
Abstract
A novel tellurium (IV) hybrid compound with 2-methylimidazole was prepared. The crystal was grown by slow evaporation method from aqueous solutions at room temperature giving birth to a new compound with formula (C4H7N2)2TeBr6. The structure was determined by single-crystal X-ray diffraction. It crystallizes in the monoclinic system, space group C 2/c, with the following parameters: a = 18.577(2)Å, b = 9.1497(10)Å, c = 13.5355(17)Å, α = 90°, β = 123.835(5)°, γ = 90° and Z = 8. The structure was solved with a final R = 0.044 for 2783 independent reflections. The crystal arrangement consists of [TeBr6]2− anions surrounded by [C4H7N2]2+ cations. The stability of the structure was ensured by hydrogen bonding contacts (N–H…Br) lengths that are in the range of 3.490–3.682 Å. The infrared spectra (FTIR) were recorded in the 4000–550 cm−1, confirming the existence of vibrational modes that correspond to the organic and inorganic groups. The crystal packing was stabilized mainly by Br…H (73.6%) interactions. Hirshfeld surface calculations were conducted to further investigate intermolecular interactions, associated 2D fingerprint plots and enrichment ratio, revealing the quantitatively relative contribution of these interactions in the crystal packing.
Similar content being viewed by others

Introduction
Hybrid organic–inorganic materials with distinctive properties have a great importance from combining both organic and inorganic components into the material [1,2,3,4]. They are characterized by a large variety of structures framework dimensionality build from chains, layers, or three-dimensional connections [5, 6] as well as a correlation between the structural features and a vast diversity of physical properties from their potential applications such as magnetic, optical, luminescence and electric [7,8,9,10,11]. Hybrid halogen-based materials have demonstrated exciting properties from the renewal of zero-dimensional structures belonging to alums and Tutton’s salts [12,13,14,15]. In addition, Tellurium metalloid, as a strong Lewis acid, has an extensive coordination chemistry [16,17,18]. A large number of salts containing tellurium halides octahedra anions [TeX6]2− (X = Cl, Br) has been prepared and structurally characterized [19, 20]. Besides, hybrids based on tellurium have caught attention as promising materials due not only to their interesting structural topologies, but also to their physical properties such as luminescence, nonlinear optical activity, ferroelectricity and semi-conductivity [21,22,23]. The introduction of tellurium in metal–organic frameworks as clusters [24, 25] or tellurium-coordination polymers [26] allows a better correlation between structural features and physical characteristics [27,28,29,30,31]. In the organic part, the use of amines as template, generally protonated, has provided many types of hybrid materials, which could exhibit interesting optical properties [32, 33]. In almost all of these materials, the amine cations interact with the inorganic part, through weak hydrogen bonds, to give supramolecular network. However, it seems that the amine group may have an influence on physical property. We recently showed that synthesized bromotellurate salt of 4-(ethylammoniummethyl)pyridinium, in its solid state, showed a maximum UV–visible absorption and photoluminescence emission based on that of the organic part [34]. The aim of this study is to introduce the influence of 2-methylimidazole diamine that includes delocalization of electrons from aromaticity in this class of materials to further explore interesting optical properties. The peculiarity of the used 2-methylimidazole, C4H6N2, is that it can be described as heterocyclic amine soluble in water characterized by the presence of a methyl group linked to an aromatic five-heterocycle which contains two nitrogen atoms. In the present study, the crystal structure of (C4H7N2)2TeBr6 is described in detail. An interest for the significant effect of the aromatic diamine template is given. We performed Hirshfeld surface analysis to further investigate and explain intermolecular interactions in the structure.
Experimental
Synthesis
The ligand 2-methylimidazole (0.082 g, 2 mmol) was added to an aqueous solution (10 ml) of Te(OH)6 (0.23 g, 1 mmol) which were dissolved in a concentrated hydrobromic acid HBr (3 ml) as shown in the following reaction:
The resulting orange solution was stirred for about 10 min and then left to slowly concentrate by solvent evaporation at room temperature for a period of 10 days. Well-formed orange parallelepiped crystals of the product appeared and were collected by filtration.
