage.
Radial arm water maze
Visible cues were returned to the walls of the room in the same positions used for MWM and RR. The RAWM was created by installing clear Plexiglas triangular inserts into the existing water maze pool (41 cm on each side × 50 cm high), which resulted in six open arms joined at the center. Each arm measured 20 cm wide × 34 cm long, and the water was maintained at a depth of 38 cm. The static platform was located 3 cm from the end of one arm and submerged 1 cm below the water’s surface. Mice were placed into a different arm at the beginning of each trial, with the order of starting positions pseudo randomly selected before training, such that no trial began in an arm adjacent to the previous start position. Mice were allowed 60 s to navigate the maze. If a mouse failed to locate the platform in the allotted time, it was gently guided there by the experimenter and allowed to remain on the platform for 15 s before being returned to its home cage between trials. Mice underwent 8 training trials in 1 day with a 15 min ITI. Animals underwent a single probe trial 3 days later for long-term memory assessment.
Tissue harvest
For T186S only, mice were euthanized by CO2, and one hemisphere was snap frozen on dry ice at -80 °C until use. For KO x tau and KI x tau animals, mice were euthanized by sodium pentobarbital overdose and transcardially perfused with ice-cold PBS containing 0.5 mM EDTA upon completion of behavioral testing at approximately 9 mo of age. Brains were removed and hemisected along the midline. One hemisphere was snap frozen for biochemistry, and the other hemisphere was immersion fixed in 4% PFA at 4 °C for 48 h. The hemi-sected brains were cryoprotected in 30% sucrose at 4 °C until equilibrated and then frozen in dry ice for histology use.
Western blotting
Tau
Frozen hemispheres were homogenized in 10 vol/wt of RIPA buffer (50 mM Tris, 150 mM NaCl, 5 mM EDTA, 0.5% sodium deoxycholate, 1% NP-40 or Igepal, 0.1% SDS, pH 8.0) containing protease and phosphatase inhibitors (Roche 05892970001 and 04906837001). Homogenates were centrifuged at 15 krpm for 10 min at 4 °C, the supernatant collected, and protein concentration measured by BCA (Fisher, 23227). 30 ug of protein was diluted with Laemmli buffer containing β-mercaptoethanol, heat denatured, loaded onto Criterion 4–15% TGX stain-free gels (BioRad, 5678084), and electrophoresed at RT in Tris–glycine SDS buffer. Gels were transferred to nitrocellulose using the Transblot system (Bio-Rad, 1704271). Blots were blocked for 5–10 min at RT in EveryBlot buffer (BioRad 12010020), and then incubated overnight at 4 °C with primary antibody diluted in blocking buffer (1:500 for all tau epitopes, 1:10,000 for GAPDH). Primary antibodies included: CP27 (human tau 130–150, gift from Peter Davies), AT8 (pSer202, Thr205, ThermoFisher MN1020), PHF-1 (pSer396/404, gift from Peter Davies), AT100 (pThr212,Ser214, ThermoFisher MN1060), AT180 (pThr231, ThermoFisher MN1040), AT270 (pThr181, ThermoFisher MN1050); TAU-5 (total tau, Santa Cruz sc-58860), and Ck anti-GAPDH (Millipore AB2302). The following day, membranes were washed in TBS containing 0.1% Tween-20 (TBSTw) before being incubated for 1 h at RT in secondary antibody diluted 1:5,000 in TBS. Secondary antibodies included Gt anti-Ms IgG 520 (BioRad 12005867) and Dk anti-Ck IgY 680RD (LI-COR 926–68075). Blots were then washed with TBSTw, visualized using a BioRad ChemiDoc MP Imaging System and quantified with Image Lab Software (version 6.1).
TMEM106B
Frozen hemispheres were homogenized in 5 or 10 vol/wt of RIPA buffer containing protease and phosphatase inhibitors as above. Homogenates were centrifuged at 15 krpm for 10 min at 4 °C, the supernatant collected, and protein concentration measured by BCA (Fisher, 23227). 30 or 50 ug of protein was diluted with Laemmli buffer containing β-mercaptoethanol and were loaded without heat denaturation onto Criterion 10–20% Tris–HCl gels (Bio-Rad, 3450043) or Novex 8–16% Tris–glycine plus gels (Fisher WXP81620BOX) and run at RT (Figs. S1, S5b) or 4 °C (Fig. S5c) in Tris–glycine SDS buffer. Gels were transferred to nitrocellulose as above. Blots were blocked for 1 h at RT in PBSTw and 5% non-fat dry milk (Fig. S1, S5b) or for 5–10 at RT in EveryBlot buffer (BioRad 12010020, Fig. S5c). Blots were then incubated overnight at 4 °C with primary antibody diluted in blocking buffer (1:1,000 for Fig. S1, S5b or 1:3000 for Fig. S5c and 1:10,000 for GAPDH). Primary antibodies included: Rb anti-TMEM106B (Invitrogen PA5-34353, Fig. S1), Rb anti-TMEM106B (Sigma HPA058342, Fig. S1, S5b), Rb anti-TMEM106B (Cell Signaling 93334S, Fig S5c), and Ck anti-GAPDH. The following day, membranes were washed in PBSTw or TBSTw before being incubated for 1 h at RT in secondary antibody diluted 1:5,000 in block or TBS. Secondary antibodies included Dk anti-Rb IgG 800 CW (LI-COR 926–32213, Fig. S1, S5b), Gt anti-Rb IgG StarBright Blue 520 (BioRad 12005869, Fig. S5c) and Dk anti-Ck IgY 680RD. Blots were then washed with PBSTw or TBSTw and visualized using an Odyssey Fc Imaging System (LI-COR) followed by quantitation in Image Studio Lite or a BioRad ChemiDoc MP with Image Lab Software.
RNA extraction
Frozen hemi-brains were used to isolate RNA using a PureLink RNA Mini Kit (Invitrogen 12183020) according to the manufacturer’s directions (n = 5 WT, 7 KO, 5 tau, and 7 KO:tau 3 males/group; n = 12 WT, 12 KI, 12 tau, and 12 KI:tau, 6 males/group). In brief, brain hemispheres were homogenized in freshly prepared lysis buffer (0.6 ml per 30 mg of brain tissue) containing 1% β-mercaptoethanol using a TissueRuptor homogenizer (Qiagen). Homogenates were centrifuged at 2,600 × g for 5 min at RT and supernatant was transferred to a clean RNase-free tube. One volume of 70% ethanol was added to the brain homogenate and 700 μl of the sample was transferred to a spin cartridge for RNA binding. This cartridge was centrifuged at 12,000 × g for 15 s and then treated with DNase (Invitrogen 12185010) to obtain a DNA-free RNA sample. After several sequential washes, RNA was eluted with 50 μl of RNase-free water. Purified RNA yield and quality was assessed using a Nanodrop spectrophotometer and stored at − 80 °C until use.
Transcriptomics
Library preparation, sequencing, read quality control, and mapping was performed by Novogene (Beijing, China). Briefly, library construction began by purifying mRNA that was then fragmented. cDNA was then synthesized from random hexamer primers and dTTP. cDNA underwent end repair, A-tailing, adapter ligation, size selection, amplification, and purification. RNA sequencing was performed by on the Illumina platform using paired end 150 bp mRNA sequencing with an expected read depth of 20 M per sample. Data were filtered by removing adapter contamination, reads with uncertain nucleotide percentage above 10%, and reads with low quality scores constituting more than 50% of the read. Reads were mapped using HISAT2 v2.0.5 [28] to mouse genome build mm10. The KO and KI analysis used separate samples arising from each colony and each contained all relevant experimental groups. Samples used in the KI analysis were generated from two different library preparations, and therefore counts from these samples were first normalized for batch effects using the Combat-Seq function in the sva package v3.44.0 with no covariates [51]. Samples from all experimental conditions and both sexes within the KI analysis were distributed evenly between the two batches. Differential gene expression analysis was performed using the edgeR pipeline v3.38.4 [35]. Differentially expressed genes from the KO and KI colonies were generated separately. Transcripts were first filtered to include only protein coding genes, and to further require cpm above 5 in 50% of any experimental group. 15,909 genes were included in analysis for the KO colony and 15,860 for the KI colony. Counts were then normalized to library size using the upper quartile method. Principle component analysis was first performed on normalized expression data to identify potential outlier samples. No samples were removed from downstream analysis. Results were calculated using the likelihood ratio testing parameter and Benjamin–Hochberg p-adjustment method. The RNA-seq data in this study was deposited at Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo/) under accession ID GSE223376.
