
Abstract
The ubiquitin proteasome system (UPS) orchestrates the turnover of innumerable cellular proteins. In the process of ubiquitination the small protein ubiquitin is attached to a target protein by a peptide bond. The ubiquitinated target protein is subsequently shuttled to a protease complex known as the 26S proteasome and subjected to degradative proteolysis. The UPS facilitates the turnover of proteins in several settings. It targets oxidized, mutant or misfolded proteins for general proteolytic destruction, and allows for the tightly controlled and specific destruction of proteins involved in development and differentiation, cell cycle progression, circadian rhythms, apoptosis, and other biological processes. In neuropathology, alteration of the UPS, or mutations in UPS target proteins may result in signaling abnormalities leading to the initiation or progression of tumors such as astrocytomas, hemangioblastomas, craniopharyngiomas, pituitary adenomas, and medulloblastomas. Dysregulation of the UPS may also contribute to tumor progression by perturbation of DNA replication and mitotic control mechanisms, leading to genomic instability. In neurodegenerative diseases caused by the expression of mutant proteins, the cellular accumulation of these proteins may overload the UPS, indirectly contributing to the disease process, e.g., sporadic Parkinsonism and prion diseases. In other cases, mutation of UPS components may directly cause pathological accumulation of proteins, e.g., autosomal recessive Parkinsonism and spinocerebellar ataxias. Defects or dysfunction of the UPS may also underlie cognitive disorders such as Angelman syndrome, Rett syndrome and autism, and muscle and nerve diseases, e.g., inclusion body myopathy and giant axon neuropathy. This paper describes the basic biochemical mechanisms comprising the UPS and reviews both its theoretical and proven involvement in neuropathological diseases. The potential for the UPS as a target of pharmacological therapy is also discussed.
Similar content being viewed by others

Introduction
The ubiquitin proteasomal system (UPS) provides both a precise and a general means for the cell to dispose of biologically non-useful proteins, including mutant, misfolded, damaged, terminally modified or over-accumulated proteins [44]. The ability of UPS to cause rapid substrate-specific proteolysis also allows it to function as a molecular switch in many signaling pathways, quickly turning off the function of a target protein.
Ubiquitin is a small 8.5 kD protein composed of 76 amino acids. In the process of ubiquitination (also known as ubiquitylation), ubiquitin is covalently attached to lysine residues within target proteins via a peptide bond [44]. In many cases, additional ubiquitin monomers are added to the first ubiquitin to form polyubiquitin chains consisting of four to seven ubiquitin monomers. These polyubiquitinated proteins are then shuttled to a multimeric protease complex known as the 26S proteasome, present in both the nucleus and cytoplasm of mammalian cells. At the proteasome, ubiquitin monomers are released from the target protein and recycled for future use. The target protein is then degraded by proteasomal protease activity.
Ubiquitination is a multistep process
Aaron Ciechanover, Avram Hershko and Irwin Rose shared the 2004 Nobel Prize in Chemistry for elucidating the multistep process of ubiquitin-mediated protein degradation [44]. Ubiquitin is first activated by an E1 ubiquitin-activating enzyme (Fig. 1). This activation is ATP-dependent and results in transient adenylation of ubiquitin. AMP is then released and a thioester bond is formed between the C-terminal carboxyl group of ubiquitin and a sulfhydryl group on an E1 cysteine residue [41]. Ubiquitin is then transferred to a cysteine residue of an E2 ubiquitin conjugating enzyme (Ubc). The ubiquitin-bound E2 Ubc then associates with an E3 ubiquitin ligase capable of recognizing a specific target protein. The Ubc facilitates transfer of activated ubiquitin to the E3 ligase–target protein complex. The E3 ligase catalyzes the formation of an isopeptide bond between a target protein lysine residue and the C-terminal glycine of ubiquitin (Gly76). In rare cases, the Ubc processes both E2 ubiquitin conjugating and E3 ubiquitin ligase activity.

Overview of the ubiquitin proteasome system. E1 ubiquitin activating enzymes transfer activated ubiquitin (aUb) to E2 ubiquitin conjugating enzymes that associate with E3 ubiquitin ligases. E3 ubiquitin ligases catalyze the formation of a isopeptide bond between ubiquitin (Ub) and the target protein. Substrate recognition often requires prior phosphorylation (PO 4 ) of the target protein. E4 enzymes promote the formation of polyubiquitin chains. Receptor proteins containing UBL and UBA domains transfer the polyubiquitinated target protein to the proteasome for cleavage in to free amino acids and small peptides. Deubiquitinating enzymes (DUBs) promote recycling of ubiquitin monomers for reactivation by E1
A more recently described E4 activity has been attributed to at least two protein factors that facilitate conjugation of additional ubiquitin monomers to form the polyubiquitin chain, usually through linkages of lysine 48 [64]. Chains formed by linkages involving other lysine residues in ubiquitin, particularly lysine 63, also occur, as do chains with mixed lysine 48 and lysine 63 linkages. The type of linkage, 48, 63, or mixed, affects the fate of the ubiquitinated protein. K48-linked polyubiquitinated proteins are generally degraded in the proteasome and K63-linked polyubiquitinated proteins are ultimately degraded in the lysosome [62, 123].
Another class of UPS proteins sometimes known as shuttle or ubiquitin receptor proteins act as carriers to deliver polyubiquitinated proteins to the 26S proteasome [8, 55]. The 26S proteasome is composed of a 20S core subunit capped by two 19S regulatory subunits, one at either end of the cylindrically shaped core to form a barrel-like structure [44]. The core subunit itself is composed of four rings. Ubiquitin-associated (UBA) domains of the ubiquitin receptor protein bind the polyubiquitin chains of target proteins and ubiquitin-like (UBL) domains of the receptor protein bind the 19S regulatory subunits of the proteasome [8, 55]. Like E1-mediated ubiquitin activation, transfer of the polyubiquitinated protein to the proteasome is an ATP-dependent process.
Proteolytic degradation of the target protein takes place within the rings of the core subunit following removal of the ubiquitin monomers by deubiquitination enzymes (DUBs) and unfolding of the substrate protein [44]. The outer rings of the core act as gates to the core and each is composed of seven α-subunits. The inner two rings contain the protease activity and are each composed of seven β-subunits.
Proteins with UBA domains also bind monoubiquitinated proteins and are involved in initiating endocytosis [62, 97]. These proteins may also protect monoubiquitinated proteins from polyubiquitination or deubiquitination. DUBs are thought to also act to deubiquinate ubiquitinated proteins to prevent their targeting to the proteasome. Ubiquitin receptor proteins effectively bind tetraubiquitin chains and may also function to protect polyubiquitinated proteins from unnecessary polyubiquitin chain expansion or deubiquitination prior to reaching the proteasome [55]. Thus, the process of ubiquitination appears to be highly regulated at various levels of the UPS.
Monoubiquitination, sumoylation and neddylation
Nuclear and cytosolic proteins generally undergo polyubiquitination and degradation by the 26S proteasome, as do some membrane proteins [129]. Membrane proteins may also be monoubiquitinated, triggering their cytoplasmic internalization and degradation in either the endosome–lysosome system or the proteasome [97]. Cbl is a plasma membrane-associated E3 ligase that along with an E2 Ubc, either monoubiquitinates or polyubiquitinates several membrane proteins [74].
More recently, a family of ubiquitin-like modifiers known as small ubiquitin-like modifier (SUMO) 1-4 has been described. These modifiers are small proteins similar in size to ubiquitin. Through a process analogous to monoubiquitination termed sumoylation, SUMO monomers are conjugated to target proteins. In some cases monoubiquitination or sumoylation may act to protect proteins from ubiquitin-dependent degradation [59] and in other cases they appear to trigger polyubiquitination [112]. Sumoylation and monoubiquitination at one or two lysines within a target protein may also alter the subcellular location of the protein [98]. Monoubiquitination of membrane proteins may trigger their cytoplasmic internalization [97]. Monoubiquitination of the p53 tumor suppressor causes it to be transported from the nucleus to the cytoplasm and transport may be further enhanced by sumoylation of p53 [71].
An additional small ubiquitin-like posttranslational modifier is known as neural precursor cell expressed, developmentally down-regulated 8 (NEDD8) [22]. The functions of neddylation are less understood; however, neddylation of cullin proteins, essential subunits of certain E3 ubiquitin ligase complexes, appears to be required for normal ligase function (Fig. 2).
Ubiquitination is substrate specific
E1 activity is nonspecific with regard to the protein targets of the UPS and only two isoforms of this enzyme exist in the cell (E1a and E1b) [18]. By comparison about 50 different E2s are known [10]. E3 ligases are the most variable and abundant with over 500 putative E3s identified so far. A given E2 can often associate with multiple different E3s providing for a large variety of functional E2/E3 activities and a relatively high degree of target substrate specificity. However, this specificity is not absolute. Many E3 proteins are able to ubiquitinate several, usually biochemically related, substrate target proteins. These related proteins often share a common small amino acid sequence that acts as a relatively substrate-specific E3 binding domain. Many target proteins are thought to be ubiquitinated by only one E3, while others, the tumor suppressor p53 for example, can be ubiquitinated by multiple E3s [71, 116, 141].
Most of the E3 ligases are classified as to whether they contain homologous to E6AP COOH-terminus (HECT) or really interesting new gene (RING) domains. A third smaller class of E3 ligases is referred to as U-box containing ligases. Both HECT and RING types may possess E3 ligase function as single proteins, for example the HECT-type ligase UBE3A and the RING-type ligase Cbl (Fig. 2). RING-type E3s, however often associate with scaffolding and other auxiliary proteins to form multimeric functional complexes. These complexes can associate with different regulatory subunits, which act as substrate-specific adaptor proteins providing for even greater substrate specificity. Examples include the Skp/cullin/F-box (SCF) and anaphase promoting complex/cyclosome (APC/C) E3 ubiquitin ligases that exhibit specificity for different substrates depending on the adaptor protein bound to the complex (Fig. 2). SCF complexes containing isoforms of the F-box protein β-TrCP can ubiquitinate several proteins, including the DNA transcriptional regulator β-catenin and the cell cycle progression regulator Emi1 [40, 43]. In some instances, substrate proteins must be phosphorylated first at specific sites before they can be recognized by the E3 ubiquitin ligase. This may provide an additional level of control as to when and where a target protein is ubiquitinated. For instance, SCF substrates generally must be phosphorylated first on the serine residues (S) of the consensus amino acid sequence motif DSGXXS before the F-box regulatory subunit can bind the substrate [43, 65]. Different substrates may be phosphorylated by different kinases to provide another layer of control, and the activity of the E3 ligase itself is often regulated by phosphorylation [105]. Lastly, the subcellular compartmentalization of the substrate and E3 ligase may add additional substrate specificity. Examples of specific subcellular locations include the cell membrane, nuclear and cytoplasmic compartments, chromatin, centrosome, mitotic spindle, and neuronal synapse. Thus, with regard to control of cell signaling and many biochemical processes, ubiquitination of specific substrates is a tightly controlled process that prevents the random destruction of proteins.