Single-crystal data collection and structure determination
A tiny single crystal was carefully chosen from the available crystals under a polarizing microscope to perform its structural analysis by X-ray diffraction. Diffraction data were collected on a Bruker APEX II CCD diffractometer, using graphite-monochromated MoKa radiation (0.71073 Å) at 296 K. Intensity measurements were made between angles, namely 2, 6 and 30°, giving rise to the following miller indices: − 24 ≤ h ≤ 25; − 12 ≤ k ≤ 12; − 19 ≤ l ≤ 17. The structure was solved by direct methods using the SHELXS-2014 program [35]. However, the positions of the tellurium atoms were determined through a three-dimensional Patterson synthesis. Bromine, carbon, nitrogen and hydrogen atoms were located by a three-dimensional Fourier function. Structure solution and refinement were carried out using SHELX programs [36] and refined by full-matrix least squares methods on F2 with all non-hydrogen atoms anisotropic SHELXL-2014 [37]. The non-hydrogen atoms were refined anisotropically. The hydrogen atoms were attributed to isotropic thermal factors close to those of the atoms to which they are linked. The hydrogen atoms of the CH, NH and NH2 groups were fixed geometrically by the appropriate instructions of the program (AFIX 43, AFIX 43 and AFIX 93, respectively). The H-atom of the N is located by difference Fourier synthesis. A final refinement on F2 with 2783 unique intensities and 89 parameters converged at wR(F2) = 0.084 (R(F) = 0.063) for 1369 observed reflections with I > 2σ (I). Calculations were performed using the WinGX crystallographic software package [38]. The crystal data collection and structure refinement results of the compound are given in Table 1. The structural graph was created with Diamond program [39].
X-ray powder diffraction
X-ray powder diffraction pattern of ground crystals of the compound (C4H7N2)2TeBr6 was recorded with a PANalytical θ/θ Bragg–Brentano Empyrean diffractometer (CuKα1+2 radiations) equipped with the PIXcel1D detector. Data were collected in the [5–100°] 2θ scattering angle range with a 0.013° step and 30 s per step for a total acquisition of 15 min.
Hirshfeld surface calculations
To define the space occupied by molecules that come into contact in the crystal, we have required the Hirshfeld surface were constructed from CIF file to the analysis of the crystal structures generated by the Crystal Explorer [40] program. It allows the visualization of the different types of intermolecular contacts in the crystal by focusing on close ones between atoms in neighboring molecules. The Hirshfeld surface was computed around the asymmetric unit using the normalized contact distance surface (dnorm). It can be generated into 2D fingerprint plots, which is a two-dimensional summary of intermolecular interactions that we take into account in the crystal. The dnorm is given by the following equation:
while di and de are the distances to the nearest atoms inside (di) and outside (de) the surface, while ridvW and redvW are the van der Waals radii of the appropriate atoms internal or external to the surface [41]. The normalized contact distance is displayed using a red-white-blue color scheme; the red color is used for highlighting shorter contacts, white for contacts around the vdW separation and blue for longer contacts [42]. Two additional colored properties (shape index and curvedness) based on the local curvature of the surface can also be specified [43]. As shown in this paper, the Hirshfeld surfaces are mapped with dnorm, shape-index, curvedness and 2D fingerprint plots (full and resolved) were performed by the Crystal-Explorer program [44].
Spectroscopic measurements
Fourier transform infrared (FT-IR) measurements were performed at room temperature on a Perkin-Elmer FT-IR Paragon 1000 PC spectrometer over the 4000–400 cm−1 region. Samples were prepared in KBr pellets. As for the Raman scattering spectrum, it was recorded using a T-64000 Raman spectrometer (ISA, JobinYvon) with standard attachments. The beam sources were argon krypton titan-sapphire and semi-conductor lasers. The spectrum was recorded in the range of 50 cm−1 to 4000 cm−1. Infrared and Raman spectra are recorded with the same resolution of 3 cm−1.
Scanning electronic microscopy (SEM)
SEM images of (C4H7N2)2TeBr6 were obtained using a JEOL microscope (JSM 6510 LV). Acceleration voltages varied between 20 and 30 kV as a function of the analyzed samples. Elementary quantitative microanalyses were performed using an energy-dispersive X-ray (EDX) OXFORD detector (Aztec software).
