
Abstract
To investigate the frequency of angiotensin-converting enzyme (ACE) gene insertion/deletion (I/D) polymorphism genotypes in adults with psoriatic arthritis (PsA), a heterogeneous chronic disease with autoimmune pathology. ACE gene I/D polymorphism influences the plasma and tissue levels of ACE and has an involvement in inflammatory mechanism. The frequency of ACE gene I/D polymorphism genotypes was determined in 51 adults with PsA from Kuwait, and compared to that in 100 ethnically matched healthy controls using polymerase chain reaction. The distribution of ACE I/D polymorphism and allele frequencies in PsA patients were not significantly different from controls (P > 0.05). Further analyses of PsA patients showed that ACE I/D gene polymorphism was not associated with family history, clinical manifestations, and disease severity. However, the frequency of II genotype was significantly higher in patients with late disease onset than in those with early onset (25 vs. 3%; P = 0.04). No difference was found between the distribution of the ACE genotype in PsA patients and the general population in Kuwait. However, the presence of II genotype may confer susceptibility to the development of late onset PsA.
Similar content being viewed by others


Introduction
Psoriatic arthritis (PsA) is a chronic systemic inflammatory arthritis disorder characterized by joint inflammation that is associated with cutaneous psoriasis. PsA is relatively common, affecting approximately 1% of the population [1]. Although previously considered to be a relatively mild form of arthritis, there has been a growing appreciation that PsA can be progressive, destructive, and deforming. PsA can have profound, deleterious effects, to an extent comparable to those of other pernicious chronic conditions, such as rheumatoid arthritis (RA) [2, 3]. The exact cause of psoriasis, and its associated arthritis, is not yet known. It seems likely, however, that genetic, immunologic, and environmental factors all may contribute [4]. It also seems reasonable to postulate that the skin and joint disease have a similar pathogenesis although the activity of these manifestations does not always change in parallel. Genetic association with PsA has been most extensively studied in human leukocyte antigen (HLA) class I and II polymorphism [5–8], immunoglobulin genes [9], CARD15 gene [10], tumor necrosis factor (TNF) genes [11, 12], and SEEK I gene [13].
The Angiotensin-converting enzyme (ACE) gene is located on the long arm of chromosome 17 [14], and contains an insertion (I) and deletion (D) polymorphism within intron 16 of the presence or absence of a 287 bp repeat sequence. Of the three possible genotypes (DD and II homozygotes and ID heterozygotes), the carriers of DD genotype have the highest levels of serum ACE, while those with the II genotype have the lowest serum levels [15].
ACE is expressed in a wide range of tissues including skin, vascular endothelium, and immune cells [16–18]. Almost half of the patients with psoriasis have been found to have an elevated serum ACE activity [19]. ACE activates Angiotensin I into Angiotensin II, inactivates bradykinin via the kallikrein–kininogen system, and plays a major role in the rennin–angiotensin system (RAS). It is suggested that inhibition of bradykinin degradation by ACE inhibitors alters the kinin–kallikrein arachidonic acid system, leading to increased concentrations of kinins and related inflammatory metabolites in the skin which might contribute to inflammatory skin lesions in psoriasis.
There was only one report in literature denoting the presence of I-allele might confer susceptibility to development of psoriasis in individuals from psoriatic families [20], but none on PsA. Therefore, the present study was undertaken to evaluate the possible role of I/D polymorphism of the ACE gene in the etiopathogenesis of PsA.