Cbl family E3 ligases and UBE3A E3 ligase are single proteins that associate with an E2 Ubc and the target substrate. The SCF and APC/C E3 ubiquitin ligases are multimeric complexes. The APC/C has only two known adapter subunits, Cdc20 and Cdh1 (not to be confused with E-cadherin, which is sometimes also referred to as Cdh1). The SCF can associate with several different substrate adapter proteins known as F-box proteins
The UPS facilitates diverse biological functions
In addition to its general wastebasket-like function, ubiquitination is also a highly specific way of controlling protein expression in signal transduction systems, in cell–cell communication during development, and at the neural synapse [113, 147]. Other diverse functions controlled by the UPS include regulation of gene transcription via monoubiquitination and deubiquitination of histones, driving circadian clocks, the auxin-mediated response to light in plants, and the regulation of cell cycle progression and apoptosis [26, 113, 131]. The UPS is also important for the functioning of the immune system and is required for MHC class I antigen presentation [39, 83, 144].
The UPS in neural development
Notch is an integral membrane protein responsive to extracellular signals. Notch has been implicated in neural stem cell proliferation, self-renewal and differentiation [73]. It has also been implicated in plasticity of adult brain hippocampus. It is cleaved by γ-secretase activity, which allows its signal transduction component to translocate to the nucleus and activate transcription. γ-Secretase is derived from the presenilin complex, the constituents of which are all UPS substrates [82]. Notch protein stability is also negatively regulated by the SCF ubiquitin ligase.
The SCF ubiquitin ligase substrate β-catenin is a transcription factor that also regulates proliferation of neural precursor cells. An amazing study by Chenn and Walsh [13] showed that transgenic mice expressing a stabilized form of β-catenin in neural precursor cells develop enlarged cerebral cortices with complex gyral patterns resembling those of primates.
Bone morphogenic proteins (BMPs) and Shh proteins are secreted proteins involved in dorsal–ventral induction in neurogenesis and other developmental and signaling processes [37]. The downstream effector proteins of BMP and Shh, Smad and Gli proteins, respectively, are both transcription factors regulated by ubiquitin-dependent proteolysis, as are BMP cell membrane surface receptors [99]. Furthermore, both Gli proteins and Smad protein complexes induce transcription of E3 ligase proteins. Additionally, the APC/C E3 ligase is involved in the regulation of axonal growth and patterning in the developing cerebellum [66].
UPS pathology in neurodegenerative disease
Alzheimer disease
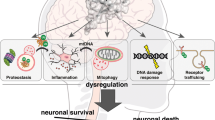
Dysregulation of the UPS appears to be both a cause and result of neurodegenerative disease processes. In Alzheimer disease, hyperphosphorylation of the microtubule-associated protein tau results in the accumulation of paired helical tau filament involved in the formation of neurofibrillary tangles and neuritic plaques. These abnormal tau aggregates are immunoreactive for anti-ubiquitin antibodies suggesting that the cell is attempting to dispose of these abnormal proteins. Kinases implicated in tau phosphorylation include those involved in phosphorylation of substrates to allow recognition by ubiquitin ligases, namely casein kinase 1 and glycogen synthase kinase 3 (GSK3) which phosphorylate SCF-β-TrCP E3 ligase substrates [24, 56]. The UPS is also involved in early stages of autophagy, and neuronal granulovacuolar degeneration structures are ubiquitin positive [15, 62]. Relative dysfunction or inhibitory overloading of the UPS may contribute to the abnormal accumulation of phosphorylated and ubiquitinated tau. Interestingly, a frameshift mutant of ubiquitin, UBB+1, found in some sporadic and hereditary Alzheimer disease and polyglutamine repeat disease patients, inhibits the UPS and enhances toxic protein aggregation in a yeast model system [30, 124]. Indeed anti-ubiquitin antibodies are good, albeit nonspecific, markers for many of the abnormal cellular inclusions and neuropil deposits found in Alzheimer disease, corticobasal degeneration, progressive supranuclear palsy, Picks disease, other frontotemporal dementias, multiple system atrophy, Parkinson disease, amyotrophic lateral sclerosis and others [15] (Fig. 3).

Anti-ubiquitin immunostaining in neurodegenerative disease. Ubiquitin antibodies highlight Alzheimer disease pathology including hippocampal plaques (a) and neurofibrillary tangles (b). Brain stem (c) and cortical Lewy bodies (d) are also ubiquitin positive. Ubiquitin-positive inclusions are found in the nucleus of cortical neurons in Huntington disease (e) and in the cytoplasm of anterior horn neurons in motor neuron disease (f). Original magnifications are ×200 (a, b) and ×400 (c–f)
The relative importance of tau neuronal pathology and β-amyloid deposition in extracellular plaques in Alzheimer disease is a perennial topic of debate. β-amyloid is derived from amyloid precursor protein (APP), a neuronal transmembrane protein that appears to function as a cell membrane receptor and is also involved in synaptic vesicle recycling [12, 14].
APP binding protein-1 (APP-BP1) binds APP intracellularly at the membrane [14]. APP-B1 also associates with UBA3 and together they act as an E1-like activating enzyme for the process of neddylation. The APP-B1/UBA3 complex and the NEDD8 conjugating enzyme Ubc12 appear to be necessary for neddylation of cullin proteins and normal cell cycle progression from S-phase to mitosis in Chinese hamster ovary cells [12]. Overexpression of APP or APP-BP1 in cultured primary rat neurons causes DNA synthesis and apoptosis by a mechanism dependent upon the presence of UBA3, Ubc12, and caspase-6 activity.
Similar to Notch, APP is subjected to intramembrane proteolysis via a presenilin-dependent γ-secretase activity. This results in release of β-amyloid, p3 and the APP intracellular cleavage domain fragment (AICD). AICD can translocate to the nucleus and result in apoptosis of cells in culture. It is postulated that APP-B1-mediated activation of neddylation, and presumably cullin-based ubiquitin ligases, signals postmitotic neurons to re-enter the cell cycle triggering their apoptosis similar to AICD [12]. Overexpression of APP increased APP-BP1 levels in cell membrane lipid rafts in cultured neurons and increased APP-B1 is found in lipid rafts in the hippocampus of Alzheimer disease patients [11]. These findings suggest that in addition to the role of β-amyloid deposition in apparently disrupting brain circuitry, cellular signaling aberrations involving APP-B1 and AICD resulting in neuronal toxicity and death may be additional mechanisms of APP-mediated Alzheimer neurodegeneration [61].
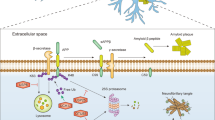
p62 is a polyubiquitin binding protein processing UBA- and UBL-like domains. It may thus act as a receptor/shuttle protein capable of transferring polyubiquitinated proteins such as polyubiquitinated tau to the 26S proteasome. Indeed p62 can be found associated with tau in neurofibrillary tangles. p62 appears to bind K63-linked polyubiquitin chains and promote the clearance of K63-linked polyubiquitinated proteins by autophagy [123]. It also may serve as a scaffold protein to promote K63-linked polyubiquitination of membrane proteins by the RING-type E3 ligase tumor necrosis factor receptor associated factor 6 (TRAF6). p62 also associates with itself and other proteins in a cytoplasmic complex known as the sequestosome and may thus be involved in delivery of proteins to the lysosome (Fig. 4).

Simplified model of the UPS, endosome–lysosome system and autophagy cellular pathways. Cell membrane proteins that are UPS substrates (S) are generally monoubiquitinated and degraded in the endosome–lysosome system, or polyubiquitinated and delivered to the 26S proteasome by ubiquitin receptor proteins (UR). Nuclear proteins are degraded in the proteasome within the nucleus or translocated to the cytosol where they can be degraded by various routes. Polyubiquitinated nuclear proteins may be deposited into neuronal intranuclear inclusions (NII) in disease states. Polyubiquitinated cytosolic proteins and UPS machinery proteins such as E2s, E3s and proteasomal subunits may be transported along microtubules (dotted lines) to be deposited into aggresomes, which may form neuronal cytoplasmic inclusions (NCI). Similar to other SCF E3 ligase substrates, tau is probably first phosphorylated by casein kinase 1, which facilitates further phosphorylation by GSK3. Hyperphosphorylated tau is deposited into neurofibrillary tangles, which like NCIs may be cleared by autophagy. K63-linked polyubiquitinated chains bind p62 and form complexes known as sequestosomes. Aggregation of insoluble misfolded proteins is prevented by heat shock proteins (HSP), which also promote the fusion of sequestosomes to autophagic vacuoles and ultimately the lysosome
A recent study of p62 knock-out mice revealed that adult animals developed age-dependent hyperphosphorylated tau, neurofibrillary tangles and neurodegeneration and a behavioral and cognitive phenotype mimicking Alzheimer disease [104]. Biochemical fractionation revealed K63-linked polyubiquitinated tau aggregates. p62 therefore binds with K63-linked polyubiquitinated tau to prevent its aggregation so that it can be sequestered and delivered to the lysosome. In support of this pathway, tau binds HSP90, which may facilitate tau phosphorylation [126], but is also important in the binding of proteins to the lysosomal membrane. Interference with this degradative pathway may produce Alzheimer disease or Alzheimer-like dementia.
Frontotemporal lobar degeneration (FTLD) includes tau immunopositive and tau immunonegative forms. FTLD with ubiquitin-immunoreactive lesions (FTLD-U) is a major type of tau negative FTLD, which can be further subdivided into TAR DNA binding protein 43 (TDP-43) immunopositive and immunonegative types [46, 53, 101, 108]. TDP-43 negative FTLD-U subtypes demonstrate either both nuclear and cytoplasmic ubiquitin-positive neuronal inclusions or cytoplasmic inclusions only. Cytoplasmic, but not nuclear inclusions are also p62 positive [46]. Thus, p62 is linked to both cytoplasmic ubiquitination and autophagy in neurons, but may not be involved in proteasome-mediated degradation of proteins. The latter may be less important compared to autophagy in Alzheimer disease and frontotemporal dementias.
Parkinson disease
Familial juvenile onset Parkinson’s disease is caused by a defect in an SCF E3 ubiquitin ligase component known as parkin (PARK2) (Fig. 2). Substrates of parkin-mediated ubiquitination include a minor glycosylated form of α-synuclein (α-sp22), the α-synuclein binding protein synphilin-1, the parkin-associated endothelin receptor-like receptor (Pael-R) and cyclin E [120]. Along with parkin, both α-sp22 and Pael-R are found in the Lewy body cytoplasmic inclusions characteristic of most forms of Parkinson disease. Overexpression of Pael-R results in the rapid loss of dopaminergic striatonigral neurons in rats with no observed effect on GABAergic neurons of the globus pallidus [25]. Since Pael-R protein would tend to accumulate in the presence of mutated parkin, this supports the possible importance of Pael-R in the pathogenesis of autosomal recessive juvenile Parkinsonism.
Parkin also ubiquitinates the ataxin-2 protein which is disrupted by expansion of its polyglutamine repeat in spinocerebellar ataxia type 2 [50]. Like the APC/C E3 ligase, parkin is believed to function as a tumor suppressor protein [33, 77].
Genes mutated in other forms of autosomal recessive Parkinson disease include phosphatase and tensin homologue deleted on chromosome ten (PTEN)-induced putative kinase 1 (PINK1, also known as PARK6) and DJ-1 (PARK7). A recent study has shown that PINK1 and DJ-1 form a complex with parkin and promote parkin’s ubiquitin ligase activity [142]. Another study showed that parkin directly interacts with PINK1 and blocks ubiquitin-dependent proteolysis of PINK1 [117]. These studies implicate dysfunction of parkin E3 ligase activity as a common etiologic mechanism for recessive forms of inherited Parkinson disease, all of which may be possibly caused by over accumulation of Pael-R. PINK1/parkin also appears to be important in mitochondrial function, which is deficient in Parkinson disease.