Results and discussion
Preliminary characterizations
Crystals taken from the stock solution, washed with ethanol and dried were studied by energy-dispersive X-ray (EDX). The data collected by this technique clearly reveal the organic/inorganic nature of the analyzed compound (Fig. 1). The deduced composition is only used as a guide to confirm the presence of tellurium, bromine and nitrogen. In addition to the DRX data of the single crystal, we can affirm that it is a hybrid material.

Analysis of a single crystal of (C4H7N2)2TeBr6 by EDX and morphology of the analyzed crystal
The recording of the X-ray powder diffraction pattern of the compound was carried out. The comparison between experimental pattern and theoretical pattern generated from the crystal structure determined by DRX from single crystal at room temperature (Fig. 2) shows that the sample of the synthesized hybrid compound presents a single phase.

X-ray powder diffraction patterns of (C4H7N2)2TeBr6. Red: simulated from the structure determined by DRX of the single crystal. black: experimental (color figure online)
Crystallographic study
The crystal structure of the title compound (C4H7N2)2TeBr6 has been solved and refined in the centrosymmetric monoclinic space group C2/c, with eight formula cell unit (Z = 8). The different parameters of the crystallographic unit cell have the values of: a = 18.577(2)Å, b = 9.1497(10)Å, c = 13.5355 (17)Å, β = 123.835(5)°, and V = 1911.1(4)Å3. Further details are reported in Table 1.
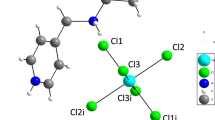
The asymmetric part of the unit cell of the title compound consists of one 2-methylimidazolium [C4H7N2]2+ cation and one hexabromotellurate (IV) anion [TeBr6]2− (Fig. 3). The Te atom is sixfold coordinated by bromide ions, forming an octahedral polyhedron. Thus, to ensure charge equilibrium, the structure associates each hexabromotellurate (IV) anions with one 2-methylimidazole cation.

Asymmetric unit of the (C4H7N2)2 TeBr6 crystal
The atomic arrangement in the structure of the title material (Fig. 1S), according to the (a, c) plan, shows an alternation of organic cations and mineral anions along the crystallographic [101] direction. Octahedral inorganic [TeBr6]2− entities are arranged along the crystallographic c axis and connected with protonated amines through N–H…Br hydrogen bonds, while the organic cations [C4H7N2]2+ are interconnected in the same direction by the π stacking between their aromatic rings in such a way to neutralize the negative charge of the inorganic part. The perspective view of the atomic arrangement of (C4H7N2)2TeBr6 compound is shown in Fig. 4. Actually, the structure is composed of parallel inorganic parts alternated by organic chains. The organic cations trapped in the spacing between the inorganic parts and connection between all molecules are made by hydrogen bonds to from an infinite zero-dimensional network. The supramolecular crystal packing is built by N–H…Br hydrogen bonds varying between 3.490 and 3.682 Å (Table 2). The anionic polyhedron [TeBr6]2− exhibits a slightly distorted octahedral coordination environment with Te–Br band lengths ranging from 2.6361 (11) Å to 2.7476 (11) Å and Br−Te−Br bans angles fall in the range of 88.58°(2) and 180° (Table 1S). The small differences of the Te–Br bond lengths are not insignificant considering the standard deviations. There appears to be no stereochemical active lone pair of electrons at Te (IV) in the hexahalogeno complex ion [45,46,47,48,49].

Hydrogen bonds between chlorine and organic entities in the crystal structure of (C4H7N2)2 TeBr6 in projection along the a-axis. (H-bonds are represented by dashed lines)
The examination of the geometric features of the organic molecule exhibits a regular spatial configuration with C–C, C–N distances and C–C–N, C–N–C, N–C–C and N–C–N angles, quite like those found in other compounds [50, 51]. The C–N band lengths vary from 1.305(8) to 1.366(8)Å, and those of C–C band vary from 1.323(10) to 1.476(9)Å and C–C–N, C–N–C, N–C–C and N–C–N angles are between 106.3°(6) and 127.2°(7) (Table 1S). The distance between two aromatic nuclei (centroids) was 3.656 Å < 3.8 Å (Fig. 5) which confirms that the π–π interaction is present in this compound [52].