Subjects and methods
Patients
ACE gene I/D polymorphism genotypes were determined in 51 patients with PsA, who were seen on regular basis in the rheumatic disease clinics of Al-Amiri teaching hospital in Kuwait (Table 1); diagnosis of PsA was made at least 6 months before the initiation of the study. The diagnosis of PsA was based on the presence of inflammatory arthritis associated with psoriasis, usually with no rheumatoid factor in the serum [21]. Detailed clinical information was available on all patients, including age, gender, age at disease onset, family history of psoriasis and/or PsA, and clinical manifestations. Patients were considered to have ‘early onset’ PsA if disease onset was at any age younger than 40 years (n = 27), and ‘late onset’ PsA if age at onset was after their fortieth birthday (n = 24). Patients were considered to have ‘familial’ PsA if they had one or more first- or second-degree relatives affected by psoriasis and/or PsA (n = 22), or to have ‘sporadic’ PsA if this was not the case (n = 29). The severity of the PsA was categorized into those who had ‘less severe’ disease (n = 32) that was controlled with only one disease modifying anti-rheumatic drug (DMARD) either methotrexate (MTX) or sulfasalazine (SSZ), and ‘severe’ disease (n = 19) which was controlled with additional anti-tumor necrosis factor (anti-TNF). In order to exclude an interaction between coexisting disease (hypertension, diabetes, etc.) and PsA in term of ACE I/D polymorphism, PsA patients with accompanying disease(s) were excluded from the study.
Controls
The incidence of ACE gene I/D polymorphism in patients with PsA was compared to that in a group of 100 healthy controls. The control subjects were of similar ethnic background; all had been previously normal and were seen at the hospital out-patient clinic for minor illnesses. They did not have a history of autoimmune or rheumatic disorders or other diseases with known genetic or hereditary predisposition. A trained Rheumatologist ascertained all controls. A randomization procedure (Pittsburg Genetic Epidemiology Group; Trucco M, personal communication) was employed for selection of controls. All the subjects enrolled in the study, both patients and controls, were unrelated to each other but were matched for age and sex.
The study was carried out in accordance with the Helsinki Declaration. The Ethics Committees of Al-Amiri Hospital and Faculty of Medicine, Kuwait University approved the study. Written informed consent was obtained from all patients and controls who participated in the study.
Genotyping
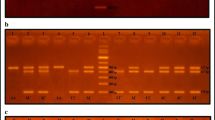
Blood samples were collected from patients and controls. Total genomic DNA was isolated by a standard procedure [22]. ACE genotypes were determined by polymerase chain reaction (PCR) using primers and conditions described previously [23]. Reactions were performed with 10 pmol of each primer: sense oligo: 5′ CTGGAGACCACTCCCATCCTTTCT 3′ and anti-sense oligo: 5′ GATGTGGCCATCACATTCGTCAGAT 3′ in a final volume of 50 μl, containing 3 mM MgCl2, 50 mM KCl, 10 mM Tris–HCl pH 8.4, 0.1 mg/ml gelatin, 0.5 mM of each dNTP (Cetus), 2.5 μl AmpliTaq DNA polymerase (Cetus). The DNA was amplified for 30 cycles with denaturation at 94°C for 1 min, anealing at 58°C for 1 min, and extension at 72°C for 2 min using a Perkin–Elmer Thermal cycler. A total of 0.6% dimethylsulfoxide is routinely added to the PCR mix to improve the amplification of the I-allele, and avoid mistyping it as a D-allele. The PCR products were analysed on 2% agarose gels after staining with ethidium bromide. In the absence of the 287 bp insertion in the intron 16 of the ACE gene, this PCR method resulted in a 190 bp product (D-allele), and in the presence of insertion, produced a 490 bp product (I-allele; Fig. 1). In heterozygous samples, two bands (490 and 190 bp) were detected along with a third fragment of intermediate size, which corresponded to a heteroduplex DNA fragment [23]. All analyses were carried out double blind, and appropriate positive and negative controls were included for each sample run.

Detection of ACE gene I/D polymorphism by polymerase chain reaction (PCR). Lane 1 HaeIII cleaved φX174 molecular size markers, lanes 2–4 PCR products from patients with DD genotype, lanes 5 and 7–8 PCR products from patients with ID genotype, lane 6 PCR products from patients with II genotype. The products of PCR amplification were analyzed on 2% agarose gel and visualized under UV light following staining with ethidium bromide
Statistical analysis
Data management and statistical analysis were carried out using SPSS (PC version 13.0). The proportion of ACE genotypes and alleles between cases and controls, and among themselves were compared using Chi-square or Fisher’s exact test. The probability values and odds ratio (OR) with 95% confidence interval (CI) were calculated and presented. The ACE genotypes and alleles were also related with PsA manifestations and severity for finding any association. A probability level (P) less than 0.05 was considered significant.