Functional enrichment analysis
To perform functional enrichment of the KO:tau vs tau mice, we input the top DEGs (FDR < 0.05) into STRINGdb (version 11.5) and applied Markov clustering followed by functional enrichment analysis of each cluster. Functional analysis was performed using STRINGdb (version 11.5) and applied Markov clustering followed by functional enrichment analysis of each cluster. Networks were analyzed and represented using Cytoscape version: 3.9.1.
Gene set enrichment analysis (GSEA) was performed using GSEA v4.3.2 [27, 39] with default settings. Raw counts from protein coding genes that were normalized using upper quartile normalization were provided as input. For signature-specific GSEA, the KEGG AD and KEGG Lysosomal pathways were run on their own (hsa05010 v2023.1 and hsa04142 v2023.1).
Integration of mouse DEGs with human AD DEGs
We used human brain RNAseq data from the AMP-AD knowledge portal to integrate DEGs identified in mice with transcriptionally dysregulated genes in AD [1, 9, 29]. The AMP-AD data used for this comparison had been re-analyzed to normalize across different studies as detailed elsewhere, with definitions for AD patients and controls identical to those defined by Wan et al. [44]. Specifically, we used datasets syn8484987, syn8466812, syn8456629 for the brain region-specific differential expression, using the data from AD vs controls only. The human brain samples were from ROSMAP (DLPFC 155 AD/ 86 Control), Mayo RNAseq (TCX 80-AD/73-control) and Mount Sinai Brain Bank (FP 167-AD/93-control, IFG 151-AD/79-control, PHG 143-AD / 82-control, STG 151-AD/89-control). P values for differential expression were adjusted for multiple hypothesis testing using false discovery rate estimation (FDR), and we selected genes with an adjusted p < 0.05 based on the analysis in Wan et al. [44]. For tangle-associated genes we used the lists published in Mostafavi et al. [29]. Significance of the overlap between human AD genes and mouse KO:tau DEGs (vs. tau alone) was calculated using Fisher's exact test. Correlation between mouse and human brain expression was calculated using Pearson’s correlation coefficient and significant differences between the regressions were calculated by ANOVA in full factorial modelling using standard least squares. Integration with single nucleus RNAseq used published data from Mathys et al. [25] (p < 0.05, FC > 10%).
Histology
Most histology for this study was performed by NeuroScience Associates using their MultiBrain technology (n = 10 tau and 10 KO:tau, 5 males/group; n = 10 tau and 10 KI:tau, 6 males/group). Frozen hemibrains were thawed and embedded into a gelatin matrix that was sectioned in the coronal plane at 35 μm thickness. Sections were stored in cryoprotect buffer at -20 °C until use. A 1 in 12 series of free-floating sections was immunostained for AT8 or Iba1 using standard protocols and then lightly counterstained with hematoxylin (AT8 only) before mounting onto subbed slides, dehydrating through alcohols, and cover slipping with Permount. A 1 in 6 series of sections was used for Campbell-Switzer staining according to the protocol available at: https://www.neuroscienceassociates.com/reference/papers/alzheimers-disease-pathology-silver-stain/.
Additional MultiBrain sections from a 1 in 12 series were immunostained at BCM for CD-68. After quenching endogenous peroxidase with hydrogen peroxide, sections were blocked in serum and then incubated in primary antibody for 2 h at RT (CD-68: 1:500, Bio-Rad clone FA-11, MCA1957GA), and then processed for secondary and HRP reagent using a Vectastain ABC kit according to the manufacturer's instructions (Rat IgG, Vector Laboratories, PK-4004). Antibody binding was visualized with DAB-nickel (VWR, 101098–434). Sections were counterstained with nuclear fast red, mounted, dehydrated, and cover slipped with Permount.
Gallyas staining was done at BCM on a 1 in 12 series of MultiBrain sections. All glassware was acid-cleaned before use. Mounted tissue was bound to the slide by 5 min in 95% EtOH/37% formalin. Slides were rehydrated and then incubated at RT sequentially in potassium permanganate, oxalic acid, and periodic acid, each followed by a water rinse. Slides were then moved to silver iodide solution followed by acetic acid, before the stain was detected with Gallyas working solution. Slides were counterstained with nuclear fast red, dehydrated, and cover slipped with Permount.
Histological quantitation
AT8, Iba1, and Campbell–Switzer-stained slides were imaged at NeuroScience Associates and uploaded into the Proscia Concentriq web-based digital pathology platform. CD68-stained slides were imaged at BCM using a Zeiss Axio Scan.Z1 at 10 × magnification (Carl Zeiss AG, Oberkochen, Germany).
For AT8 and Iba1 area analyses, 2 plane matched sections containing the dorsal hippocampus and spanning approximately − 1.5 to − 2.1 mm from bregma (tau and KO:tau), or 4–5 sections spanning approximately -1.5 to -2.8 mm (tau and KI:tau), were used for analysis. For AT8, Iba1, and CS silver, images were screen-captured from the Concentriq website at 3 × magnification, saved as png files, and imported into ImageJ v1.53 s (NIH) as png files that were converted to 8-bit greyscale. Default (AT8) or Yen (Iba1) threshold was applied, then the maximum value was manually adjusted to better match the stain before calculating % area. For CD68 staining, six tif files per mouse, spanning approximately − 1.82 to − 4.24 mm from bregma, were imported into ImageJ, and quantification performed as for Iba1 using the Yen threshold and manually adjusting to match the stain before calculating area. Values were averaged across sections to calculate % area for each animal.
Area measurements were taken from a 1 in 6 series of Campbell–Switzer silver-stained sections that spanned the full rostro-caudal extent of the hippocampus for each animal; cortex and lateral ventricle areas were measured from the same sections (n = 19–21 sections/mouse). Images were screen captured from the Concentriq website at 1.5 × magnification and imported into ImageJ as png files. Regions of interest (cortex, hippocampus, or ventricle) were manually traced and each area was measured in pixel2. The area of each region was then summed across all sections to generate the total area for cortex, hippocampus, or lateral ventricle for each individual animal. Summed values were then converted from pixels2 to mm2. Although not done here, converting from summed area to regional volume could be done by multiplying by the section thickness (35 µm) and interval spacing (1 in 6).
Electrophysiology
Female KI:tau and tau mice ~ 8 mo of age were anesthetized with isoflurane and transcardially perfused with a modified ice-cold sucrose-based cutting solution (pH 7.4; 10 mM NaCl, 25 mM NaHCO3, 195 mM sucrose, 5 mM glucose, 2.5 mM KCl, 1.25 mM NaH2PO4, 2 mM sodium pyruvate, 0.5 mM CaCl2, and 7 mM MgCl2, bubbled continuously with 95% O2 / 5% CO2). The mice were then decapitated, the brain removed, and submerged in cutting solution. Transverse slices through the hippocampus (300 μm) were cut with a Microm HM 650 V vibratome (Thermo Scientific). Slices were recovered for ~ 30 min at 32 °C and then maintained at RT for another hour in oxygenated artificial cerebrospinal fluid before recording (ACSF, pH 7.4; 126 mM NaCl, 2.5 mM KCl, 2.4 mM CaCl2, 1.2 mM NaH2PO4, 1.2 mM MgCl2, 11.1 mM glucose, and 21.4 mM NaHCO3). Slices were transferred to the recording chamber at 32 °C and perfused continuously with oxygenated ACSF at a flow rate of 1.8–2.0 ml/min. Slices were allowed to equilibrate for at least 5 min before recording. The recording site was visualized using infrared–differential interference contrast imaging (IR-DIC) on an upright microscope (Eclipse FN-1, Nikon) equipped with a moveable stage (MP-285, Sutter Instruments). Recordings were made using a MultiClamp 700B amplifier (Axon Instruments), sampled using a Digidata 1440A, and analyzed offline with pClamp 10.3 software (Axon Instruments). Field EPSPs were elicited by stimulating the Schaffer collateral and recording with a borosilicate glass electrode filled with ACSF placed in CA1 stratum radiatum. Baseline and tetanic stimulations were delivered by a bipolar stimulating electrode (CE2C75, FHC Inc.) placed 200–300 μm away from the recording electrode. The stimulation intensity was adjusted to elicit an EPSP with an amplitude of 30–50% of the maximum. Baseline fEPSPs were evoked at 0.05 Hz and recorded for at least 10 min (response variability < 10%). Two trains of 1 s stimulation at 100 Hz separated by 20 s were delivered to induce LTP. Recordings were amplified, filtered at 1 kHz, and digitized at 10 kHz.