View of the p...p interactions between ring centroids
Molecular Hirshfeld surface analysis
The Hirshfeld surfaces have been made to explore the type of interactions, such as (C…H, H…H, C…C etc.) or hydrogen bonds and to study the various intermolecular interactions that play an important role in a molecular crystal. The molecular Hirshfeld surface, 3D dnorm, shape index and curvedness for (C4H7N2)2TeBr6 are illustrated in Fig. 6, showing surfaces that have been napped over dnorm ranging from − 0.246 to 1.026 Å, shape index ranging from − 1.000 to 1000 Å, and curvedness ranging from − 4.000 to 0.400 Å, respectively. The dnorm mapping indicates that strong hydrogen bond interactions, such as N–H…Br hydrogen bonding between amino group and bromide, appear as the main interaction between the complexes and are seen as a bright red area in the Hirshfeld surfaces (Fig. 6a). The brightest and largest red spots observed in the dnorm refer to the shorted intermolecular contacts indicating the existence of hydrogen bonds N–H…Br in the crystal packing. The superseding H…Br interactions viewed in Hirshfeld surfaces by the bright red are in Fig. 6a, which appear as distinct spike in the 2D fingerprints plots (Fig. 2S).

Hirshfeld surfaces of (4-ethylaminomethyl pyridinum Telluric chlorid VI): a 3D dnorm surface, b surface index, c curvedness
We can decompose 2D fingerprints plots to highlight close contacts. This decomposition enables the separation of each contribution of the different interactions from the full fingerprint. Visible complementary regions are visible in the fingerprint plot where one molecule acts as a donor (de > di) and the other as an acceptor (de < di). The 2D fingerprint plots of the title compound (Fig. 2S) indicate the strong intermolecular contacts, which are Br-H, H–H, C–H, Br–Br, N–Br, C–N, C–Br, N–H. The H–H…Br/Br…H contacts exhibit the characteristic shape of two “wings” at the top left and the bottom right of the fingerprint plots (Fig. 2Sa). In fact, the H…Br/Br…H constitutes the most important interactions in the crystal and their relative contribution extends to 73.6% (indicated as red areas) due to the existence of N–H…Br hydrogen bonds. In contrast, the H–H contacts are rather homogeneously extended over a large range of (di, de) pairs with an average contribution of 12.4% (Fig. 2Sb). The C…H/H…C contacts are also broadly distributed with accumulation comprising 2.8% (Fig. 2Sc). However, Br…Br and N…Br/Br…N contacts display relatively a sharp and acicular distribution with an average of 2.7 and 2.7%, respectively (Fig. 2Sd,e). The intermolecular C…Br/Br…C and C…N/N…C interactions appear as short blue-colored patches with the proportion of 1.7 and 1.3%, respectively (Fig. 2Sf,g). In addition, N…H/H…N interactions are negligible and exhibit only 1.1% (Fig. 2S,h).
The dnorm values were mapped onto the Hirshfeld surface (Fig. 6a) using red, blue and white schemes as follows: red regions representing closer contacts and a negative dnorm value; blue regions representing longer contacts and a positive dnorm value; and white regions representing the contacts distance that is exactly equal to the vdW separation with a dnorm value of zero. These normalized contact distances (dnorm) disclose the adjacent contacts of hydrogen bond donors and acceptors as well as evident other close contacts. Indeed, Fig. 6a shows that the huge circular depressions are indicative of hydrogen bonding contacts and the prevailing interactions are Br-H. Nonetheless, other noticeable spots are due to H–H contacts, based on both de and di. The shape index is extremely susceptible to very delicate changes in the surface shape. Indeed, shape index (S) is 0D dimensionless surface property described in terms of the two major surface curvatures and can be used to differentiate complementary hollows (S− 1.0; concave region; red colored) and bumps (S+ 1.0; convex region; blue colored) where two molecular Hirshfeld surfaces come into contact [53,54,55,56,57]. In particular, adjacent red/orange and blue triangle like patches on a shape index map (Fig. 6b) provide information about π–π stacking interactions [53, 56, 57]. However, in this compound, the π–π interactions were present as indicated in Fig. 6b; thus, the Hirshfeld surface confirms the presence of the π–π stacking interactions in organic cations. The curvedness is a measure of the shape of the molecule surface area. As can be seen in Fig. 6c, while the flat surface areas correspond to the curvedness values, the sharp curvature areas correspond to the high curvedness values and usually tend to divide the surface into patches, representing interactions between neighboring molecules. The big flat region that is described by a blue outline refers to the π–π staking interactions.