Rare autosomal dominant forms of Parkinson disease in German and Japanese kindred have been linked to polymorphisms in the DUB enzyme known as ubiquitin carboxy-terminal hydrolase L1 (UCH-L1) [38, 115]. UCH-L1 is an abundant protein in neurons and can also act as an E3 ubiquitin ligase when dimerized. In addition to α-synuclein and synphilin-1, Lewy bodies found in most forms of Parkinson disease contain UCH-LI and mice lacking UCH-L1 show neurodegeneration [115]. Furthermore, similar to parkin, overexpression of UCH-L1 leads to aggresome formation in response to proteasomal inhibition [2].
Aggresomes are organized cellular structures in which nonfunctional aggregated proteins are shuttled along microtubules by dynein-dependent transport [2, 67] (Fig. 4). Lewy bodies are though to form by growth or coalescence of perinuclear aggresomes. Neurons in general may be more susceptible to mutations in UPS components such as E3 ligases and DUBs because they have lower overall cellular UPS activity capacity compared to glia [127].
It has been suggested that ubiquitination and sequestration of proteins such as α-synuclein and synphilin-1 in aggresomes and large cytoplasmic inclusions is a mechanism used by the cell to protect itself from the toxicity of these proteins [80]. This is plausible considering evidence that small oligomers of prions can overload the proteasome and are toxic to the cell [68]. This may also explain why Parkinson disease occurs in the autosomal recessive form that harbors defective parkin E3 ligase despite that dopaminergic nigral neurons do not develop Lewy bodies in these patients.
Yet, there is conflicting evidence for the pathogenesis of α-synuclein aggregation. α-Synuclein is mono- and di-ubiquitinated by the E3 ubiquitin ligase SIAH-1. This reportedly promotes α-synuclein aggregation and enhances its neural toxicity [75, 109]. Ubiquitination of synphilin-1 by SIAH-1 also promotes its aggregation into inclusions [28, 80]. Interestingly, SIAH-1-mediated α-synuclein ubiquitinization was inhibited by the Parkinson disease-associated A30P α-synuclein mutation supporting a role for toxicity of the unaggregated form of mutant α-synuclein in inhibiting UPS function.
Leucine-rich repeat kinase 2 (LRRK2) (PARK8) is mutated in about 2–5% of sporadic and autosomal dominant Parkinson disease, respectively [34, 149]. LRRK2 is ubiquitinated by the E3 ubiquitin ligase carboxy terminus of Hsc70-interacting protein (CHIP), which associates with HSP70 [63]. CHIP and parkin both can bind HSP70 and ubiquitinate polyglutamine repeat expanded proteins such as ataxin-3 [95]. LRRK2 is found associated with α-synuclein in Lewy bodies and in glial cytoplasmic inclusions in multisystem atrophy [49]. LRRK2 has been shown to coimmunoprecipitate with parkin consistent with it also being a parkin substrate. One study showed that LRRK2 aggregates in LRRK2-transfected cells were increased by parkin coexpression, which increased ubiquitination of aggregated LRRK2 [142]. LRRK2 also associates with microtubules and induces protein aggregation at the cell membrane, synapses, and membrane-bound organelles suggesting that it has a physiological role in autophagy. Indeed cytoplasmic neuronal inclusions resulting from proteasomal inhibition can be cleared by autophagy in model systems.
Prion diseases
Spongiform encephalopathies such as sporadic and variant Creutzfeldt–Jakob disease are neurodegenerative conditions caused by mutant hereditary or transmissible prion proteins. The unique property of prions is their ability to induce abnormal conformational changes in normal prion proteins. In some forms of prion disease, e.g., Kuru, abnormal prion proteins form ubiquitin immunopositive amyloid plaques. As misfolded mutant proteins pathological prions are “born” UPS substrates. Although disease-associated prions are deposited into perinuclear cytoplasmic aggresomes in prion infected cultured neurons and infected mouse brains, evidence suggests that prion-mediated neuronal toxicity is due to inhibition of proteasome proteolytic activity by prion oligomers [68, 69].
Additional evidence linking prion pathology to disruption of the UPS has come from mouse models. Mutation of the RING-type ubiquitin ligase mahogunin results in a spongiform encephalopathy in mice histologically mimicking human prion disease [58]. Mahogunin monoubiquitinates tumor susceptibility gene 101 (TSG101), a protein involved in endosomal sorting. Mahogunin knockdown by siRNAs interferes with endosome to lysosome trafficking of EGFR receptors in cultured cells. Study of mice strains exhibiting variable incubation times for prion diseases identified an E3 ligase known as HECT domain containing 2 (HECTD2) as a quantitative trait gene associated with decreased disease incubation periods. Furthermore, analysis of human prion disease patient samples identified specific HECTD2 haplotypes associated with variant CJD and Kuru, further implicating a role for HECTD2 ubiquitin ligase in prion disease [84]. The above studies link both ubiquitin ligase or proteasome dysfunction and endosome–lysosome dysfunction to the pathogenesis of prion diseases.
Motor neuron disease
Amyotrophic lateral sclerosis (ALS) is a rapidly progressive fatal motor neuron disease that occurs in sporadic and inherited forms. Approximately 20% of the familial cases harbor mutations in superoxide dismutase-1. In ALS, spinal motor neurons demonstrate ubiquitin immunopositive cytoplasmic inclusions (Fig. 3). Two recent studies have revealed that these inclusions are also immunopositive for TDP-43 in all cases of ALS not associated with superoxide dismutase-1 mutations [87]. This finding is accompanied by decreased normal expression of TDP-43 in the nucleus. TDP-43 is a DNA binding protein involved in the pathogenesis of HIV infection and has no known functional role in the UPS. Its presence in both FTLD and ALS inclusions strongly indicates a common link in the neurodegenerative process in these disorders which is at least in part mediated by the UPS [60, 103]. Furthermore, the amyotrophic lateral sclerosis/parkinsonism–dementia complex of Guam (ALS/PDC) shows histopathological overlap with all major types of neurodegeneration further illustrating the central role of the UPS in the pathophysiology of neurodegeneration [91, 122].
ALS2 is mutated in a juvenile autosomal recessive form of ALS and codes for an endosomal membrane associated protein involved in endosomal membrane fusion and endosomal trafficking along microtubules. Mutant ALS2 is unstable and is rapidly degraded by the proteasome [143]. Thus, like other neurodegenerative conditions, motor neuron disease has been linked to dysfunction of both the ubiquitin proteasomal and endosome/lysosome systems.
Polyglutamine repeat diseases
Huntington disease
Huntington disease is characterized by expression of toxic polyglutamine repeat-expanded mutated forms of the protein huntingtin and its aggregation in intranuclear and cytoplasmic inclusions. These polyglutamine repeat expanded proteins are degraded by the UPS, components of which are also found within the inclusions, e.g., ubiquitin, proteasomal subunits, chaperones and autophagy proteins [30, 51] (Fig. 3). Decreased UPS function appears to be associated with huntingtin neuronal toxicity. Although the mechanism for this is not known [30, 140], neuronal toxicity may, at least in part, be due to synaptic dysfunction [133]. It is possible that as postulated for other mutant UPS substrates, such as prions, misfolded mutant huntingtin protein effectively inhibits the proteasome machinery allowing toxicity from over-accumulated mutated huntingtin or other proteins.
Lysine 48-linked polyubiquitin is increased in the brains of mouse Huntington models and Huntington disease patients further suggesting proteasome dysfunction [4]. In cell culture, induction of neuronal death by mutated huntingtin depends on the amount of mutant protein and the length of the polyglutamine expansion. Neuron death can occur in the absence of inclusions, which are associated with increased neuronal survival consistent with toxicity from unaggregated mutant huntingtin [3]. Interestingly, p53 binds a p53-responsive element in the huntingtin gene activating its transcription [29]. This potentially links huntingtin expression to p53-mediated stress responses and implicates control of p53 stability as a possible effector of the expression of mutant huntingtin.
Spinocerebellar ataxias
The spinocerebellar ataxias (SCA) are characterized by progressive cerebellar ataxia, often accompanied by other long tract and variable ophthalmic signs [5, 74, 107]. Most are autosomal dominant disorders and many are mediated by pathological expansion of polyglutamine repeats within functional proteins. Spinocerebellar ataxia type 1 (SCA1) results from polyglutamine repeat expansion of the protein encoded by the gene ataxin-1. Ataxin-1 protein is a substrate of the E3 ubiquitin ligase UBE3A and mutant ataxin-1 is resistant to ubiquitin-dependent proteolysis. An ataxin-1 interacting protein known as ataxin-1 interacting ubiquitin-like protein (A1Up) possesses N-terminal UBL and C-terminal UBA domains [107]. It also contains heat shock protein chaperonin-binding motifs. Heat shock chaperones, e.g., HSP70 and HSP90, and chaperonin proteins prevent protein aggregation, especially of misfolded proteins and are involved in the ubiquitin-dependent degradation of misfolded and otherwise damaged proteins. The UBL is necessary for A1Up’s association with the 19S proteasome and this association is inhibited by misfolded mutant polyglutamine repeat ataxin-1 [107]. Thus, disruption of the delivery of ubiquitinated ataxin-1 to the proteasome may account for ubiquitin immunopositive nuclear inclusions observed in Purkinje, pontine and inferior olive neurons in SCA1. The interaction of ataxin-1 with the deubiquitinating enzyme USP7 is also weakened by ataxin-1 mutation. This may additionally contribute to abnormal accumulation of ubiquitinated mutant ataxin-1 [47].
As previously noted, the SCA2 gene product ataxin-2 is ubiquitinated by parkin [50]. Ataxin-2 binds membrane-associated proteins and transmembrane cell surface receptors. A recent study has shown that ataxin-2 can also associate with endophilin, a protein involved in synaptic vesicle endocytosis, and the Cbl E3 ubiquitin ligase involved in membrane receptor internalization [102]. Ataxin-2 thus appears to be involved in endocytic cycling of membrane proteins.
The polyglutamine expanded mutant protein of SCA3 (Machado–Joseph disease) contains an ubiquitin-interacting motif [5]. Ataxin-3 has been shown to preferentially bind K63-linked polyubiquitin chains and act as a deubiquitinating enzyme (DUB) [125]. Ubiquitination of ataxin-3 itself may enhance its deubiquitinating activity.
SCA7 is also a polyglutamine repeat disorder. Ataxin-7 was found to associate with the R85 splice variant of the Cbl-associated protein (CAP), which contains a nuclear localization signal domain and is therefore translocated to the nucleus [74]. Mutant ataxin-7 interferes with the nuclear translocation of the transcription factor NF-κB resulting in decreased BCL-X(L) expression and induction of apoptosis in cultured cerebellar neurons [132]. Both translocation of NF-kB to the nucleus and the nuclear activity of NF-κB are dependent on the ubiquitin-mediated destruction of inhibitors of κB (IκBs). More recently ataxin-7 mutants have been shown to inhibit gene transcription and proteasome activity [121, 132]. Inhibition of NF-κB-mediated gene transcription by mutant, polyglutamine repeat expanded ataxin-7 can therefore be explained by its apparent ability to inhibit ubiquitin-dependent proteolysis. Ataxin-7 mutants may selectively interfere with the UPS-mediated degradation of proteins bound for the nucleus.