Statistical analyses: behavior, histology, and electrophysiology
Statistical analyses were performed using GraphPad Prism 9. Comparison of two groups was done using unpaired two-tailed Student's t test or Mann–Whitney for non-parametric distribution. Comparison of three or more groups, or of two groups across time, was done using one- or two-way ANOVA followed by Tukey or Bonferroni post-hoc testing. Comparison of three or more groups with non-parametric distributions was done by Kruskal–Wallis followed by Dunn post-hoc testing. Survival curves were analyzed by Mantel–Cox log-rank test.
Statistical outliers were identified using either Grubb's or ROUT testing in Prism. In only two cases were 2 animals removed from the same genotype (OF: 2 tau from the KI colony, and lateral ventricle area: 2 KI:tau). In all other cases, no more than 1 animal was removed as an outlier per genotype. One KO:tau animal that was impaired in visible platform testing was eliminated from all behavioral analyses.
Results
TMEM106B deletion worsens cognitive impairment and accelerates paralysis caused by pathogenic tau
Because so many of the diseases influenced by TMEM106B are associated with dementia, we began by examining how the presence or absence of TMEM106B influenced cognitive function in the context of tauopathy. We generated TMEM106B null mice using embryonic stem cells from the KnockOut Mouse Project (KOMP), which were crossed with ubiquitous Cre and Flp recombinase lines to remove the lacZ cassette and delete the essential exon 4 (Supplementary Fig. 1a, online resource). Unlike past work with KOMP mice in which both the lacZ and exon remained intact [22, 32], our strategy produced nearly complete loss of TMEM106B protein (Supplementary Fig. 1b, online resource). The TMEM106B deletion mice were intercrossed with the PS19 model of tauopathy carrying the P301S MAPT mutation to generate four genotypes for study (WT, KO, tau, and KO:tau). This model has been extensively used for cognitive testing in past studies with behavioral deficits that roughly parallel the timing of tau pathology [3, 4, 10, 26, 40, 52]. Behavioral testing began at 8 mo of age with several assays of locomotor function. Animals that showed evidence of hind limb paralysis were removed from further testing. KO:tau animals were hyperactive in the open-field arena (Supplementary Fig. 2a-b, online resource) but swam at the same speed and efficiency as other genotypes in the straight swim test (Supplementary Fig. 2c-d, online resource), which allowed for cognitive testing using water-based tasks.
We performed a cognitive test battery that started with Morris water maze (MWM) to assess spatial reference memory (Supplementary Fig. 3a-c, online resource), followed by repeated reversal learning (RRL) to assess cognitive flexibility (Supplementary Fig. 3d, e, online resource), and ended with radial arm water maze (RAWM) to assess spatial working memory (Supplementary Fig. 3f, g, online resource). Each task identified significant deficits in tau and KO:tau relative to WT. The KO:tau animals tended to be the poorest performing genotype of the four, but were never distinct enough from the tau animals to reach significance.
We initially chose to perform behavioral testing at 8 mo based on past studies of cognitive dysfunction in this model. While aging animals to 8 mo, a subset of the tau animals developed hind-limb paralysis that required euthanasia, which is a known phenotype of the PS19 model [48]. By recording the age of paralysis onset, we realized that TMEM106B deletion was accelerating this phenotype. KO:tau mice developed paralysis as early as 5.9 mo, while tau animals first showed hind limb weakness at 8.4 mo (Fig. 1a,), and the effect was more pronounced in females than in males (Supplementary Fig. 4a, online resource). The median age at euthanasia due to paralysis was 9.2 months for KO:tau vs 9.9 mo for tau. The accelerated paralysis was surprising given the locomotor hyperactivity seen in KO:tau mice prior to cognitive testing at 8 mo. While limb paralysis is not found in AD, it provides a complementary readout of tau neurotoxicity in this model. This discovery led us to wonder if our behavioral outcomes at 8 mo may have reflected a survivorship bias, such that only the healthiest animals lived long enough to undergo cognitive testing.

TMEM106B deletion accelerates paralysis and worsens cognitive behavior. a Onset of hind limb paralysis requiring euthanasia was measured for tau animals, excluding mice used in behavioral testing. The age at paralysis onset was significantly earlier in KO:tau than tau mice. (tau = 27, KO:tau = 43). b Spatial learning in the Morris water maze was assessed by the distance traveled to reach the escape platform. KO:tau mice performed significantly worse than tau animals on days 3 and 5 of training. c KO:tau mice recalled the escape location less well than tau animals during daily short term memory testing (c). but this difference did not persist during long-term memory testing (d). e Cognitive flexibility measured by changing the platform location each day showed that KO:tau mice performed more poorly than tau mice on trial 5. Graph shows average performance across days as a function of trial number (8 trials/day). f Short-term memory for the changing platform location was no different for any given location (left), but was worse overall in KO:tau mice when averaged across the three locations. g-h During the learning phase of radial arm water maze, KO:tau mice made more incorrect arm entries (g reference memory, left) and more repeated arm entries (h working memory, right and middle graphs). Long-term memory was also impaired in KO:tau mice compared to tau when measured as incorrect arm entries (g right), but did not reach significance for repeated arm errors (h, right). See Supplementary Table 1 for ANOVA statistics. (b-h tau = 10, KO:tau = 10) For all scatter plots: open circles = female, closed circles = male. * p < 0.05, ** p < 0.01, *** p < 0.001
To overcome the possible survivorship effect, we examined a younger cohort of animals starting at 6 mo of age, when > 90% of the KO:tau mice were still healthy. We focused these experiments on tau and KO:tau groups because we had seen no significant differences between WT and KO tested at 8 mo of age (Supplementary Fig. 3, online resource). We repeated the same 3 cognitive tasks in the same order as before (MWM, RRL, and RAWM), preceded by basic locomotor testing in open field. In contrast to what we had seen at 8 mo, KO:tau mice showed no hyperactivity or thigmotaxis in the open-field chamber. Further, KO:tau mice swam as well as tau animals in the cued test (Supplementary Fig. 2e, f, online resource). Unlike what we had seen in the older animals, cognitive performance differed between genotypes for all three tasks at 6 mo. In Morris water maze, the KO:tau mice learned the hidden platform location less efficiently than tau animals. KO:tau mice swam farther to the escape platform than tau mice (Fig. 1b). KO:tau mice also remembered the trained location less well than tau mice when tested at the start of each training session (Fig. 1c), but recalled the escape location as well as tau mice when tested 3 d after training was completed (Fig. 1d). In the repeated reversal task, KO:tau mice learned each of three new platform locations less efficiently than tau mice (Fig. 1e). When recall for each new platform location was tested the following day, KO:tau mice also remembered the trained locations less precisely than tau mice (Fig. 1f). Finally, in the radial arm water maze, KO:tau learned to find the escape arm less efficiently than tau mice. KO:tau made more incorrect arm entries (reference memory errors) and reentered incorrect arms more often than tau mice (working memory errors), both while learning the radial arm task and when recall for the escape arm was tested 3 d later (Fig. 1g, h). These findings indicate that TMEM106B deletion accelerated cognitive decline in the tau mice and that the effect was more pronounced at 6 mo than at later ages.
Expression of T186S coding variant protects against cognitive decline in tau mice
The only common coding variant found in TMEM106B is a relatively conservative change from threonine to serine at position 185 in the human gene, or position 186 in the mouse. This variant is in linkage disequilibrium with multiple non-coding SNPs that associate with differential risk for various neurodegenerative disorders including FTLD and AD. We set out to directly test whether the T185S variant could influence disease pathogenesis by introducing the equivalent coding substitution in mice using CRISPR/Cas9. We confirmed the mouse T186S variant by Sanger sequencing of the full exon and high-resolution melting point analysis to check for off-target mutations (Supplementary Fig. 5a, online resource). Three lines were expanded for basic characterization and one was selected for further study. Because past work had suggested that the in vitro expression of the human T185S variant reduced TMEM106B protein half-life and steady-state levels [30], we tested TMEM106B expression in our T186S mice but found no differences from WT at any age examined (Supplementary Fig. 5b, online resource). T186S mice were intercrossed with PS19 tau to generate four genotypes for study (WT, KI, tau, and KI:tau); both KI and KI:tau mice were homozygous for the T186S variant. All four genotypes expressed similar levels of monomeric and presumptive dimeric TMEM106B protein (Supplementary Fig. 5c, online resource). Animals underwent the same behavioral testing procedure as for KO x tau, starting at 8 mo of age.