Infrared and Raman spectroscopy
In order to obtain more information on the crystal structure, we studied the vibrational properties using Raman scattering and infrared absorption. IR and Raman spectroscopy is a proficient method to confirm the functional groups present in the crystal and to study the structural consequences such as in-plane or out-of-plane vibrations [58]. At this level, we have discussed the vibrational analyses of the compound and tried to give the most precise assignment of the detected bands.
In fact, the Raman bands correspond to the translational, vibrational and external modes of the inorganic groups [TeBr6]2− in the low-frequency range 50–250 cm−1 (Fig. 7). As for the bands observed between 250 and 4000 cm−1 in the Raman spectra and those in the IR spectra (Fig. 8), they are assigned to the internal modes of the cation. The assignment of the internal and lattice modes of the organic cations and inorganic anion is based on the comparison with the well-documented spectra of the homologous compounds [59,60,61,62,63].

Experimental Infrared spectrum of the (C4H7N2)2 TeBr6

Exprimental Raman spectrum of the (C4H7N2)2 TeBr6
In the following, the IR assignments will be described in detail. The Raman spectrum of this crystal confirms also the presence of the majority of vibration mode function for the organic molecule (Fig. 7, Table 3). We note that the broadening of the band is caused by hydrogen bonding interactions which influence the band frequency as well as the intensity of the peak [64].
The IR spectrum of this compound (Fig. 8 and Table 3) (in Supporting information) shows the characteristic absorption peaks of the [C4H7N2]2+ cation [65, 66]. The peaks at 3456 cm−1 and 3436 cm−1 correspond to the (N–H) stretching symmetric and asymmetric vibrations. However, the scissoring asymmetric and symmetric vibrations δ(N–H) have been identified as broad and weak bands at 1578 and 1521 cm−1 in the IR spectrum, respectively. In aromatic compounds, the C–H stretching wave numbers appear in the range of 3000–3100 cm−1. Accordingly, in the present study, the aromatic =C–H stretching gives bands at 3081 cm−1 in the IR spectrum. The symmetric and asymmetric stretching vibrations of (C–H) are observed at 2827 and 2525 cm−1. The bands observed at 1475 and 1456 cm−1 in FT-IR spectra are attributed to the scissoring symmetric and asymmetric vibrations δ(C–H), respectively. Moreover, the N–H and C–H in plane bending in aromatic groups are located at the interval 1120–1106 cm−1.
The vibrations modes between 3176 and 3126 cm−1 are due to the (CH3) asymmetric and symmetric stretching vibrations. The deformation asymmetric and symmetric of (CH3) bond is observed at 1563 and 1503 cm−1 in IR spectrum. The C=N aromatic stretches are predicted at 1618 cm−1 in IR spectra [67]. In the present work, the C=C aromatic stretch is observed at 1603 cm−1 in the FT-IR. The aromatic ring carbon–nitrogen υ(C–N) stretching vibrations are detected at 1395 cm−1. The strong bands identified at 1408 cm−1 in the IR spectrum are assigned to υ(C–C) stretching vibrations in the ligand. The bands relating to the deformation mode symmetric and asymmetric of the C–N group appear in IR spectrum at 1138 and 943 cm−1. The deformation of (C–C) bond is observed at 733 cm−1. Besides, the absorption bonds located at 1485 and 1436 cm−1 in IR spectrum are assigned to the (C–N–H) symmetric and asymmetric scissoring vibration.
The bands relating to the in-plane deformation modes of the C–C–N, N–C–C, N–C–N and C–N–C groups appear in the spectral range between 1077 and 1037 cm−1. The bands corresponding to the δ(C–C–N) scissoring in-plane mode appear at 524 cm−1. The band detected at 619 cm−1 corresponds to the in-plane bending of C–N–C. The β(C–C–H) in-plane bending mode is located at 1006 cm−1. The peak located in IR spectrum at 854 cm−1 is generated by the out-of-plane bending mode of γ(C–C–H) groups.