Results
A total of 51 patients with PsA were enrolled in the study. The over all mean age ± standard deviation (SD) was 46.6 ± 8.4 years. Sixty-three percentage of the patients were females (Table 1). There were 35 males and 65 females (ratio 1:1.8) in the control group with a mean age (±SD) of 46.2 (±7.9) years.
All patients with psoriatic arthritis had peripheral joint involvement (39 had asymmetrical polyarthritis, 7 had symmetrical polyarthritis, and 5 had oligo-arthritis). Ten patients had associated spondylitis, 24 dactylitis, and 10 enthesitis. Data on the frequencies of ACE gene I/D polymorphism genotypes in Kuwaiti PsA patients and controls are presented in Table 2. No significant difference was found in frequencies of ACE genotypes and I/D alleles between patients and controls (P > 0.05). In addition, when a comparison was made between genotype and subgroups of PsA patients, no association was found, either for age at disease onset or with familial type of PsA (P > 0.05). However, the frequency of the genotype II was statistically found to be significantly higher in patients with late age at onset of PsA (>40 years) as compared to those with early onset (≤40 years), P = 0.04 (Table 3).
Further, comparison was made on the homozygous and the heterozygous states of patients. No statistically significant differences were observed in ACE allele and genotype frequencies with the subgroups of PsA patients, compared to control group and within each subgroup (P > 0.05) with regard to those having associated spondylitis, dactylitis, or enthesitis. Similar findings were noticed on comparing ACE allele and genotype frequencies in PsA patients for disease severity with that of controls (P > 0.05).
Discussion
A great deal has been learned about the immunopathogenesis of PsA in recent years. Clearly, the disease has a notable genetic component. This is evidenced by family studies that show a genetic predisposition for psoriasis and PsA that is actually larger than that for a number of other autoimmune conditions. A number of studies have demonstrated that genes play an important role in the etiology of PsA [24, 25]. Multiple genes are likely to be involved, interacting not only with each other but also with the environment to cause disease expression. Information on the significance of ACE gene polymorphism in psoriasis and PsA is scarce. To our knowledge; this is the first such study in PsA patients. We did not find any association between controls and PsA patients in term of ACE gene polymorphism. However, a clear differential distribution was revealed when patients were stratified according to disease onset. There was a positive association of the I-allele with late onset disease. ACE takes part in the renin–angiotensin and kallikrein–kininogen systems by creating angiotensin II and inactivating bradykinin. The I-allele of the ACE gene has recently been shown to be associated with lower ACE activity and kinin degradation [26]. For this reason, we speculate that changes in ACE activity due to I/D polymorphism, leading to reduced ACE levels in plasma and/or in the joints may increase bradykinin levels to trigger an alternative pathway that would cause joint inflammation in PsA patients, especially those with late onset disease.
Angiotensin converting enzyme is expressed in the synovial membranes from patients with the adult form of rheumatoid arthritis (RA), and a role for this enzyme in the pathophysiology has been suggested [27–29]. However, a recent study did not find a significant difference between the prevalence of ACE gene polymorphism in adults with rheumatoid arthritis (RA) and controls, and between severe and non-severe RA [30]. In our study, we found for the first time no correlation with different manifestations of PsA, and even with the severity of the disease.
There are reports concerning the induction and/or exacerbation of psoriasis by ACE inhibitors, which have been attributed to the ACE inhibitor-induced augmentation of kinin levels in skin [31, 32]. Whether this is also applied to joints has to be investigated in future studies.
Molecular genetic studies indicate that there is a multiple PsA susceptibility loci present throughout the human genome, and the genetic factors are likely to be of fundamental importance in the expression of the disease. Although, the exact gene(s) involved in genetic susceptibility have not been identified, some candidate genes are located on chromosome 6p [11, 12, 33, 34]. However, these genes are too distant from the ACE gene (17q23) to explain the association observed.