Initial assessment in the open field and straight swim showed no difference between genotypes (Supplementary Fig. 6a, b, online resource), so we proceeded to the cognitive test battery. The three cognitive tasks revealed significant differences in performance between the two tau genotypes. Both KI:tau and tau mice learned the escape location equally well in the Morris water maze; however, there were pronounced differences in both short- and long-term memory for the trained location (Fig. 2a-c, see Supplementary Fig. 6, online resource, for graphs including all four genotypes). The preservation of short-term memory in KI:tau mice compared to tau alone reached significance in the MWM day 4 probe trial, and approached significance when averaged across days (p = 0.06, Fig. 2b). The effect became more pronounced for long-term memory, with KI:tau mice performing as well as WT (Fig. 2c). Repeated reversal testing confirmed a protective effect of the KI allele, with KI:tau mice learning the new platform locations more efficiently and recalling them more accurately than tau animals (Fig. 2d, e). Cognitive preservation was also observed in the RAWM, where KI:tau mice made fewer incorrect arm entries than tau animals during training and showed better memory for the escape arm during long-term probe tests (Fig. 2f, g). Collectively, these results demonstrate that the serine variant imparts resilience against cognitive impairments caused by pathogenic tau.

The T186S coding variant protects against cognitive decline and delays paralysis in tau mice. a No difference was noted between KI:tau and tau groups during the learning phase of Morris water maze, measured as distance to the escape platform. b Short-term memory for the escape location was better in KI:tau mice than tau on day 4 (left). When short-term memory in daily probe trials was averaged across days, the improvement in KI:tau vs tau mice was nearly significant at p = 0.065 (right). c Long-term memory for the escape location was better in KI:tau mice than tau animals. Track plots show example search paths for one animal of each genotype during the long-term probe trial (right). (a-c: WT = 18, KI = 21, tau = 20, KI:tau = 20). d Cognitive flexibility measured by changing the platform location each day showed that KI:tau mice performed better than tau mice on trials 5 and 7. Graph shows average performance across days as a function of the trial number (8 trials/day). e Short-term memory for each new platform location was better in KI:tau than tau mice by the second day of testing (platform location NW), and this difference widened with the third day of testing (platform location SE, left). On average, KI:tau mice showed better short-term memory for the changing platform locations than tau mice (right). (d, e: WT = 18, KI = 21, tau = 19, KI:tau = 19). f, g During the learning phase of radial arm water maze, the number of incorrect arm entries made by KI:tau was lower than in tau mice on trial 7 (f reference memory, right), but did not reach significance when averaged across trials (f, middle). Both genotypes made the same number of repeated arm entries during training (g, working memory, right and middle graphs). Remarkably, KI:tau mice recalled the trained escape arm better than tau animals during long-term testing, both when measured as incorrect arm entries (f, reference errors, right) and when measured as repeated arm entries (g working errors, right). (f, g: WT = 16, KI = 18, tau = 17, KI:tau = 19). h Onset of hind limb paralysis requiring euthanasia occurred significantly later in KI:tau than tau mice. tau = 23, KI:tau = 23. Significant values shown here reflect ANOVA testing with all four genotypes. For all scatter plots: open circles = female, closed circles = male. * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001. See Supplementary Table 1 for ANOVA statistics
Because we had noted such a profound impact of TMEM106B deletion on the onset of paralysis in the PS19 mice, we extended this analysis to the KI colony. In contrast to the accelerated phenotype observed in KO:tau animals, expression of the T186S allele slowed the onset of hind limb paralysis from a median survival of 9.7 mo in the tau animals to 11.9 mo in KI:tau mice, with a stronger effect in females than males (Fig. 2h, Supplementary Fig. 4b, online resource). The minor coding substitution again had a pronounced impact on neurobehavioral phenotypes in this model.
Loss of TMEM106B enhances neurodegeneration and pathologic markers of tauopathy
Our next challenge was to understand if and how each of our TMEM106B manipulations affected neuropathology to evoke such profound differences in cognitive function. Starting with the KO, we considered multiple mechanisms by which loss of TMEM106B may have accelerated cognitive decline and paralysis; however, the most obvious is by hastening tau aggregation and its downstream consequences. We examined this question in a subset of mice that were behaviorally tested at 8 mo and began by asking whether the loss of TMEM106B exacerbated neurodegeneration in tau mice. The Campbell–Switzer silver stain provided good tissue contrast for this analysis and allowed us to measure ventricular, hippocampal, and cortical areas. We found that deleting TMEM106B in tau mice profoundly worsened neurodegeneration (Fig. 3a, b). Total ventricular area was substantially increased, while hippocampal and cortical areas were both decreased in KO:tau relative to tau. Ventricular area was enlarged by 2.3 fold; hippocampal area diminished by ~ 30%; and cortical area reduced by 10% in KO:tau relative to tau animals.

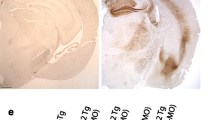
TMEM106B deletion worsens degeneration and tau pathology. a Campbell–Switzer silver staining did not identify tangles, but did reveal marked ventricular enlargement in the KO:tau animals compared to tau alone. b Silver-stained sections were used to measure the summed area of the hippocampus (HPC), overlying cortex (CTX) and adjacent lateral ventricle (LV) in tau and KO:tau mice to demonstrate a quantitative difference in the summed area of all three regions upon TMEM106B deletion. c-h Immunostaining and quantitation of AT8 (c, d), Iba1 (e, f), and CD68 (g, h) all demonstrate marked worsening of tau pathology and microgliosis upon TMEM106B deletion. Graphs in d, f, g show the percent area stained by each marker within hippocampus (HPC) and cortex (CTX). (a-h: tau = 10, KO:tau = 10). i, j Western blots for phospho-tau epitopes AT8 (S202/T205), PHF1 (S396/S404), AT100 (T212/S214), AT270 (T181), and AT180 (T231), total human tau CP27, and total tau Tau5 (i) and quantitation of band intensity (j). (i, j: WT = 2, KO = 2, tau = 6, KO:tau = 6). Open circles = female, closed circles = male. * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001
Tau pathology was also worsened by loss of TMEM106B. Phospho-tau AT8 staining occupied roughly 30% of the hippocampus in tau mice at 9 mo, but more than three times this area in KO:tau animals (Fig. 3c, d). Even at 6 mo of age when phospho-tau accumulation was just beginning, KO:tau mice harbored twice as much AT8 signal as tau siblings (Supplementary Fig. 8, online resource). Neurofibrillary tangles identified by Gallyas staining were also more frequent in the KO:tau mice (data not shown), and appeared as early as 6 mo of age in both male and female KO:tau animals (Supplementary Fig. 8, online resource). Microgliosis identified by Iba1 staining tracked with pathological tau accumulation. Iba1 staining occupied ~ 40% of the hippocampus in tau mice, but nearly twice that area in KO:tau mice (Fig. 3e, f). CD68 immunostaining as a marker of phagocytic microglia also rose dramatically in KO:tau mice (Fig. 3g, h).
Biochemical assessment of phospho-tau epitopes AT8 (S202/T205), PHF1 (S396/S404), AT100 (T212/S214), AT270 (T181), and AT180 (T231) confirmed the accumulation of pathogenic tau markers in KO:tau mice compared with tau alone, while overall levels of transgenic human tau (CP27) and total tau (Tau5) were unchanged (Fig. 3i, j). Conversely, overexpression of pathogenic tau did not affect TMEM106B levels (Supplementary Fig. 5c, online resource). Overall, the loss of TMEM106B accelerated tau accumulation and worsened neurodegeneration resulting from tauopathy.
TMEM106B T186S protects against degeneration without changing tau pathology
Given that the loss of TMEM106B dramatically worsened both cognitive impairment and neuropathology, we predicted that the T186S variant would preserve cognitive function by diminishing neuropathology. Instead, we found that the coding variant protected against neurodegeneration without influencing phospho-tau pathology or microgliosis. Cortical area was 13% larger while the ventricular area was ~ 50% smaller in KI:tau mice compared to tau, without changing hippocampal area (Fig. 4a, b). Preservation of cortical size was not accompanied by any difference in AT8 (Fig. 4c, d), Iba1 (Fig. 4e, f), or CD68 burden (Fig. 4g, h). Biochemical assessment of phospho-tau epitopes AT8, PHF1, AT100, AT270, and AT180 confirmed the equivalence of pathogenic tau between genotypes; accumulation of human tau and total tau were also unchanged (Fig. 4i, j). These findings suggest that the T186S variant protects against degeneration downstream of tau, but does not alter tau burden itself.