The above-mentioned SCA types all involve mutation of UPS component proteins (Table 1). Thus, this group of diseases associated with similar clinical manifestations appears to be linked by unique disruptions of the UPS, particularly with regard to turnover of membrane and/or synaptic proteins in SCA2 and nuclear proteins in SCA1 and SCA7. Mutant ataxin-2 may also inhibit parkin possibly contributing to the SCA2 disease process. SCA3 appears to differ in that the role of ataxin-3 in the deubiquitination of K63-linked ubiquitin chains suggests dysregulation of autophagy.
Angelman, Prader–Willi, Rett and Autism
Angelman syndrome is characterized by microcephaly, severe developmental delay, seizures, ataxia, a stereotypical hand flapping, and a happy excitable demeanor. It is associated with deletions of chromosomal segment 15q11-13 and is caused by loss of function of the ubiquitin ligase alternately known as UBE3A or E6AP. UBE3A is the same E3 ligase that mediates the human papillomavirus E6 protein-induced ubiquitination of the p53 tumor suppressor protein. Loss or mutation of the maternally inherited UBE3A allele is sufficient to cause Angelman disease [111]. Differences in expression patterns of paternally and maternally inherited UBE3A alleles are due to differential gene methylation (imprinting). Unlike paternally derived UBE3A, maternal UBE3A is active in the cerebellum and hippocampus. It is needed to maintain CA1 synaptic plasticity, which is lost in UBE3A −/− mice and is necessary for normal experience-based maturation of the visual cortex [146]. Known substrates of UBE3A have not been linked to the pathogenesis of Angelman syndrome. The disease can be prevented, however, in an Angelman mouse model by mutation of the inhibitory phosphorylation site of the alpha subunit of calcium-calmodulin-dependent protein kinase II (α-CaMKII), suggesting Angelman syndrome may result from increased inhibitory phosphorylation of α-CaMKII [27]. The important target of UBE3A in Angelman disease may thus be a direct or indirect regulator of α-CaMKII phosphorylation. Since α-CaMKII is also important in maintaining hippocampal plasticity, this is a highly plausible hypothesis [27].
Prader–Willi syndrome, characterized by hyperphagia, cognitive impairment and hypogonadism, results from loss of genetic material from the same chromosomal region as in Angelman, but differs in that the paternally derived segment is lost. UBE3A has not been associated with Prader–Willi in which multiple different genes have been implicated. A common misconception is that Angelman and Prader–Willi are the same disease manifesting differently in the two sexes. They are of course genetically distinct disorders and both can occur in either boys or girls.
Rett syndrome shows similar clinical manifestations to Angelman including microcephaly, cognitive impairment, hypotonia, seizures, ataxia and abnormal hand movements; however, it occurs nearly exclusively in girls. Misdiagnoses between these two conditions in girls are therefore not uncommon. Unlike Angelman, Rett patient behavior is characterized by frequent loss of speech and other previously attained skills, and poor socialization similar to autism. Most cases are due to sporadic mutations of methyl-CpG-binding protein-2 (MECP2) on the X chromosome. Male fetuses with the mutation are usually nonviable or die before 2 years of age. Surviving males are 46, XXY. MECP2 binds methylated DNA and acts as a transcriptional repressor and its mutation can lead to disruption of methylation imprinting of the UBE3A gene [88]. Decreased UBE3A has indeed been observed in the brain tissue of MECP2-deficient mice and Rett syndrome patients.
Further evidence of the global importance of UBE3A, other ubiquitin ligases and related UPS proteins in cognitive function has come in a very recent publication identifying these proteins along with neuronal adhesion proteins as exhibiting copy number variation in a cohort of over 800 patients suffering from autism spectrum disorders [36]. UBE3A dysfunction appears to produce a large spectrum of pediatric cognitive functional levels depending on the mechanism and severity of UBE3A loss, i.e., autism to Rett to Angleman syndromes.
The UPS in diseases of the PNS and skeletal muscle
The UPS appears be involved in the initiation of Wallerian degeneration and a side effect of proteasomal inhibitors is peripheral neuropathy [148]. Altered ubiquitin-dependent degradation of Schwann cell proteins may contribute to the pathogenesis of certain neuropathies [32]. In the Trembler J mouse model of Charcot–Marie–Tooth disease type 1A demyelinating neuropathy the turnover rate of nascent peripheral myelin protein 22 (PMP22) is decreased. These animals show proteasome impairment correlating with increased levels of polyubiquitinated PMP22 in nerves [32].
Giant axon neuropathy is a severe autosomal recessive motor and sensory disorder presenting in infants and children that is associated with disruption of neurofilaments [145]. It is caused by the mutation of an ubiquitin ligase substrate adapter known as gigaxonin, which targets the light chain of microtubule-associated protein 1B (MAP1B), MAP8 and the tubulin folding chaperone tubulin folding cofactor B (TBCB) [1]. MAP1B over accumulation is toxic to neurons although the mechanism of neurofilament disruption is not known [16].
A significant rare untoward effect of statin-type cholesterol lowering drugs such as atorvastatin is a necrotizing myopathy often accompanied by an inflammatory response, so called statin-induced myopathy. Selective vulnerability of type IIb muscle fibers occurs in statin-induced myopathy [135]; however, the precise pathophysiologic mechanisms of the condition remain poorly understood. Mitochondrial dysfunction has been implicated because statins inhibit both cholesterol and ubiquinone synthesis. The latter can potentially interfere with mitochondrial oxidative phosphorylation.
Increased turnover of muscle proteins by the UPS may be important in the activation of events that lead to statin-induced myopathy. The muscle-specific F-box protein MAFbx (atrogin-1) is part of a SCF E3 ligase induced in muscle atrophy. Hanai et al. [42] found that MAFbx is also induced by lovostatin and that knockdown of MAFbx prevented lovastatin-induced muscle damage in zebrafish embryos. SCF-MAFbx targets the muscle transcription factor MyoD [110]. MyoD is important in both muscle development and maintenance of the differentiated phenotype of type IIb fibers. MAFbx gene silencing by RNA interference inhibits MyoD proteolysis and expression of a stabilized K133R MyoD mutant not recognized by SCF-MAFbx prevents atrophy of mouse skeletal muscle fibers in model systems [72]. These findings support the hypothesis that increased ubiquitination of skeletal muscle proteins, particularly MyoD, may be an important final common pathway in toxic myopathies caused by statins, and perhaps other drugs.
Overloading or other perturbations of ubiquitin-dependent proteolysis are probably involved in inclusion myopathies, such as inclusion body myositis, the myofiber inclusions of which are strongly ubiquitin immunopositive. IBM degenerating fibers contain hyperphosphorylated tau and casein kinase 1α, a kinase that phosphorylates tau and other SCF E3 ligase substrates [56].
Hereditary inclusion body myopathy with Paget disease of bone and frontotemporal dementia (IBMPFD) is a rare autosomal dominant disease associated with mutations in the gene encoding valosin-containing protein (VCP) [134]. VCP is a chaperone protein involved in the ubiquitin-dependent degradation of membrane and cytoplasmic proteins and in translocation of misfolded proteins from the endoplasmic reticulum into the cytoplasm for proteasomal degradation [96, 137]. As a chaperone, VCP prevents aggregation of UPS substrates prior to their presentation to the proteasome [118, 138]. Frontotemporal dementia is further linked to IBM by the finding of TDP-43 immunopositive sarcoplasmic inclusions in both IBMPFD and sporadic IBM muscle [134]. Additionally, VCP is also found in inclusions of both IBMPFD and sporadic IBM.
The UPS in CNS tumors
von Hippel–Lindau disease
Perhaps the most direct example of misregulation of the UPS in the etiology of neurological tumors is in the vascular tumors of the hereditary cancer syndrome von Hippel–Lindau disease (VHLD) [54]. In VHLD, CNS and retinal hemangioblastomas grow in response to overexpression of vascular endothelial growth factor (VEGF). VEGF transcription is driven by hypoxia-inducible factor-1alpha (HIF-1α). pVHL protein is the substrate recognition component of the ubiquitin ligase that ubiquitinates HIF-1α [17] (Fig. 2). The mutant form of pVHL in VHDL is unable to ubiquitinate HIF-1α leading to its over accumulation. This in turn results in increased VEGF transcription, promoting the formation of vascular tumors. VHLD patients also often develop pheochromocytoma, pancreatic carcinoma and clear cell renal cell carcinoma, and pVHL is mutated in most sporadic clear cell renal cell cancers. Thus, the von Hippel–Lindau ubiquitin ligase complex acts as a tumor suppressor by indirectly regulating levels of the growth-promoting signal VEGF.
p53 and PTEN
The tumor suppressors p53 and PTEN are commonly mutated in human neoplasms including glial tumors, particularly high-grade astrocytomas and secondary glioblastomas. The p53 protein has many functions within the cell [7, 19–21, 70]. Its most well-described functions are in the triggering of the G1/S checkpoint and induction of apoptosis. p53 activation of INK4a gene results in transcription of the cyclin-dependent kinase inhibitor p16 which interferes with the ability of these kinases to phosphorylate and inactivate the pRb tumor suppressor. p53 also induces transcription of the BAX family of proteins responsible for inducing apoptosis via disruption of the mitochondrial membrane, and may initiate apoptosis by direct interaction with mitochondria in the cytosol. Loss of p53 function may thus lead to uncontrolled cellular proliferation by allowing unregulated cell cycling through the G/S checkpoint. It may also contribute to survival and proliferation of genetically damaged cells by removing the signal to initiate apoptosis in such cells. Thus, p53 loss is thought to be a key mechanism in tumorigenesis.
A persistent misconception is that positive nuclear p53 immunohistochemical reactivity indicates p53 mutation. p53 transcription is induced by several forms of cellular stress and nuclear p53 reactivity can be detected in both benign and malignant cells with wild-type p53. However, some p53 mutants are resistant to ubiquitin-dependent degradation rendering them inherently more stable than wild-type p53 protein. Such mutants tend to accumulate, enabling their detection by immunohistochemical staining.
Human double minute 2 (HDM2) (homologous to MDM2 in mice) is transcriptionally induced by p53, and is an E3 ubiquitin ligase targeting p53. p14ARF (homologous to the mouse p19ARF) is encoded by the INK4a locus induced by p53. p14ARF binds HDM2 and prevents its association with p53. Loss of p14ARF therefore leads to uninhibited HDM2 mediated p53-ubiquitination and functional loss of p53.
The AKT (protein kinase B) pathway is activated by the phosphorylation of AKT kinases by phosphoinositide-dependent kinase (PDK). Among their many functions AKT serine–threonine kinases inhibit apoptosis by phosphorylation of the pro-apoptotic BCL-2/BAX family protein BAD, and thus promote tumor cell survival. PTEN is a lipid phosphatase that inhibits the AKT pathway by dephosphorylating phosphatidylinositol (3,4,5)-trisphosphate (PIP3), the phosphate donor used by PDK. PTEN loss in tumors thus leads to upregulation of the AKT activating pathway.
Research has also uncovered a link between PTEN and p53 destruction. PTEN inhibits HDM2-mediated p53 destruction (or translocation) in the cell nucleus. Specifically, through inhibition of the AKT pathway, PTEN inhibits the translocation of HDM2 from the cytoplasm to the nucleus [90]. PTEN loss therefore leads to increased ubiquitination of nuclear p53. The resultant decreased nuclear p53 protects tumor cells from DNA damaging chemotherapy [76, 90]. PTEN is ubiquitinated by the pro-oncogenic HECT-type E3 ligase Nedd4-1. Thus, in addition to p53 stabilizing mutations, loss of the UPS regulatory and functional components p14ARF, HDM2 and PTEN may lead to p53 misregulation in tumors.