In our study, we found for the first time no association between ACE gene polymorphism and PsA in Kuwaiti population. Our results suggest that the ACE gene may be a genetic factor contributing to the development of late onset PsA. Further work is required to confirm these findings in different ethnic and racial groups.
References
Gladman D, Espinoza L (1992) International symposium on psoriatic arthritis. J Rheumatol 19:290–291
McHugh N, Balachrishnan C, Jones SM (2003) Progression of peripheral joint disease in psoriaric arthritis: a 5 year prospective study. Rheumatology 42:778–783
Gladman D, Farewell V, Wong K, Husted J (1998) Mortality studies in psoriatic arthritis: results from a single outpatient center. II. Prognostic indicators for death. Arthritis Rheum 41:1103–1110
Gladman DD (1992) Psoriatic arthritis: recent advances in pathogenesis and treatment. Rheum Dis Clin North Am 18:247–256
Gladman DD, Anhorn KA, Schachter RK, Mervart H (1986) HLA antigens in psoriatic arthritis. J Rheumatol 13:586–592
Eastmond CJ (1994) Genetics HLA antigens. Baillières Clin Rheumatol 8:263–276
Gladman DD, Cheung C, Ng CM, Wade JA (1999) HLA-C locus alleles in patients with psoriatic arthritis. Hum Immunol 60:259–261
Gladman DD, Farewell VT, Kopciuk K, Cook RJ (1998) HLA markers and progression in psoriatic arthritis. J Rheumatol 25:730–733
Sakkas LI, Jachesoni A, Kerr LA, Ranza R, Colombo B, Welsh KI, Panayi GS (1991) Immunoglobulin heavy chain gene polymorphism in Italian patients with psoriasis and psoriatic arthritis. Br J Rheumatol 30:449–450
Rahman P, Bartlett S, Siannis F, Pellett FJ, Farewll VT, Peddle L, Schentag CT, Alderdice CA, Hamilton S, Khraishi M, Tobin Y, Hefferton D, Gladman DD (2003) CARD15: a pleiotropic autoimmune gene that confers susceptibility to psoriatic arthritis. Am J Hum Genet 73:677–681
Balding J, Kane D, Livingstone W, Mynett-Johnson L, Bresniham B, Smith O, Fitz-Gerald O (2003) Cytokine gene polymorphisms: association with psoriatic arthritis susceptibility and severity. Arthritis Rheum 48:1408–1413
Hohler T, Grossmann S, Stradmann-Bellinghausen B, Kaluza W, Reuss E, dev Lam K, Veys E, Marker-Hermann E (2002) Differential association of polymorphisms in the TNFalpha region with psoriatic arthritis but not psoriasis. Ann Rheum Dis 61:213–218
Rahman P, Butt C, Siannis F, Farewell VT, Peddle L, Pellett FJ, Schentag C, Gladman DD (2005) Association of SEEK1 and psoriatic arthritis in two distinct Canadian populations. Ann Rheum Dis 64:1370–1372
Mattei MG, Hubert C, Alhenc-Gelas F, Roeckel N, Corvol P, Soubrier F (1989) Angiotensin I converting enzyme gene is on chromosome 17. Cytogenet Cell Genet 51:1041
Rigat B, Hubert C, Alhenc-Gelas F, Cambien F, Corvol P, Soubrier F (1990) An insertion/deletion polymorphism in the Angiotensin I-converting enzyme gene accounting for half the variance of serum enzyme levels. J Clin Invest 86:1343–1346
Smallridge RC, Gamblin GT, Eil C (1986) Angiotensin-converting enzyme characteristics in human skin fibroblasts. Metabolism 10:899–904
Petrov V, Fagard R, Lijnen P (2000) Effect of protease inhibitors on Angiotensin-converting enzyme activity in human T-lymphocytes. Am J Hypertens 13:535–539
Noveral JP, Mueller SN, Levine EM (1987) Release of Angiotensin I-converting enzyme by endothelial cells in vitro. J Cell Physiol 131:1–5