TMEM106B T186S protects against degeneration without changing tau pathology. a Campbell–Switzer silver stain hinted at ventricular preservation in T186S KI:tau mice compared with tau alone. b Summed areas of hippocampus (HPC), cortex (CTX), and lateral ventricle (LV) were measured from silver-stained sections spanning the full extent of hippocampus. Tau animals carrying the T186S variant retained more cortical tissue and showed less ventricular enlargement than tau alone. c-h Immunostaining and percent area quantitation of AT8 (c, d), Iba1 (e, f), and CD68 (g, h) were no different between genotypes in hippocampus or cortex. (a-h: tau = 10, KI:tau = 10). i, j Western blots for phospho-tau epitopes AT8 (S202/T205), PHF1 (S396/S404), AT100 (T212/S214), AT270 (T181), and AT180 (T231), total human tau CP27, and total tau with Tau5 (i) and quantitation of band intensity (j). (i, j: WT = 2, KI = 2, tau = 6, KI:tau = 6). Open circles = female, closed circles = male. * p < 0.05
TMEM106B deletion increases transcriptional overlap with AD
We next turned to RNA sequencing to see if a broad view of transcriptional changes between genotypes might provide clues about how TMEM106B protects from tauopathy. We performed mRNA sequencing on hemi-forebrain tissue from 5–7 behaviorally tested mice of each genotype. We found few differences between WT and KO, in line with past studies on other TMEM106B deletion models ([13, 46] but see [54], Fig. 5a, Supplementary Table 2, online resource). We found far more differentially expressed genes (DEGs) between KO:tau and tau, where 150 genes were downregulated and 321 upregulated at an FDR cutoff < 0.05 (Fig. 5a, Supplementary Table 2, online resource).

TMEM deletion significantly alters the transcriptional signature of tauopathy. a Volcano plot showing FDR log10 p value as a function of log2 fold change in forebrain gene expression between KO and WT (left) and KO:tau vs. tau (right). Mice were 9–10 mo of age at time of harvest. 16 DEGs distinguish KO from WT. TMEM106B deletion had a far greater effect on transcriptional signature in the context of tauopathy. n = 5–7/genotype. b GSEA for the KEGG AD signature approached significant enrichment for KO:tau vs tau with an ES of 1.71 and FDR q = 0.06 (left). GSEA for the KEGG lysosome signature was significantly enriched for KO:tau vs. tau with an ES of 1.55 and FDR q = 0.034 (right)
To gain functional insight into the DEGs distinguishing KO:tau from tau, we first performed a GSEA enrichment score (ES) analysis against the KEGG Alzheimer’s disease (AD) signature, which revealed significant enrichment of genes in this pathway (Fig. 5b). Consistent with the lysosomal localization of TMEM106B and lysosomal dysfunction in AD, KO:tau was also significantly enriched for genes in the KEGG lysosomal signature (Fig. 5b).
Next, we directly compared the KO:tau vs tau DEGs against multiple AMP-AD human brain transcriptional datasets [1, 2, 9, 29, 44, 45]. A significant number of mouse DEGs overlapped and were concordant with human DEGs identified from comparison of AD vs healthy control (HC) postmortem brain tissue (55 downregulated and 127 upregulated, overlap Fisher p < 0.0112, Fig. 6a, Supplementary Table 3, online resource). The correspondence between KO:tau and human AD was more clearly visualized by plotting the expression level of each tau genotype against the expression level of AMP-AD human AD for all DEGs identified from comparison of KO:tau vs tau. The resulting correlation between KO:tau and AD was significantly stronger than for tau and AD, and was consistent when plotting expression from human whole brain (Fig. 6b) or specific regions such as temporal cortex, parahippocampal gyrus or superior temporal gyrus (not shown). Because we found increased tau aggregation upon TMEM106B deletion, we assessed whether there was also increased correlation with neurofibrillary tangle-associated genes from human AD [29]. We found a stronger correlation with expression in human AD brains of NFT-associated genes in the KO:tau vs tau alone (Fig. 6c).

TMEM deletion enhances functional overlap with AD. a Venn diagrams illustrating the number and overlap of DEGs identified by comparison of human AD vs healthy controls (HC, turquoise) against KO:tau vs. tau (red; left) b Relationship between human AD and mouse brain for DEGs identified by comparison of KO:tau vs tau. Values for human AD gene expression (relative to HC) were derived from the AMP-AD database on the X axis (log2 fold change) against the corresponding expression level for KO:tau vs WT (red) or tau vs WT (grey). Correlation (Pearson’s r value) and linear regression with human AD expression is significantly higher in the KO:tau animals than in mice expressing tau alone. c Relationship between human AD and mouse brain for human NFT-associated genes. Correlation with human NFT-associated gene expression is again increased by TMEM106B deletion. d Network clustering showing several enriched modules among the KO:tau vs tau DEGs that overlap and change in the same direction as human AD brain. Colors distinguish separate, but related, functional modules. Outlines indicate genes concordant with single cell analyses testing the effect of NFT burden in human AD brain. e Heat map illustrating DEGs used to generate functional modules in d, plotted to show the degree of change against WT (mouse, left) or HC (human, right) controls. TMEM106B deletion increases the magnitude of change towards a more AD-like expression pattern compared to tau alone. DEG expression is shown for both KO:tau and tau groups against 4 different human AD brain regions. TCX = temporal cortex, PHG = parahippocampal gyrus, STG = superior temporal gyrus, DLPFC = dorsolateral prefrontal cortex. n = 5–7 mice per genotype. **** p < 0.0001
We also saw a high degree of overlap and concordance of mouse DEGs with human AMP-AD functional pathways (Fig. 6e, Supplementary Table 4, online resource). Examples of this concordance show that expression differences between tau and WT were more pronounced in the absence of TMEM106B across multiple functional pathways. This analysis revealed enrichment in the TYROBP causal network associated with microglia, which also included synapse pruning genes (Fig. 6d, Supplementary Fig. 9 and Supplementary Table 5, online resource). Other pathways related to microglia and inflammation included chemokine signaling, cytokine-cytokine receptor interaction, and microglia phagocytosis. In line with TMEM106B function in autophagy and endo-lysosomal trafficking, we observed enrichment in pathways related to lysosomal lumen peptidases and hydrolases, chaperone mediated autophagy, and exosome/secretory granules (Fig. 6d and Supplementary Fig. 9, online resource). Many of these genes were also concordant with single cell AD data, suggesting that expression changes caused by TMEM106B deletion were not due solely to differences in cell type proportions within our bulk RNA datasets (Fig. 6d, Supplementary Fig. 9, online resource; [25]).
Given the overlap of microglial signatures from KO:tau and AD, we next tested their correlation with disease-associated microglial genes (DAMs) indicative of microglial activation in disease models [21]. We again found that TMEM106B deletion increased the correlation between mouse tau and human AD both overall and within several of the individual brain regions tested (Supplementary Fig. 10, online resource). Taken together, this data suggests that loss of TMEM106B pushes tau mice farther along the AD transcriptional spectrum than tau alone.
TMEM106B T186S elicits only mild transcriptional changes, but preserves synaptic plasticity downstream of tauopathy
We next performed similar transcriptomic analysis with the KI:tau mice. As observed previously for KO vs WT, we also found few DEGs that distinguished KI from WT (22 genes, Supplementary Table 2, online resource). Comparison of KI:tau vs tau yielded a similarly small number of DEGs that met an FDR cutoff of 0.05 (10 genes, Supplementary Table 2, online resource).
The limited results of our transcriptional analyses hinted that the beneficial effects of TMEM106B impact synaptic function in the KI:tau mice to preserve cognitive function. We used acute hippocampal slices from 8 mo old KI:tau and tau mice to test basal synaptic transmission along with synaptic plasticity in the Schaffer collateral pathway. Basal synaptic transmission measured from I–V curves was identical between the two genotypes (Fig. 7a). In contrast, long-term synaptic plasticity measured from the postsynaptic response to tetanic stimulation was significantly greater in KI:tau than tau (Fig. 7b). The postsynaptic potentiation measured during the last 5 min of recording was nearly two-fold larger in KI:tau than tau (153.8% vs 127.6% of baseline, Fig. 7c). These findings suggest that the TMEM106B T186S coding variant may protect cognitive function by enhancing synaptic plasticity even while leaving basal synaptic function unchanged.