SCF ubiquitin ligase targets and medulloblastoma
Medulloblastoma is the most common pediatric brain tumor and is thought to arise from cerebellar neural progenitor cells. β-catenin protein binds TCF and LEF transcription factors resulting in transcription of several genes, and has been shown to regulate neural progenitor cell proliferation. Transgenic mice overexpressing β-catenin in the CNS developed enlarged brains because a greater number of their periventricular neural progenitor cells re-entered the cell cycle [13]. Up-regulation of the β-catenin signaling pathway occurs in many human neoplasms. It has been most widely recognized in colon tumors, particularly in familial adenomatosus polyposis coli syndrome, but has also been implicated in medulloblastomas. The adenomatous polyposis coli (APC) protein is an essential participant in the ubiquitination of β-catenin. Like other SCF target proteins, β-catenin must be phosphorylated before it can be recognized by the SCF. APC is essential for this to occur. Together with the protein axin, APC is thought to act as a scaffold necessary for the binding and phosphorylation of β-catenin by GSK3. APC mutations thus prevent phosphorylation and subsequent ubiquitin-dependent degradation of β-catenin. Mutated β-catenin resistant to phosphorylation by GSK3 is therefore abnormally stabilized. Such mutations are found in some sporadic colon cancers, some medulloblastomas and adamantinomatous craniopharyngiomas [9, 114]. Interestingly, c-Myc, Notch and Gli (a downstream effecter of Shh) have also been found to be mutated or dysregulated in medulloblastomas, and like β-catenin, all are involved in brain development and are regulated by SCF-mediated ubiquitin-dependent proteolysis. This suggests that SCF controlled signaling pathways are particularly important in controlling the fate of cerebellar stem cells. Patch, a negative regulator of Shh signaling, is also mutated in medulloblastoma, but appears to ubiquitinated by the Nedd4 E3 ligase [23, 85].
Anaphase promoting complex/cyclosome pathway
The anaphase promoting complex/cyclosome (APC/C) (not to be confused with the fore mentioned APC protein) is an E3 ubiquitin ligase that ubiquitinates several important cell cycle regulatory proteins. The APC/C is associated with two different activator subunits that impart general substrate specificity, Cdh1 and Cdc20 (Fig. 2). The APC/C regulates the ubiquitin-dependent degradation of several proteins that drive the cell cycle including cyclins A and B. The APC/C substrates geminin, survivin, polo and aurora kinases are overexpressed in ependymomas and high-grade gliomas [35, 77]. Securin or pituitary tumor transforming gene-1 (PTTG-1) is an APC/C target frequently overexpressed in pituitary adenomas and is a key component of the mitotic spindle checkpoint, which controls the initiation of chromosome segregation at the metaphase plate of dividing cells. Securin inhibits an enzyme that initiates chromosome segregation known as separase. Upon formation of a mature metaphase mitotic spindle the APC/C ubiquitinates securin. This results in release of separase inhibition and segregation of chromosomes to daughter cells. Radiation or microtubule destabilizing drugs such as pacitaxel can interfere with the checkpoint and arrest cell division. Overexpression or knockdown of levels of APC/C substrate proteins leads to genomic instability and cellular neoplastic transformation [77, 79]. The APC/C also acts as a tumor suppressor by regulating the levels of these cell cycle proteins.
Emil protein is a pseudosubstrate inhibitor of the APC/C and is overexpressed in ependymomas and high-grade astrocytomas [48, 77, 93, 105]. Emi1 regulates APC/C E3 ligase activity during S-phase and early mitosis [48, 130]. The protein product of the RASSF1A gene takes over the negative regulation of APC/C activity following the SCF-mediated ubiquitin-dependent degradation of Emi1 in early mitosis [119]. The RASSF1A gene is silenced by promoter methylation in a large percentage of glial tumors and medulloblastomas [45, 86, 92]. Thus, dysregulation of the APC/C pathway may be a common feature of glial tumors.
An interesting form of biochemical crosstalk occurs between the SCF and APC/C ubiquitin ligases. p27 is a cyclin-dependent kinase inhibitor whose ubiquitin-dependent degradation serves as an important trigger to allow both the G1/S and G2/M cell cycle transitions. Ubiquitination of p27 is accomplished by the SCF E3 ligase complexed with the F-box protein Skp2. Skp2 is in turn ubiquitinated by the APC/C. Tumors overexpressing Skp2, e.g., because of dysregulated Emi1 over-inhibiting the APC/C, tend to have defective regulation of the G1/S or G2/M transitions due to heightened targeting of p27 by SCFSkp2 [78] (Fig. 5).

Cross-talk between the SCF and APC/C regulates cell cycle progression. The SCF E3 ubiquitin ligase F-box adapter protein Skp2 is expressed at levels undetectable by immunohistochemistry in benign schwannomas while its target the cyclin-dependent kinase inhibitor p27 is expressed at detectable levels. Skp2 is a substrate target of the APC/C-Cdh1 E3 ligase, the activity of which may be decreased in many malignant tumors. The opposite pattern of p27 and Skp2 expression may therefore be found in malignant neoplasms [77, 78]. Immunostained tissue microarray cores are shown (×100)
The UPS as a target of therapy
Several proteasome inhibitors have undergone clinical trials as anti-cancer agents. The proteasome inhibitor bortezomib (PS-341) has been approved for the treatment of multiple myeloma and mantle cell lymphoma [106]. Bortezomib is thought to have many pharmacological effects including inducing aggresome formation; however, its antineoplastic action may be attributable to induction of apoptosis. This may define a mechanistic pathway of drug action because aggresome formation can itself lead to apoptosis.
A pharmacologic inhibitor of the HDM2 E3 ubiquitin ligase targeting p53, known as MI-63, showed antiproliferative activity and induction of apoptosis in mantle cell lymphoma cells, and was synergistic with rapamycin and bortezomib in vitro [52]. SCF and APC/C E3 ligases are intriguing additional possible pharmacological targets. Small molecule inhibitors of the APC/C substrates aurora A and aurora B kinases have shown antineoplastic activity in animal tumor models and several are currently in clinical trials [89, 128]. Some of these anti-UPS drugs or novel agents targeting other components of the UPS may eventually prove efficacious against CNS neoplasms. For instance, aurora A (STK15) expression was found to be an independent predictor of survival in medulloblastoma [100].
In situations where UPS components are mutated, as in autosomal recessive forms of Parkinson disease and Angelman disease, gene therapy to replace the specific lost E3 ligase activity may be possible in the future. New therapies for disease in which mutant misfolded proteins are produced such as polyglutamine repeat disorders may be particularly challenging. Anti-aggresome therapy for all neurogenerative diseases involving abnormal protein accumulation has also been postulated. One general form of such therapy would be potentiation of the UPS, which has been proposed to treat neurodegenerative diseases. Finkbeiner and Mitra [30] warn that such treatment would have to target components specific to UPS-mediated degradation of specific substrates, or perhaps groups of substrates to prevent numerous potential off target effects. An interesting study showed that the anti-acidosis drug benzamil enhanced UPS activity, and that blocking the expression of an isoform of the drug’s target the acid-sensing ion channel also enhanced UPS activity and decreased mutant huntingtin aggregation in the brains of a mouse model of Huntington disease [140]. Other potential drug targets might be components of the UPS relatively specific to neurons such as synaptic endocytosis factors, ubiquitin-like proteins, E2 Ubc’s or E3 ligases. Small changes in the activity of such factors might selectively affect neurons because of their lower overall UPS capacity compared to glia [127]. One possible candidate target is UBE3A. Mutation or loss of function of UBE3A and or its substrates is found in Angelman syndrome, Rett syndrome, autism and SCA1 (Table 1). Overexpression of UBE3A protected against the toxicity of polyglutamine repeat expanded proteins in models of Huntington disease and spinocerebellar ataxia [94]. Thus, UBE3A potentiating therapy may be a means to treat multiple polyglutamate repeat diseases as well as Angelman and Rett syndromes and autisms. A possible approach would be to screen for a compound that inhibits the phosphorylation and/or subsequent ubiquitination of UBE3A. In the absence of small molecule potentiators or gene therapy, perhaps intrathecal delivery of UBE3A might be transiently effective. Since viral E6 can activate UBE3A to target p53, this suggests an intriguing possibility of rationally designed viral-mediated therapy to target other UBE3A substrates.
A different approach might have to be taken for tauopathies, ALS and Parkinsonian conditions which show some commonalities in their UPS pathology. Pharmacologic strategies have been proposed for Parkinson disease based on the effects of the LRRK2 mutation. Reduction in LRRK2 results in decreased neuronal toxicity. Such a reduction could be achieved by antagonizing the protective role of HSP90 in blocking LRRK2 ubiquitination by CHIP E3 ligase [63]. Since many neurodegenerative cytoplasmic inclusions can be cleared by autophagy, upregulation of autophagy has been proposed as a general treatment for Parkinson diseases, polyglutamine repeat disorders, and tauopathies [136]. γ-Secretase inhibitors are in clinical trial for Alzheimer disease and effectively lower serum β-amyloid [31, 139]. Because the same γ-secretase that acts on APP also acts on Notch, γ-secretase inhibition results in some off target effects, namely alterations in lymphopoesis and intestinal epithelial cell differentiation [139].
Summary
Research into the possible and established roles of components of the UPS in human diseases, particularly in neurological disorders, is appearing in the literature in an explosive manner. Common themes of UPS disruption are emerging in related disorders such as among the autosomal recessive forms of Parkinson disease, the spinocerebellar ataxias, and the cognitive disorders Angelman disease, Rett syndrome and autism.
This review covers only basic UPS components and provides simplified overviews of specific UPS-related diseases. New levels of biochemical, genetic and epigenetic regulation of the UPS, as well as new interconnections between UPS regulated signaling pathways are continually being discovered. The UPS and its substrates are thus topics of growing complexity. Yet there is little doubt that the UPS and its components will come to represent key factors in our future understanding, diagnosis and perhaps even treatment of neuropathological diseases. The discovery of disease producing defects in the UPS will likely lead to routine molecular subtyping of neurodegenerative, myopathic, neuropathic and neoplastic diseases. Some of these defects might be detectable by simple immunohistochemical methods, e.g., when a protein is absent, overexpressed, or sequestered in aggresomes. In some instances the neuropathologist may be responsible for providing proper neuropathological context and tissue samples to the molecular pathologist, e.g., for gene mutational or methylation status analysis. Familiarity with the basic components of the UPS and knowledge of UPS defects in neurological diseases will definitely be of future importance in neuropathology.