Ryder KW, Epinette WW, Jay SJ, Ransburg RC, Glick MR (1985) Serum angiotensin-converting enzyme activity in patients with psoriasis. Clin Chim Acta 153:143–146
Ozkur M, Erbagci Z, Nacak M, Tuncel AA, Alasehirli B, Aynacioglu AS (2004) Association of insertion/deletion polymorphism of the angiotensin-converting enzyme gene with psoriasis. Br J Dermatol 151:792–795
Moll JMH, Wright V (1973) Psoriatic arthritis. Semin Arthritis Rheum 3:55–78
Sambrook J, Freitsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual, 2nd edn. Cold spring Harbor Laboratory, NY
Rigat B, Hubert C, Corvol P, Soubrier F (1992) PCR-detection of the insertion/deletion polymorphism of the human Angiotensin converting enzyme gene (DCPI) (dipeptidyl carboxypeptidase I). Nucleic Acids Res 20:1433
Korendowych E, McHugh N (2005) Genetic factors in psoriatic arthritis. Curr Rheumatol Rep 7:306–312
Rahman P, Elder JT (2005) Genetic epidemiology of psoriasis and psoriatic arthritis. Ann Rheum Dis 64(suppl 2):ii37–ii39
Murphey LJ, Gainer JV, Vaughan DE, Brown NJ (2000) Angiotensin converting enzyme insertion/deletion polymorphism modulates the human in vivo metabolism of bradykinin. Circulation 102:829–832
Goto M, Fujisawa M, Yamada A, Okabe T, Takaku F, Sasano M, Nishioka K (1990) Spontaneous release of angiotensin converting enzyme and interleukin 1-beta from peripheral blood monocytes from patients with rheumatoid arthritis under a serum-free condition. Ann Rheum Dis 49:172–176
Goto M, Sasano M, Fuzisawa M, Okabe T, Nishizawa K (1992) Constitutive production of angiotensin converting enzyme from rheumatoid nodule cell under serum-free conditions. Ann Rheum Dis 51:741–742
Vaele D, Yanni G, Bresnihan B, Fitz-Gerald O (1992) Production of angiotensin converting enzyme by rheumatoid synovial membrane. Ann Rheum Dis 51:476–480
Zapico I, Coto E, Rodriguez A, Alvarez C, Torre JC, Alvarez V (2000) A DNA polymorphism at the α2-macrogloulin gene is associated with the severity of rheumatoid arthritis. J Rheumatol 27:2308–2311
Coulter DM, Pillans PI (1993) Angiotensin-converting enzyme inhibitors and psoriasis. N Z Med 106:392–393
Wolf R, Tamir A, Brenner S (1990) Psoriasis related to angiotensin-converting enzyme inhibitors. Dermatologica 181:51–53
Gonzalez S, Martinez-Borra J, Torre-Alonso JC, Gonzalez-Roces S, sanchez del Rio J, Rodriguez Perez A, Braulbar C, Lopez-Larrea C (1999) The MICA-A9 triplet repeat polymorphism in the transmembrane region confers additional susceptibility to the development of psoriatic arthritis and is independent of the association of Cw*0602 in psoriasis. Arthritis Rheum 42:1010–1016
Gonzalez S, Martinez-Borra J, Torre-Alonso JC, Lopez-Vazquez A, Segal R, Blanco-Gelaz MA, Enk CD, Safriman C, Lopez-Larrea C (2001) Polymorphism in MICA rather than HLA-B/C genes is associated with psoriatic arthritis in the Jewish population. Hum Immunol 62:632–638
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Al-Awadhi, A.M., Hasan, E.A., Sharma, P.N. et al. Angiotensin-converting enzyme gene polymorphism in patients with psoriatic arthritis. Rheumatol Int 27, 1119–1123 (2007). https://doi.org/10.1007/s00296-007-0349-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00296-007-0349-y