TMEM106B T186S coding variant enhances hippocampal plasticity in tauopathy mice. Acute hippocampal slices from 8 mo old KI:tau and tau mice were used to measure synaptic responses in the Schaeffer collateral pathway. a Input–output (or I–V) curve plotting amplitude of the field excitatory postsynaptic potential (fEPSP) as a function of simulation intensity. Basal synaptic transmission was unchanged by TMEM106B coding variant in this setting. n = KI:tau 9 slices from 4 mice, tau 10 slices from 3 mice. b fEPSP amplitude was normalized to baseline and plotted as a function of time, before and after tetanic stimulation, to induce long-term potentiation. Potentiation was significantly greater in slices from KI:tau mice than tau. c Average normalized fEPSP amplitude measured over the last 5 min of recording (55–60 min after tetanus) highlights the difference in synaptic potentiation between genotypes. n = KI:tau 16 slices from 6 mice, tau 18 slices from 5 mice. **p < 0.01
Discussion
Past work to understand the role of TMEM106B in the brain has focused primarily on mouse models of FTLD-TDP, driven by the early, consistent, and robust GWAS associations with this subtype of FTD [31]. Since these seminal GWAS studies in FTLD-TDP, TMEM106B has been found to modify risk for various other neurological disorders, many of which share cognitive decline as a key feature [11]. More recently, TMEM106B fibrillar aggregates have been discovered in a range of dementias, including FTLD, AD, and PSP, that are characterized by tauopathy [33]. Here we present the first experimental test of how TMEM106B manipulation affects disease progression and cognitive decline in a mouse model of tauopathy. Our work offers four pivotal insights to the growing literature on this enigmatic protein. First, we demonstrate that endogenous TMEM106B protects against tau pathology and tau-induced cognitive dysfunction. Loss of TMEM106B in MAPT P301S mice profoundly worsened every outcome we measured. Second, TMEM106B deletion shifted the mouse tau transcriptional profile towards greater concordance with human AD. To the best of our knowledge, this represents the first instance of TMEM106B manipulation enhancing transcriptional alignment with human disease. Third, our findings demonstrate that the T186S coding variant affects TMEM106B function to limit both neurodegeneration and cognitive decline due to tauopathy. Finally, and most broadly, our study expands the protective scope of TMEM106B to models of proteinopathy beyond TDP-43.
Prior studies of TMEM106B deletion in the GRN null model of FTLD-TDP produced conflicting outcomes. The initial study reported TMEM106B deletion to be protective against a subset of lysosomal abnormalities, locomotor hyperactivity, and behavioral disinhibition caused by GRN deletion, without impacting microgliosis, lipofuscinosis, or retinal degeneration [22]. In contrast, three subsequent studies in the same GRN model found that TMEM106B deletion either worsened existing abnormalities, or more often, synergized with GRN deletion to induce new deficits in lysosomal function, locomotor behavior, gliosis, neurodegeneration, and neuroinflammation [13, 46, 53]. Our findings align more closely with these latter three studies in concluding that TMEM106B is normally protective against multiple aspects of neurodegeneration associated with proteinopathy. We show that TMEM106B deletion accelerated or exacerbated cognitive decline, hind limb paralysis, phospho-tau and NFT accumulation, microgliosis, and neurodegeneration. Loss of TMEM106B significantly impacted the transcriptional profile of tauopathy, particularly affecting signatures of microglial function. Collectively, we interpret this to suggest that endogenous TMEM106B acts upstream of tau pathology to slow aggregation and thus delay downstream effects on behavior, gliosis, and degeneration. This protective function was unmasked only in the context of cellular stress. Similar to prior studies, we found only a small number of transcriptional changes between WT and KO mice [13, 22, 24, 46] but see [54].
Past work has also shown that TMEM106B can influence myelination, lysosomal pH, size, and movement [11, 12], but it is under the stress of aging and disease that this protein appears to have greatest impact. Our transcriptional analysis of KO:tau mice confirmed and expanded our understanding of the broad impact TMEM106B deletion had on tau-associated pathological processes. TMEM106B deletion shifted the mouse tau transcriptional signature to more closely align with that of human AD, making it a more aggressive model of disease than tau alone. Network modules enriched for mouse DEGs overlapped significantly with modules altered in human AD. These modules encompassed a wide range of functions connected to the TYROBP signaling pathway, consistent with the idea that TMEM106B plays a major role in the microglial response to neurological insult [50]. Yet, several neuronal pathways were also downregulated, complicating attribution of the primary cell type driving disease. The upregulation of exosome-related genes suggests a mechanism by which loss of TMEM106B might promote tau spreading, yet provides no hint as to the cell type responsible. The broad range of transcriptional modules and cell types affected by TMEM106B deletion supports the idea that endogenous TMEM106B gates an early pathological event to slow disease progression.
Our study is the first to demonstrate a biological effect of the T186S variant in a disease model. This finding was surprising, as prior work suggested the causal SNP associated with disease risk was located in the 3' UTR where it was predicted to increase transcription by altering chromatin architecture [16]. According to this model, disease protection associated with the minor allele was independent of TMEM106B protein sequence. We suspect that changes in transcription due to non-coding variants may act in concert with functional effects of coding substitution in the protein. Another possibility raised by prior overexpression experiments combined the two hypotheses, suggesting that the serine substitution diminished TMEM106B expression through a change in protein half-life [30]. In vitro the T185S variant reduced TMEM106B protein stability during cyclohexamide treatment, which resulted in lower steady-state TMEM106B levels in untreated cells [30]. Although we do not directly test protein stability here, the amount of TMEM106B protein detected in KI mice is identical to WT, suggesting that the variant did not affect stability in our model. Instead, the T186S variant appears to safeguard against cognitive decline, hind limb paralysis, and neurodegeneration, without altering TMEM106B steady-state protein levels.
Our work also uncovered a stark contrast between disease acceleration resulting from TMEM106B deletion and neuroprotection afforded by the T186S coding variant. Several phenotypic changes in KI:tau mice directly contrasted those in KO:tau. The difference was particularly pronounced for the two functional outcomes of cognition and mobility, which arose without any obvious change in tau pathology. This lack of obvious neuropathological influence is consistent with a recent study of T186S in a GRN deletion model of FTLD-TDP which also found no change in the primary lipofuscin neuropathology caused by GRN deletion [5]. T186S also had limited effect on transcriptional signatures of GRN deletion or tau overexpression. Instead, the cognitive, motor, and neuroprotective outcomes we observed in the PS19 mice suggest that T186S acts downstream of tau aggregation and transcription here, and perhaps downstream of lysosomal pathology in the GRN model.
We acknowledge several limitations to the current study that should be taken into account when interpreting our results. First, our data exhibits significant inter-animal variability stemming from two main sources. The PS19 model was generated on a mixed C3B6 genetic background, which we maintained through intercrossing with TMEM106B KO and KI lines. This choice facilitated breeding with increased litter sizes and fewer litter losses, but it also meant that no two animals had the same genetic background. The PS19 model is known to exhibit a marked sex difference, with earlier pathological onset in males than in females [47, 49]. All of our experimental groups included both sexes, yet few of our outcomes stratified by sex. These two factors mean that we likely missed subtle differences between genotypes, but also that the significant differences we found were robust to sex and genetic variables. Another experimental limitation of our work was the use of intact forebrain tissue for transcriptional analyses, rather than individual brain regions or single nuclei. This decision arose in part out of expediency during the COVID-19 pandemic when we had limited time to collect tissue from many of our initial animals. In retrospect, we might have more effectively focused transcriptional analyses on dissected hippocampal tissue, where pathological changes were most pronounced, and where circuits responsible for spatial learning and memory are centered. By including unaffected brain areas in our RNA sequencing, we likely missed small focal transcriptional differences between groups, but it also means that the differences we found were robust enough to emerge through substantial background noise.
Moving from GWAS to mechanistic understanding can be challenging under the best of circumstances, and has been particularly elusive for TMEM106B. The conservative coding substitution T185S yielded no immediate insight into its potential functional impact [31]. TMEM106B lacks the clear functional clues that have come from gene deletion studies for TREM2 [8], from protein structure analyses for LILRB2 [42], or from identification of binding partners for CR1 [55]. Our understanding of TMEM106B has been further slowed by conflicting results in seemingly similar experimental models, hinting at complex biology we have yet to unravel [11]. We suspect that TMEM106B may play different roles depending on the cell type and the disease context in which it is tested. Future studies have much yet to discover.
Data availability
RNA-seq data from this study was deposited at Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo/) under accession ID GSE223376.