Abbreviations
- A1Up:
-
Ataxin-1 interacting ubiquitin-like protein
- APC:
-
Protein mutated in adenomatous polyposis coli syndrome
- APC/C:
-
Anaphase promoting complex/cyclosome multimeric E3 ubiquitin ligase
- BMP:
-
Bone morphogenic protein, an extracellular patterning signal in CNS development
- Cbl:
-
Casitas B-lineage lymphoma, a small family of RING-type E3 ubiquitin ligases
- CDK:
-
Cyclin-dependent kinase
- CHIP:
-
Carboxy terminus of Hsc70-interacting protein, a U-box E3 ubiquitin ligase
- DUB:
-
Deubiquitination enzyme
- E1:
-
Ubiquitin activating enzyme
- E2:
-
Ubiquitin conjugating enzyme
- E3:
-
Ubiquitin ligase
- EGFR:
-
Epidermal growth factor receptor
- FTLD-U:
-
Frontotemporal lobar degeneration with ubiquitin-positive inclusions
- G76:
-
Terminal glycine residue of ubiquitin
- GSK3:
-
Glycogen synthase kinase 3
- HDM2:
-
Human double minute 2, human MDM2 homolog, an E3 ubiquitin ligase for p53
- HECTD2:
-
HECT domain containing 2, a HECT-type E3 ubiquitin ligase
- HIF-1α:
-
Hypoxia-inducible factor 1, alpha subunit
- HSP70:
-
Heat shock protein 70, a chaperone protein and ATPase
- HSP90:
-
Heat shock protein 90, a chaperone protein and ATPase
- IBM:
-
Inclusion body myopathy
- IBMPFD:
-
Hereditary inclusion body myopathy with Paget disease of bone and frontotemporal dementia
- K48:
-
Lysine 48 of ubiquitin
- K63:
-
Lysine 63 of ubiquitin
- LRRK2:
-
Leucine-rich repeat kinase-2
- MAFbx:
-
Atrogin-1, a muscle-specific F-box ubiquitin ligase adapter protein
- MAP8:
-
Microtubule-associated protein 8
- MAP1B:
-
Microtubule-associated protein 1B
- MDM2:
-
Mouse double minute 2, an E3 ubiquitin ligase for p53
- NII:
-
Neuronal intranuclear inclusion
- NCI:
-
Neuronal cytoplasmic inclusion
- NEDD4:
-
Neural precursor cell expressed developmentally down-regulated 4, an E3 ubiquitin ligase
- NEDD8:
-
Neural precursor cell expressed developmentally down-regulated 8, a small ubiquitin-like posttranslational modifier protein
- PIP3:
-
Phosphatidylinositol (3,4,5)-trisphosphate
- PMP22:
-
Peripheral myelin protein 22
- PTTG-1:
-
Pituitary tumor transforming gene-1, securin, an APC/C E3 ubiquitin ligase substrate
- pVHL:
-
von Hippel–Lindau protein, an E3 ubiquitin ligase substrate adaptor protein
- SCF:
-
Skp/cullin/F-box E3 ubiquitin ligase complex
- Shh:
-
A cholesterol-dependent extracellular patterning signal in CNS development
- STK15:
-
Serine-threonine kinase 15, aurora A kinase, an APC/C E3 ubiquitin ligase substrate
- RASSF1A:
-
Ras association (RalGDS/AF-6) domain family member 1
- siRNAs:
-
Small interfering RNAs (ribonucleic acid oligomers)
- SUMO:
-
Small ubiquitin-like posttranslational modifier protein
- TDP-43:
-
TAR DNA binding protein 43
- TRAF6:
-
Tumor necrosis factor receptor associated factor 6, a RING-type E3 ligase
- TSG101:
-
Tumor susceptibility gene 101
- Ubc:
-
Ubiquitin conjugating enzyme (E2)
- UBA:
-
Ubiquitin-associated domain
- UBE3A:
-
E6AP, a HECT-type E3 ubiquitin ligase
- UBL:
-
Ubiquitin-like domain
- UCH-L1:
-
Ubiquitin carboxy-terminal hydrolase L1, a deubiquitination enzyme (DUB), and E3 ubiquitin ligase when dimerized
- UPS:
-
Ubiquitin proteasome system
- USP7:
-
Ubiquitin specific protease 7, a deubiquitinating enzyme (DUB), its targets include ubiquitinated forms of p53, histones and ataxin-1
- VEGF:
-
Vascular endothelial growth factor
- VHLD:
-
von Hippel–Lindau disease
References
Allen E, Ding J, Wang W et al (2005) Gigaxonin-controlled degradation of MAP1B light chain is critical to neuronal survival. Nature 438:224–228
Ardley HC, Scott GB, Rose SA, Tan NG, Robinson PA (2004) UCH-L1 aggresome formation in response to proteasome impairment indicates a role in inclusion formation in Parkinson’s disease. J Neurochem 90:379–391
Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S (2004) Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature 431:805–810
Bennett EJ, Shaler TA, Woodman B et al (2007) Global changes to the ubiquitin system in Huntington’s disease. Nature 448:704–708
Berke SJ, Chai Y, Marrs GL, Wen H, Paulson HL (2005) Defining the role of ubiquitin-interacting motifs in the polyglutamine disease protein, ataxin-3. J Biol Chem 280:32026–32034
Boland B, Kumar A, Lee S et al (2008) Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer’s disease. J Neurosci 28:6926–6937
Braun CJ, Zhang X, Savelyeva I et al (2008) p53-Responsive microRNAs 192 and 215 are capable of inducing cell cycle arrest. Cancer Res 68:10094–10104
Buchberger A (2002) From UBA to UBX: new words in the ubiquitin vocabulary. Trends Cell Biol 12:216–221
Buslei R, Nolde M, Hofmann B et al (2005) Common mutations of beta-catenin in adamantinomatous craniopharyngiomas but not in other tumours originating from the sellar region. Acta Neuropathol 109:589–597
Chen C, Seth AK, Aplin AE (2006) Genetic and expression aberrations of E3 ubiquitin ligases in human breast cancer. Mol Cancer Res 4:695–707
Chen Y, Liu W, McPhie DL, Hassinger L, Neve RL (2003) APP-BP1 mediates APP-induced apoptosis and DNA synthesis and is increased in Alzheimer’s disease brain. J Cell Biol 163:27–33
Chen Y, McPhie DL, Hirschberg J, Neve RL (2000) The amyloid precursor protein-binding protein APP-BP1 drives the cell cycle through the S-M checkpoint and causes apoptosis in neurons. J Biol Chem 275:8929–8935
Chenn A, Walsh CA (2002) Regulation of cerebral cortical size by control of cell cycle exit in neural precursors. Science 297:365–369
Chow N, Korenberg JR, Chen XN, Neve RL (1996) APP-BP1, a novel protein that binds to the carboxyl-terminal region of the amyloid precursor protein. J Biol Chem 271:11339–11346
Chu CT, Caruso JL, Cummings TJ, Ervin J, Rosenberg C, Hulette CM (2000) Ubiquitin immunochemistry as a diagnostic aid for community pathologists evaluating patients who have dementia. Mod Pathol 13:420–426
Cleveland DW, Yamanaka K, Bomont P (2009) Gigaxonin controls vimentin organization through a tubulin chaperone-independent pathway. Hum Mol Genet 18:1384–1394
Cockman ME, Masson N, Mole DR et al (2000) Hypoxia inducible factor-alpha binding and ubiquitylation by the von Hippel–Lindau tumor suppressor protein. J Biol Chem 275:25733–25741
Cook JC, Chock PB (1992) Isoforms of mammalian ubiquitin-activating enzyme. J Biol Chem 267:24315–24321
Cully M, You H, Levine AJ, Mak TW (2006) Beyond PTEN mutations: the PI3K pathway as an integrator of multiple inputs during tumorigenesis. Nat Rev Cancer 6:184–192
Cuesta A, Zambrano A, Royo M, Pascual A (2009) The tumour suppressor p53 regulates the expression of amyloid precursor protein (APP). Biochem J 418:643–650
Dayal S, Sparks A, Jacob J, Allende-Vega N, Lane DP, Saville MK (2009) Suppression of the deubiquitinating enzyme USP5 causes the accumulation of unanchored polyubiquitin and the activation of p53. J Biol Chem 284:5030–5041
Dil Kuazi A, Kito K, Abe Y, Shin RW, Kamitani T, Ueda N (2003) NEDD8 protein is involved in ubiquitinated inclusion bodies. J Pathol 199:259–266
Dong J, Gailani MR, Pomeroy SL, Reardon D, Bale AE (2000) Identification of PATCHED mutations in medulloblastomas by direct sequencing. Hum Mutat 16:89–90
Duka T, Duka V, Joyce JN, Sidhu A (2009) α-Synuclein contributes to GSK-3β-catalyzed Tau phosphorylation in Parkinson’s disease models. FASEB J (in press)
Dusonchet J, Bensadoun JC, Schneider BL, Aebischer P (2009) Targeted overexpression of the parkin substrate Pael-R in the nigrostriatal system of adult rats to model Parkinson’s disease. Neurobiol Dis 35(1):32–41
Eide EJ, Woolf MF, Kang H et al (2005) Control of mammalian circadian rhythm by CKIepsilon-regulated proteasome-mediated PER2 degradation. Mol Cell Biol 25:2795–2807
Elgersma Y, Sweatt JD, Giese KP (2004) Mouse genetic approaches to investigating calcium/calmodulin-dependent protein kinase II function in plasticity and cognition. J Neurosci 24:8410–8415
Engelender S (2008) Ubiquitination of alpha-synuclein and autophagy in Parkinson’s disease. Autophagy 4:372–374
Feng Z, Jin S, Zupnick A et al (2006) p53 tumor suppressor protein regulates the levels of huntingtin gene expression. Oncogene 25:1–7
Finkbeiner S, Mitra S (2008) The ubiquitin–proteasome pathway in Huntington’s disease. Sci World J 8:421–433
Fleisher AS, Raman R, Siemers ER et al (2008) Phase 2 safety trial targeting amyloid beta production with a gamma-secretase inhibitor in Alzheimer disease. Arch Neurol 65:1031–1038
Fortun J, Li J, Go J, Fenstermaker A, Fletcher BS, Notterpek L (2005) Impaired proteasome activity and accumulation of ubiquitinated substrates in a hereditary neuropathy model. J Neurochem 92:1531–1541
Fujiwara M, Marusawa H, Wang HQ et al (2008) Parkin as a tumor suppressor gene for hepatocellular carcinoma. Oncogene 27:6002–6011
Gaig C, Marti MJ, Ezquerra M, Rey MJ, Cardozo A, Tolosa E (2007) G2019S LRRK2 mutation causing Parkinson’s disease without Lewy bodies. J Neurol Neurosurg Psychiatry 78:626–628
Gibson SE, Zeng WF, Weil RJ, Prayson RA (2008) Aurora B kinase expression in ependymal neoplasms. Appl Immunohistochem Mol Morphol 16:274–278
Glessner JT, Wang K, Cai G (2009) Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature 459:569–573
Golden JA, Bracilovic A, McFadden KA, Beesley JS, Rubenstein JL, Grinspan JB (1999) Ectopic bone morphogenetic proteins 5 and 4 in the chicken forebrain lead to cyclopia and holoprosencephaly. Proc Natl Acad Sci USA 96:2439–2444
Gong B, Leznik E (2007) The role of ubiquitin C-terminal hydrolase L1 in neurodegenerative disorders. Drug News Perspect 20:365–370
Grant EP, Michalek MT, Goldberg AL, Rock KL (1995) Rate of antigen degradation by the ubiquitin–proteasome pathway influences MHC class I presentation. J Immunol 155:3750–3758