References
Allen M, Carrasquillo MM, Funk C, Heavner BD, Zou F, Younkin CS et al (2016) Human whole genome genotype and transcriptome data for Alzheimer’s and other neurodegenerative diseases. Sci Data 3:160089. https://doi.org/10.1038/sdata.2016.89
Bennett DA, Schneider JA, Arvanitakis Z, Wilson RS (2012) Overview and findings from the religious orders study. Curr Alzheimer Res 9:628–645
Briggs DI, Defensor E, Memar Ardestani P, Yi B, Halpain M, Seabrook G et al (2017) Role of endoplasmic reticulum stress in learning and memory Impairment and Alzheimer’s disease-Like neuropathology in the PS19 and APP(Swe) mouse models of tauopathy and amyloidosis. ENeuro. https://doi.org/10.1523/ENEURO.0025-17.2017
Brody AH, Nies SH, Guan F, Smith LM, Mukherjee B, Salazar SA et al (2022) Alzheimer risk gene product Pyk2 suppresses tau phosphorylation and phenotypic effects of tauopathy. Mol Neurodegener 17:32. https://doi.org/10.1186/s13024-022-00526-y
Cabron AS, Borgmeyer U, Richter J, Peisker H, Gutbrod K, Dormann P et al (2023) Lack of a protective effect of the Tmem106b “protective SNP” in the Grn knockout mouse model for frontotemporal lobar degeneration. Acta Neuropathol Commun 11:21. https://doi.org/10.1186/s40478-023-01510-3
Chang A, Xiang X, Wang J, Lee C, Arakhamia T, Simjanoska M et al (2022) Homotypic fibrillization of TMEM106B across diverse neurodegenerative diseases. Cell 185:1346-1355.e1315. https://doi.org/10.1016/j.cell.2022.02.026
Chen-Plotkin AS, Unger TL, Gallagher MD, Bill E, Kwong LK, Volpicelli-Daley L et al (2012) TMEM106B, the risk gene for frontotemporal dementia, is regulated by the microRNA-132/212 cluster and affects progranulin pathways. J Neurosci 32:11213–11227. https://doi.org/10.1523/JNEUROSCI.0521-12.2012
Colonna M (2023) The biology of TREM receptors. Nat Rev Immunol. https://doi.org/10.1038/s41577-023-00837-1
De Jager PL, Ma Y, McCabe C, Xu J, Vardarajan BN, Felsky D et al (2018) A multi-omic atlas of the human frontal cortex for aging and Alzheimer’s disease research. Sci Data 5:180142. https://doi.org/10.1038/sdata.2018.142
Etelainen TS, Silva MC, Uhari-Vaananen JK, De Lorenzo F, Jantti MH, Cui H et al (2023) A prolyl oligopeptidase inhibitor reduces tau pathology in cellular models and in mice with tauopathy. Sci Transl Med. https://doi.org/10.1126/scitranslmed.abq2915
Feng T, Lacrampe A, Hu F (2021) Physiological and pathological functions of TMEM106B: a gene associated with brain aging and multiple brain disorders. Acta Neuropathol 141:327–339. https://doi.org/10.1007/s00401-020-02246-3
Feng T, Luan L, Katz II, Ullah M, Van Deerlin VM, Trojanowski JQ et al (2022) TMEM106B deficiency impairs cerebellar myelination and synaptic integrity with Purkinje cell loss. Acta Neuropathol Commun 10:33. https://doi.org/10.1186/s40478-022-01334-7
Feng T, Mai S, Roscoe JM, Sheng RR, Ullah M, Zhang J et al (2020) Loss of TMEM106B and PGRN leads to severe lysosomal abnormalities and neurodegeneration in mice. EMBO Rep 21:e50219. https://doi.org/10.15252/embr.202050219
Feng T, Sheng RR, Solé-Domènech S, Ullah M, Zhou X, Mendoza CS et al (2020) A role of the frontotemporal lobar degeneration risk factor TMEM106B in myelination. Brain. https://doi.org/10.1093/brain/awaa154
Fowler SW, Chiang AC, Savjani RR, Larson ME, Sherman MA, Schuler DR et al (2014) Genetic modulation of soluble abeta rescues cognitive and synaptic impairment in a mouse model of Alzheimer’s disease. J Neurosci 34:7871–7885. https://doi.org/10.1523/JNEUROSCI.0572-14.2014
Gallagher MD, Posavi M, Huang P, Unger TL, Berlyand Y, Gruenewald AL et al (2017) A dementia-associated risk variant near TMEM106B alters chromatin architecture and gene expression. Am J Hum Genet 101:643–663. https://doi.org/10.1016/j.ajhg.2017.09.004
Gotzl JK, Mori K, Damme M, Fellerer K, Tahirovic S, Kleinberger G et al (2014) Common pathobiochemical hallmarks of progranulin-associated frontotemporal lobar degeneration and neuronal ceroid lipofuscinosis. Acta Neuropathol 127:845–860. https://doi.org/10.1007/s00401-014-1262-6
Jiang YX, Cao Q, Sawaya MR, Abskharon R, Ge P, DeTure M et al (2022) Amyloid fibrils in FTLD-TDP are composed of TMEM106B and not TDP-43. Nature 605:304–309. https://doi.org/10.1038/s41586-022-04670-9
Jiao HS, Yuan P, Yu JT (2023) TMEM106B aggregation in neurodegenerative diseases: linking genetics to function. Mol Neurodegener 18:54. https://doi.org/10.1186/s13024-023-00644-1
Jun MH, Han JH, Lee YK, Jang DJ, Kaang BK, Lee JA (2015) TMEM106B, a frontotemporal lobar dementia (FTLD) modifier, associates with FTD-3-linked CHMP2B, a complex of ESCRT-III. Mol Brain 8:85. https://doi.org/10.1186/s13041-015-0177-z
Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK et al (2017) A unique microglia type associated with restricting development of Alzheimer’s disease. Cell 169:1276-1290.e1217. https://doi.org/10.1016/j.cell.2017.05.018
Klein ZA, Takahashi H, Ma M, Stagi M, Zhou M, Lam TT et al (2017) Loss of TMEM106B ameliorates lysosomal and frontotemporal dementia-related phenotypes in progranulin-deficient mice. Neuron 95(281–296):e286. https://doi.org/10.1016/j.neuron.2017.06.026
Lewandoski M, Meyers EN, Martin GR (1997) Analysis of Fgf8 gene function in vertebrate development. Cold Spring Harb Symp Quant Biol 62:159–168
Luningschror P, Werner G, Stroobants S, Kakuta S, Dombert B, Sinske D et al (2020) The FTLD risk factor TMEM106B Regulates the transport of lysosomes at the axon initial segment of motoneurons. Cell Rep 30:3506-3519.e3506. https://doi.org/10.1016/j.celrep.2020.02.060
Mathys H, Peng Z, Boix CA, Victor MB, Leary N, Babu S et al (2023) Single-cell atlas reveals correlates of high cognitive function, dementia, and resilience to Alzheimer’s disease pathology. Cell 186:4365-4385.e4327. https://doi.org/10.1016/j.cell.2023.08.039
Min SW, Chen X, Tracy TE, Li Y, Zhou Y, Wang C et al (2015) Critical role of acetylation in tau-mediated neurodegeneration and cognitive deficits. Nat Med 21:1154–1162. https://doi.org/10.1038/nm.3951
Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J et al (2003) PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 34:267–273. https://doi.org/10.1038/ng1180
Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B (2008) Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 5:621–628. https://doi.org/10.1038/nmeth.1226
Mostafavi S, Gaiteri C, Sullivan SE, White CC, Tasaki S, Xu J et al (2018) A molecular network of the aging human brain provides insights into the pathology and cognitive decline of Alzheimer’s disease. Nat Neurosci 21:811–819. https://doi.org/10.1038/s41593-018-0154-9
Nicholson AM, Finch NA, Wojtas A, Baker MC, Perkerson RB 3rd, Castanedes-Casey M et al (2013) TMEM106B p. T185S regulates TMEM106B protein levels: implications for frontotemporal dementia. J Neurochem 126:781–791. https://doi.org/10.1111/jnc.12329
Nicholson AM, Rademakers R (2016) What we know about TMEM106B in neurodegeneration. Acta Neuropathol 132:639–651. https://doi.org/10.1007/s00401-016-1610-9
Nicholson AM, Zhou X, Perkerson RB, Parsons TM, Chew J, Brooks M et al (2018) Loss of Tmem106b is unable to ameliorate frontotemporal dementia-like phenotypes in an AAV mouse model of C9ORF72-repeat induced toxicity. Acta Neuropathol Commun 6:42. https://doi.org/10.1186/s40478-018-0545-x
Perneel J, Rademakers R (2022) Identification of TMEM106B amyloid fibrils provides an updated view of TMEM106B biology in health and disease. Acta Neuropathol 144:807–819. https://doi.org/10.1007/s00401-022-02486-5