Guardavaccaro D, Kudo Y, Boulaire J et al (2003) Control of meiotic and mitotic progression by the F box protein beta-Trcp1 in vivo. Dev Cell 4:799–812
Haas AL, Warms JV, Hershko A, Rose IA (1982) Ubiquitin-activating enzyme mechanism and role in protein–ubiquitin conjugation. J Biol Chem 257:2543–2548
Hanai J, Cao P, Tanksale P et al (2007) The muscle-specific ubiquitin ligase atrogin-1/MAFbx mediates statin-induced muscle toxicity. J Clin Invest 117:3940–3951
Hart M, Concordet JP, Lassot I et al (1999) The F-box protein beta-TrCP associates with phosphorylated beta-catenin and regulates its activity in the cell. Curr Biol 9:207–210
Hershko A, Ciechanover A (1998) The ubiquitin system. Annu Rev Biochem 67:425–479
Hesson L, Bièche I, Krex D et al (2004) Frequent epigenetic inactivation of RASSF1A and BLU genes located within the critical 3p21.3 region in gliomas. Oncogene 23:2408–2419
Holm IE, Englund E, Mackenzie IR, Johannsen P, Isaacs AM (2007) A reassessment of the neuropathology of frontotemporal dementia linked to chromosome 3. J Neuropathol Exp Neurol 66:884–891
Hong S, Kim SJ, Ka S, Choi I, Kang S (2002) USP7, a ubiquitin-specific protease, interacts with ataxin-1, the SCA1 gene product. Mol Cell Neurosci 20:298–306
Hsu JY, Reimann JD, Sorensen CS, Lukas J, Jackson PK (2002) E2F-dependent accumulation of hEmi1 regulates S phase entry by inhibiting APC (Cdh1). Nat Cell Biol 4:358–366
Huang Y, Song YJ, Murphy K et al (2008) LRRK2 and parkin immunoreactivity in multiple system atrophy inclusions. Acta Neuropathol 116:639–646
Huynh DP, Nguyen DT, Pulst-Korenberg JB, Brice A, Pulst SM (2007) Parkin is an E3 ubiquitin-ligase for normal and mutant ataxin-2 and prevents ataxin-2-induced cell death. Exp Neurol 203:531–541
Iwata A, Nagashima Y, Matsumoto L et al (2009) Intranuclear degradation of polyglutamine aggregates by the ubiquitin–proteasome system. J Biol Chem 284:9796–9803
Jones RJ, Chen Q, Voorhees PM et al (2008) Inhibition of the p53 E3 ligase HDM-2 induces apoptosis and DNA damage-independent p53 phosphorylation in mantle cell lymphoma. Clin Cancer Res 14:5416–5425
Josephs KA, Lin WL, Ahmed Z, Stroh DA, Graff-Radford NR, Dickson DW (2008) Frontotemporal lobar degeneration with ubiquitin-positive, but TDP-43-negative inclusions. Acta Neuropathol 116:159–167
Kaelin WG (2002) Molecular basis of the VHL hereditary cancer syndrome. Nat Rev Cancer 2:673–682
Kang Y, Vossler RA, Diaz-Martinez LA, Winter NS, Clarke DJ, Walters KJ (2006) UBL/UBA ubiquitin receptor proteins bind a common tetraubiquitin chain. J Mol Biol 356:1027–1035
Kannanayakal TJ, Mendell JR, Kuret J (2008) Casein kinase 1 alpha associates with the tau-bearing lesions of inclusion body myositis. Neurosci Lett 431:141–145
Kawashima T, Furuta A, Doh-ura K, Kikuchi H, Iwaki T (2000) Ubiquitin-immunoreactive skein-like inclusions in the neostriatum are not restricted to amyotrophic lateral sclerosis, but are rather aging-related structures. Acta Neuropathol 100:43–49
Kim BY, Olzmann JA, Barsh GS, Chin LS, Li L (2007) Spongiform neurodegeneration-associated E3 ligase Mahogunin ubiquitylates TSG101 and regulates endosomal trafficking. Mol Biol Cell 18:1129–1142
Kim MJ, Chia IV, Costantini F (2008) SUMOylation target sites at the C terminus protect Axin from ubiquitination and confer protein stability. FASEB J 22:3785–3794
Kim SH, Shi Y, Hanson KA et al (2009) Potentiation of amyotrophic lateral sclerosis (ALS)-associated TDP-43 aggregation by the proteasome-targeting factor, ubiquilin 1. J Biol Chem 284:8083–8092
Kinoshita A, Whelan CM, Berezovska O, Hyman BT (2002) The gamma secretase-generated carboxyl-terminal domain of the amyloid precursor protein induces apoptosis via Tip60 in H4 cells. J Biol Chem 277:28530–28536
Kirkin V, McEwan DG, Novak I, Dikic I (2009) A role for ubiquitin in selective autophagy. Mol Cell 34:259–269
Ko HS, Bailey R, Smith WW et al (2009) CHIP regulates leucine-rich repeat kinase-2 ubiquitination, degradation, and toxicity. Proc Natl Acad Sci USA 106:2897–2902
Koegl M, Hoppe T, Schlenker S, Ulrich HD, Mayer TU, Jentsch S (1999) A novel ubiquitination factor, E4, is involved in multiubiquitin chain assembly. Cell 96:635–644
Koepp DM, Schaefer LK, Ye X et al (2001) Phosphorylation-dependent ubiquitination of cyclin E by the SCFFbw7 ubiquitin ligase. Science 294:173–177
Konishi Y, Stegmüller J, Matsuda T, Bonni S, Bonni A (2004) Cdh1-APC controls axonal growth and patterning in the mammalian brain. Science 303:1026–1030
Kopito RR (2000) Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol 10:524–530
Kristiansen M, Deriziotis P, Dimcheff DE et al (2007) Disease-associated prion protein oligomers inhibit the 26S proteasome. Mol Cell 26:175–188
Kristiansen M, Messenger MJ, Klohn PC et al (2005) Disease-related prion protein forms aggresomes in neuronal cells leading to caspase activation and apoptosis. J Biol Chem 280:38851–38861
Kruse JP, Gu W (2009) Modes of p53 regulation. Cell 137:609–622
Kruse JP, Gu W (2009) MSL2 promotes Mdm2-independent cytoplasmic localization of p53. J Biol Chem 284:3250–3263
Lagirand-Cantaloube J, Cornille K, Csibi A, Batonnet-Pichon S, Leibovitch MP, Leibovitch SA (2009) Inhibition of atrogin-1/MAFbx mediated MyoD proteolysis prevents skeletal muscle atrophy in vivo. PLoS ONE 4:e4973
Lathia JD, Mattson MP, Cheng A (2008) Notch: from neural development to neurological disorders. J Neurochem 107:1471–1481
Lebre AS, Jamot L, Takahashi J et al (2001) Ataxin-7 interacts with a Cbl-associated protein that it recruits into neuronal intranuclear inclusions. Hum Mol Genet 10:1201–1213
Lee JT, Wheeler TC, Li L, Chin LS (2008) Ubiquitination of alpha-synuclein by Siah-1 promotes alpha-synuclein aggregation and apoptotic cell death. Hum Mol Genet 17:906–917
Lehman NL, Danenberg PV (2000) Modulation of RTX cytotoxicity by thymidine and dipyridamole in vitro: implications for chemotherapy. Cancer Chemother Pharmacol 45:142–148
Lehman NL, Tibshirani R, Hsu JY et al (2007) Oncogenic regulators and substrates of the anaphase promoting complex/cyclosome are frequently overexpressed in malignant tumors. Am J Pathol 170:1793–1805
Lehman NL, van de Rijn M, Jackson PK (2005) Screening of tissue microarrays for ubiquitin proteasome system components in tumors. Methods Enzymol 399:334–355
Lehman NL, Verschuren EW, Hsu JY, Cherry AM, Jackson PK (2006) Overexpression of the anaphase promoting complex/cyclosome inhibitor Emi1 leads to tetraploidy and genomic instability of p53-deficient cells. Cell Cycle 5:1569–1573
Liani E, Eyal A, Avraham E et al (2004) Ubiquitylation of synphilin-1 and alpha-synuclein by SIAH and its presence in cellular inclusions and Lewy bodies imply a role in Parkinson’s disease. Proc Natl Acad Sci USA 101:5500–5505
Liscic RM, Grinberg LT, Zidar J, Gitcho MA, Cairns NJ (2008) ALS and FTLD: two faces of TDP-43 proteinopathy. Eur J Neurol 15:772–780
Liu S, Bromley-Brits K, Xia K, Mittelholtz J, Wang R, Song W (2008) TMP21 degradation is mediated by the ubiquitin–proteasome pathway. Eur J Neurosci 28:1980–1988
Liu YC (2004) Ubiquitin ligases and the immune response. Annu Rev Immunol 22:81–127
Lloyd SE, Maytham EG, Pota H et al (2009) HECTD2 is associated with susceptibility to mouse and human prion disease. PLoS Genet 5:e1000383
Lu X, Liu S, Kornberg TB (2006) The C-terminal tail of the Hedgehog receptor Patched regulates both localization and turnover. Genes Dev 20:2539–2551
Lusher ME, Lindsey JC, Latif F, Pearson AD, Ellison DW, Clifford SC (2002) Biallelic epigenetic inactivation of the RASSF1A tumor suppressor gene in medulloblastoma development. Cancer Res 62:5906–5911
Mackenzie IR, Bigio EH, Ince PG et al (2007) Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann Neurol 61:427–434
Makedonski K, Abuhatzira L, Kaufman Y, Razin A, Shemer R (2005) MeCP2 deficiency in Rett syndrome causes epigenetic aberrations at the PWS/AS imprinting center that affects UBE3A expression. Hum Mol Genet 14:1049–1058
Manfredi MG, Ecsedy JA, Meetze KA et al (2007) Antitumor activity of MLN8054, an orally active small-molecule inhibitor of Aurora A kinase. Proc Natl Acad Sci USA 104:4106–4111
Mayo LD, Dixon JE, Durden DL, Tonks NK, Donner DB (2002) PTEN protects p53 from Mdm2 and sensitizes cancer cells to chemotherapy. J Biol Chem 277:5484–5489
Miklossy J, Steele JC, Yu S et al (2008) Enduring involvement of tau, beta-amyloid, alpha-synuclein, ubiquitin and TDP-43 pathology in the amyotrophic lateral sclerosis/parkinsonism-dementia complex of Guam (ALS/PDC). Acta Neuropathol 116:625–637
Michalowski MB, de Fraipont F, Michelland S et al (2006) Methylation of RASSF1A and TRAIL pathway-related genes is frequent in childhood intracranial ependymomas and benign choroid plexus papilloma. Cancer Genet Cytogenet 166:74–81
Miller JJ, Summers MK, Hansen DV et al (2006) Emi1 stably binds and inhibits the anaphase-promoting complex/cyclosome as a pseudosubstrate inhibitor. Genes Dev 20:2410–2420
Mishra A, Dikshit P, Purkayastha S, Sharma J, Nukina N, Jana NR (2008) E6-AP promotes misfolded polyglutamine proteins for proteasomal degradation and suppresses polyglutamine protein aggregation and toxicity. J Biol Chem 283:7648–7656
Morishima Y, Wang AM, Yu Z, Pratt WB, Osawa Y, Lieberman AP (2008) CHIP deletion reveals functional redundancy of E3 ligases in promoting degradation of both signaling proteins and expanded glutamine proteins. Hum Mol Genet 17:3942–3952
Morreale G, Conforti L, Coadwell J, Wilbrey AL, Coleman MP (2009) Evolutionary divergence of valosin-containing protein/cell division cycle protein 48 binding interactions among endoplasmic reticulum-associated degradation proteins. FEBS J 276:1208–1220
Mosesson Y, Shtiegman K, Katz M et al (2003) Endocytosis of receptor tyrosine kinases is driven by monoubiquitylation, not polyubiquitylation. J Biol Chem 278:21323–21326