Rademakers R, Nicholson AM, Ren Y, Koga S, Nguyen HP, Brooks M et al (2021) Loss of Tmem106b leads to cerebellum Purkinje cell death and motor deficits. Brain Pathol 31:e12945. https://doi.org/10.1111/bpa.12945
Robinson MD, McCarthy DJ, Smyth GK (2010) edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26:139–140. https://doi.org/10.1093/bioinformatics/btp616
Rodriguez CI, Buchholz F, Galloway J, Sequerra R, Kasper J, Ayala R et al (2000) High-efficiency deleter mice show that FLPe is an alternative to Cre-loxP. Nat Genet 25:139–140. https://doi.org/10.1038/75973
Schweighauser M, Arseni D, Bacioglu M, Huang M, Lovestam S, Shi Y et al (2022) Age-dependent formation of TMEM106B amyloid filaments in human brains. Nature 605:310–314. https://doi.org/10.1038/s41586-022-04650-z
Stroobants S, D’Hooge R, Damme M (2021) Aged Tmem106b knockout mice display gait deficits in coincidence with Purkinje cell loss and only limited signs of non-motor dysfunction. Brain Pathol 31:223–238. https://doi.org/10.1111/bpa.12903
Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA et al (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102:15545–15550. https://doi.org/10.1073/pnas.0506580102
Takeuchi H, Iba M, Inoue H, Higuchi M, Takao K, Tsukita K et al (2011) P301S mutant human tau transgenic mice manifest early symptoms of human tauopathies with dementia and altered sensorimotor gating. PLoS ONE 6:e21050. https://doi.org/10.1371/journal.pone.0021050
Van Deerlin VM, Sleiman PM, Martinez-Lage M, Chen-Plotkin A, Wang LS, Graff-Radford NR et al (2010) Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP-43 inclusions. Nat Genet 42:234–239. https://doi.org/10.1038/ng.536
van der Touw W, Chen HM, Pan PY, Chen SH (2017) LILRB receptor-mediated regulation of myeloid cell maturation and function. Cancer Immunol Immunother 66:1079–1087. https://doi.org/10.1007/s00262-017-2023-x
van der Zee J, Van Langenhove T, Kleinberger G, Sleegers K, Engelborghs S, Vandenberghe R et al (2011) TMEM106B is associated with frontotemporal lobar degeneration in a clinically diagnosed patient cohort. Brain 134:808–815. https://doi.org/10.1093/brain/awr007
Wan YW, Al-Ouran R, Mangleburg CG, Perumal TM, Lee TV, Allison K et al (2020) Meta-analysis of the Alzheimer’s disease human brain transcriptome and functional dissection in mouse models. Cell Rep 32:107908. https://doi.org/10.1016/j.celrep.2020.107908
Wang M, Beckmann ND, Roussos P, Wang E, Zhou X, Wang Q et al (2018) The Mount Sinai cohort of large-scale genomic, transcriptomic and proteomic data in Alzheimer’s disease. Sci Data 5:180185. https://doi.org/10.1038/sdata.2018.185
Werner G, Damme M, Schludi M, Gnorich J, Wind K, Fellerer K et al (2020) Loss of TMEM106B potentiates lysosomal and FTLD-like pathology in progranulin-deficient mice. EMBO Rep 21:e50241. https://doi.org/10.15252/embr.202050241
Yanamandra K, Kfoury N, Jiang H, Mahan TE, Ma S, Maloney SE et al (2013) Anti-tau antibodies that block tau aggregate seeding in vitro markedly decrease pathology and improve cognition in vivo. Neuron 80:402–414. https://doi.org/10.1016/j.neuron.2013.07.046
Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC et al (2007) Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 53:337–351. https://doi.org/10.1016/j.neuron.2007.01.010
Zhang B, Carroll J, Trojanowski JQ, Yao Y, Iba M, Potuzak JS et al (2012) The microtubule-stabilizing agent, epothilone D, reduces axonal dysfunction, neurotoxicity, cognitive deficits, and Alzheimer-like pathology in an interventional study with aged tau transgenic mice. J Neurosci 32:3601–3611. https://doi.org/10.1523/JNEUROSCI.4922-11.2012
Zhang T, Pang W, Feng T, Guo J, Wu K, Nunez Santos M et al (2023) TMEM106B regulates microglial proliferation and survival in response to demyelination. Sci Adv. https://doi.org/10.1126/sciadv.add2676
Zhang Y, Parmigiani G, Johnson WE (2020) ComBat-seq: batch effect adjustment for RNA-seq count data. NAR Genom Bioinform. https://doi.org/10.1093/nargab/lqaa078
Zhang Z, Song M, Liu X, Kang SS, Kwon IS, Duong DM et al (2014) Cleavage of tau by asparagine endopeptidase mediates the neurofibrillary pathology in Alzheimer’s disease. Nat Med 20:1254–1262. https://doi.org/10.1038/nm.3700
Zhou X, Brooks M, Jiang P, Koga S, Zuberi AR, Baker MC et al (2020) Loss of Tmem106b exacerbates FTLD pathologies and causes motor deficits in progranulin-deficient mice. EMBO Rep 21:e50197. https://doi.org/10.15252/embr.202050197
Zhou X, Nicholson AM, Ren Y, Brooks M, Jiang P, Zuberi A et al (2020) Loss of TMEM106B leads to myelination deficits: implications for frontotemporal dementia treatment strategies. Brain 143:1905–1919. https://doi.org/10.1093/brain/awaa141
Zhu XC, Yu JT, Jiang T, Wang P, Cao L, Tan L (2015) CR1 in Alzheimer’s disease. Mol Neurobiol 51:753–765. https://doi.org/10.1007/s12035-014-8723-8
Acknowledgements
We thank Philip De Jager for sharing then-unpublished data on the link between TMEM106B and cognitive resilience that sparked our original interest in this protein. We thank Chelsea Jiaqi Zong, Zoe Lai, Jennifer Saldana, and Melissa Comstock for support with the mouse colonies; Gabriella Perez for help with imaging, Cecilia Ljungberg for help with slide scanning, I-Chih Tan for equipment support and repair, and Denise Lanza, Lan Liao, and Jason Heaney for help with mouse creation and cryorecovery. TMEM106B knockout germplasm was provided by the KnockOut Mouse Project (KOMP) Repository and the Mouse Biology Program at the University of California Davis from ES cells generated by the Wellcome Trust Sanger Institute. The results published here are in whole or in part based on data obtained from the AD Knowledge Portal (https://adknowledgeportal.org). The specific human AD RNAseq datasets used here were generated by: 1) the Rush Alzheimer’s Disease Center, Rush University Medical Center, Chicago. 2) Dr. Allan Levey from Emory University based on postmortem brain tissue collected by Dr. Eric Schadt from the Mount Sinai School of Medicine through the Mount Sinai VA Medical Center Brain Bank, and 3) the Mayo RNAseq study led by Dr. Nilüfer Ertekin-Taner, Mayo Clinic, Jacksonville, FL.
Funding
This work was supported by National Institutes of Health grants R21AG056028, RF1AG058188, and P01AG066606-01A1 and -S1 (JLJ), R01AG074009 (IAR and JLJ), F31AG067676-01A1 (CAW), and T32NS043124 (support for GAE), Alzheimer’s Association grant ZEN-19–591129 (JLJ), and by Federal Work Study funds to BCM (support for QN and PJK). This work was assisted by several advanced technology core laboratories at BCM, including the RNA In Situ Hybridization Core, Bioengineering Core, and Genetically Engineered Rodent Models Core. BCM core laboratories were funded in part by National Institutes of Health grants S10 OD016167 and P50 HD103555 (RNA ISH), P30 CA125123 (GERM), and P30 EY002520 (Bioeng).
Author information
Authors and Affiliations
Contributions
JLJ and IAR conceptualized the study; GAE, YH, QN, KWP, RGG, CZ, and PJK performed experimental investigation; GAE, CAW, QN, and IAR contributed to formal analysis; JLJ, CAW, and IAR wrote the paper; JLJ, CAW, YX, and IAR obtained funding for the work.
Corresponding author
Ethics declarations
Conflict of interest
CZ is a paid employee of NeuroScience Associates. The remaining authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Edwards, G.A., Wood, C.A., He, Y. et al. TMEM106B coding variant is protective and deletion detrimental in a mouse model of tauopathy. Acta Neuropathol 147, 61 (2024). https://doi.org/10.1007/s00401-024-02701-5
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00401-024-02701-5