Muller S, Matunis MJ, Dejean A (1998) Conjugation with the ubiquitin-related modifier SUMO-1 regulates the partitioning of PML within the nucleus. EMBO J 17:61–70
Murakami G, Watabe T, Takaoka K, Miyazono K, Imamura T (2003) Cooperative inhibition of bone morphogenetic protein signaling by Smurf1 and inhibitory Smads. Mol Biol Cell 14:2809–2817
Neben K, Korshunov A, Benner A et al (2004) Microarray-based screening for molecular markers in medulloblastoma revealed STK15 as independent predictor for survival. Cancer Res 64:3103–3111
Neumann M, Mackenzie IR, Cairns NJ et al (2007) TDP-43 in the ubiquitin pathology of frontotemporal dementia with VCP gene mutations. J Neuropathol Exp Neurol 66:152–157
Nonis D, Schmidt MH, van de Loo S et al (2008) Ataxin-2 associates with the endocytosis complex and affects EGF receptor trafficking. Cell Signal 20:1725–1739
Parkinson N, Ince PG, Smith MO et al (2006) ALS phenotypes with mutations in CHMP2B (charged multivesicular body protein 2B). Neurology 67:1074–1077
Ramesh Babu J, Lamar Seibenhener M, Peng J et al (2008) Genetic inactivation of p62 leads to accumulation of hyperphosphorylated tau and neurodegeneration. J Neurochem 106:107–120
Reimann JD, Freed E, Hsu JY, Kramer ER, Peters JM, Jackson PK (2001) Emi1 is a mitotic regulator that interacts with Cdc20 and inhibits the anaphase promoting complex. Cell 105:645–655
Richardson PG, Sonneveld P, Schuster M et al (2007) Extended follow-up of a phase 3 trial in relapsed multiple myeloma: final time-to-event results of the APEX trial. Blood 110:3557–3560
Riley BE, Xu Y, Zoghbi HY, Orr HT (2004) The effects of the polyglutamine repeat protein ataxin-1 on the UbL-UBA protein A1Up. J Biol Chem 279:42290–42301
Roeber S, Mackenzie IR, Kretzschmar HA, Neumann M (2008) TDP-43-negative FTLD-U is a significant new clinico-pathological subtype of FTLD. Acta Neuropathol 116:147–157
Rott R, Szargel R, Haskin J et al (2008) Monoubiquitylation of alpha-synuclein by seven in absentia homolog (SIAH) promotes its aggregation in dopaminergic cells. J Biol Chem 283:3316–3328
Sadeh R, Breitschopf K, Bercovich B et al (2008) The N-terminal domain of MyoD is necessary and sufficient for its nuclear localization-dependent degradation by the ubiquitin system. Proc Natl Acad Sci USA 105:15690–15695
Sartori S, Anesi L, Polli R et al (2008) Angelman syndrome due to a novel splicing mutation of the UBE3A gene. J Child Neurol 23:912–915
Schimmel J, Larsen KM, Matic I et al (2008) The ubiquitin–proteasome system is a key component of the SUMO-2/3 cycle. Mol Cell Proteomics 7:2107–2122
Schwartz AL, Ciechanover A (2009) Targeting proteins for destruction by the ubiquitin system: implications for human pathobiology. Annu Rev Pharmacol Toxicol 49:73–96
Sekine S, Shibata T, Kokubu A et al (2002) Craniopharyngiomas of adamantinomatous type harbor beta-catenin gene mutations. Am J Pathol 161:1997–2001
Setsuie R, Wada K (2007) The functions of UCH-L1 and its relation to neurodegenerative diseases. Neurochem Int 51:105–111
Sheng Y, Laister RC, Lemak A et al (2008) Molecular basis of Pirh2-mediated p53 ubiquitylation. Nat Struct Mol Biol 15:1334–1342
Shiba K, Arai T, Sato S et al (2009) Parkin stabilizes PINK1 through direct interaction. Biochem Biophys Res Commun 383:331–335
Song C, Wang Q, Li CC (2007) Characterization of the aggregation-prevention activity of p97/valosin-containing protein. Biochemistry 46:14889–14898
Song SJ, Song MS, Kim SJ et al (2009) Aurora A regulates prometaphase progression by inhibiting the ability of RASSF1A to suppress APC-Cdc20 activity. Cancer Res 69:2314–2323
Staropoli JF, McDermott C, Martinat C, Schulman B, Demireva E, Abeliovich A (2003) Parkin is a component of an SCF-like ubiquitin ligase complex and protects postmitotic neurons from kainate excitotoxicity. Neuron 37:735–749
Ström AL, Forsgren L, Holmberg M (2005) A role for both wild-type and expanded ataxin-7 in transcriptional regulation. Neurobiol Dis 20:646–655
Tai HC, Schuman EM (2008) Ubiquitin, the proteasome and protein degradation in neuronal function and dysfunction. Nat Rev Neurosci 9:826–838
Tan JM, Wong ES, Dawson VL, Dawson TM, Lim KL (2007) Lysine 63-linked polyubiquitin potentially partners with p62 to promote the clearance of protein inclusions by autophagy. Autophagy 4(2) (in press)
Tank EM, True HL (2009) Disease-associated mutant ubiquitin causes proteasomal impairment and enhances the toxicity of protein aggregates. PLoS Genet 5:e1000382
Todi SV, Winborn BJ, Scaglione KM, Blount JR, Travis SM, Paulson HL (2009) Ubiquitination directly enhances activity of the deubiquitinating enzyme ataxin-3. EMBO J 28:372–382
Tortosa E, Santa-Maria I, Moreno F, Lim F, Perez M, Avila J (2009) Binding of Hsp90 to tau promotes a conformational change and aggregation of tau protein. J Alzheimers Dis (in press)
Tydlacka S, Wang CE, Wang X, Li S, Li XJ (2008) Differential activities of the ubiquitin–proteasome system in neurons versus glia may account for the preferential accumulation of misfolded proteins in neurons. J Neurosci 28:13285–13295
VanderPorten EC, Taverna P, Hogan JN, Ballinger MD, Flanagan WM, Fucini RV (2009) The Aurora kinase inhibitor SNS-314 shows broad therapeutic potential with chemotherapeutics and synergy with microtubule-targeted agents in a colon carcinoma model. Mol Cancer Ther 8:930–939
Varghese B, Barriere H, Carbone CJ et al (2008) Polyubiquitination of prolactin receptor stimulates its internalization, postinternalization sorting, and degradation via the lysosomal pathway. Mol Cell Biol 28:5275–5287
Verschuren EW, Ban KH, Masek MA, Lehman NL, Jackson PK (2007) Loss of Emi1-dependent anaphase-promoting complex/cyclosome inhibition deregulates E2F target expression and elicits DNA damage-induced senescence. Mol Cell Biol 27:7955–7965
Vissers JH, Nicassio F, van Lohuizen M, Di Fiore PP, Citterio E (2008) The many faces of ubiquitinated histone H2A: insights from the DUBs. Cell Div 3:8
Wang HL, He CY, Chou AH, Yeh TH, Chen YL, Li AH (2007) Polyglutamine-expanded ataxin-7 decreases nuclear translocation of NF-kappaB p65 and impairs NF-kappaB activity by inhibiting proteasome activity of cerebellar neurons. Cell Signal 19:573–581
Wang J, Wang CE, Orr A, Tydlacka S, Li SH, Li XJ (2008) Impaired ubiquitin–proteasome system activity in the synapses of Huntington’s disease mice. J Cell Biol 180:1177–1189
Weihl CC, Temiz P, Miller SE et al (2008) TDP-43 accumulation in inclusion body myopathy muscle suggests a common pathogenic mechanism with frontotemporal dementia. J Neurol Neurosurg Psychiatry 79:1186–1189
Westwood FR, Scott RC, Marsden AM, Bigley A, Randall K (2008) Rosuvastatin: characterization of induced myopathy in the rat. Toxicol Pathol 36:345–352
Williams A, Jahreiss L, Sarkar S et al (2006) Aggregate-prone proteins are cleared from the cytosol by autophagy: therapeutic implications. Curr Top Dev Biol 76:89–101
Wojcik C, Rowicka M, Kudlicki A et al (2006) Valosin-containing protein (p97) is a regulator of endoplasmic reticulum stress and of the degradation of N-end rule and ubiquitin–fusion degradation pathway substrates in mammalian cells. Mol Biol Cell 17:4606–4618
Wojcik C, Yano M, DeMartino GN (2004) RNA interference of valosin-containing protein (VCP/p97) reveals multiple cellular roles linked to ubiquitin/proteasome-dependent proteolysis. J Cell Sci 117:281–292
Wong GT, Manfra D, Poulet FM et al (2004) Chronic treatment with the gamma-secretase inhibitor LY-411, 575 inhibits beta-amyloid peptide production and alters lymphopoiesis and intestinal cell differentiation. J Biol Chem 279:12876–12882
Wong HK, Bauer PO, Kurosawa M et al (2008) Blocking acid-sensing ion channel 1 alleviates Huntington’s disease pathology via an ubiquitin–proteasome system-dependent mechanism. Hum Mol Genet 17:3223–3235
Xia Y, Padre RC, De Mendoza TH, Bottero V, Tergaonkar VB, Verma IM (2009) Phosphorylation of p53 by IkappaB kinase 2 promotes its degradation by beta-TrCP. Proc Natl Acad Sci USA 106:2629–2634
Xiong H, Wang D, Chen L et al (2009) Parkin, PINK1, and DJ-1 form a ubiquitin E3 ligase complex promoting unfolded protein degradation. J Clin Invest 119:650–660
Yamanaka K, Vande Velde C, Eymard-Pierre E, Bertini E, Boespflug-Tanguy O, Cleveland DW (2003) Unstable mutants in the peripheral endosomal membrane component ALS2 cause early-onset motor neuron disease. Proc Natl Acad Sci USA 100:16041–16046
Yang B, Hahn YS, Hahn CS, Braciale TJ (1996) The requirement for proteasome activity class I major histocompatibility complex antigen presentation is dictated by the length of preprocessed antigen. J Exp Med 183:1545–1552
Yang Y, Allen E, Ding J, Wang W (2007) Giant axonal neuropathy. Cell Mol Life Sci 64:601–609
Yashiro K, Riday TT, Condon KH et al (2009) Ube3a is required for experience-dependent maturation of the neocortex. Nat Neurosci (in press)
Yi JJ, Ehlers MD (2005) Ubiquitin and protein turnover in synapse function. Neuron 47:629–632
Zhai Q, Wang J, Kim A et al (2003) Involvement of the ubiquitin–proteasome system in the early stages of wallerian degeneration. Neuron 39:217–225
Zimprich A, Biskup S, Leitner P et al (2004) Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 44:601–607
Acknowledgments
The author thanks Kathy Roszka for expert performance of anti-ubiquitin immunohistochemical stains, and Nandita Mani and Theresa Hobbs for assistance with formatting the manuscript.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Lehman, N.L. The ubiquitin proteasome system in neuropathology. Acta Neuropathol 118, 329–347 (2009). https://doi.org/10.1007/s00401-009-0560-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-009-0560-x