Conclusion
The present research work reported the synthesis and characterization of the new centrosymmetric hybrid compound (C4H7N2)2TeBr6. Thus, single crystals of (C4H7N2)2TeBr6 were collected from an aqueous solution by a slow evaporation technique. The obtained material crystallizes in the space group C2/c with monoclinic system. The structure of this compound consists of isolated [TeBr6]2− octahedral anions and 2-methylimidazole cations. The crystal structure is stabilized by N–H…Br hydrogen bonds and \(\pi-\pi \) stacking interactions. Hirshfeld surface allowed us to investigate the stabilization of the crystal packing and quantify the propensity of the intermolecular interactions to form the supramolecular assembly. The study of the vibration spectroscopy by IR, Raman at room temperatures proved the homogeneity and purity of this synthesis.
References
Sanchez C, Julián B, Belleville P, Popall M (2005) Application of hybrid organic–sinorganic nanocomposites. J Mater Chem 15:3559
Sanchez C, Belleville P, Popall M, Nicole L (2011) Crystal structure, thermal behavior and vibrational spectra of 4,4′ diamoniumdiphenylmethan sulfate hydrate. Chem Soc Rev 40:696
Sanchez C, Ribot F (1994) Hybrid organic-inorganic copolymers based on oxo-hydroxo organotin nanobuilding blocks. New J Chem 18:1007
Cheetham AK, Férey G, Loiseau Angewandte T (1999) Synthesis, characterization and X-ray structure of a Ba(II)/Ag(I)/Cr(III)-oxalate salt with water-filled nanochannels. Chemie; International Edition in English 3268
Ozin GA (1992) Microwave-assisted wet chemical synthesis: advantages, significance, and steps to industrialization. Nanochemistry Adv Mater 4:612
Teraski O, Barry JC, Thomas JM (1987) On crystal structure imaging of silicalite by HREM. Nature (London) 330:58
Fleck M, Ohat L, Tillmanns E (2004) Structural characterisation of MII guanidinium sulphate hydrates (MII=Mn, Fe Co, Ni, Cd, VO). Solid State Sci 6:469
Wang YJ, Xu L (2008) Synthesis, crystal structure, and characterization of a new organic-inorganic hybrid material. J Mol Struct 875:570
Arjunan V, Marchewka MK, Kalaivani M (2012) Spectrochim. Acta Part A Mol Biomol Spectrosc 96:744
Santos SS, Schulz Lang E, Manzoni de Oliveira G (2007) Raman and infrared spectra of thymine. A matrix isolation and DFT study. J. Organomet. Chem. 692:3081–3088
Schulz Lang E, de Oliveira GM, Fernandes Jr RM E.M. V_azquezeL_opez Z; Anorg (2004)Synthesis, crystal structure, and optical characteristics of [Pd2Hg4Cl6{Te(DMB)}6]·2DMF, [HgClTe(DMB)]4, and the ring-forming cluster [Pd12(TePh)24]·2DMF. Allg Chem 630:717
Wang G-E, Wang M-S, Zhang M-J, Cai L-Z, Liu B-W, Zhang C-J, Guo G-C, Huang J-S (2012) Two viologen-based photoluminescent compounds: excitation-wavelength-dependent and photoirradiation-time-dependent photoluminescent switches. Inorg Chem Commun 23:137
Leblanc N, Mercier N, Zorina L, Simonov S, Auban-Senzier P, Pasquier C (2011) Large spontaneous polarization and clear hysteresis loop of a room-temperature hybrid ferroelectric based on mixed-Halide [BiI3Cl2] polar chains and methylviologen dication. JACS 133:14924
Hrizi C, Chaker C, Chaabouni S (2011) Synthesis, crystal structure, thermal and dielectric properties of bis(p-phenylenediammonium) chloride hexachlorobismuthate(III) monohydrate [C6H4(NH3)2]2ClBiCl6·H2O. Ionics 17:545
Phanon D, Gautier-Luneau I, Angew (2007) Frequency conversion in the acentric tetragonal nonlinear crystal NaI3O8. Chem Int Ed Engl 46:8488
Levason W, Reid G, Victor M, Zhang W (2009) Tellurium(II) and tellurium(IV) complexes of phosphine chalcogenide ligands, synthesis and X-ray structures. Polyhedron 28:4010
Mironov YV (2000) Preparation, structures, and redox and emission characteristics of the isothiocyanate complexes of hexarhenium(III) clusters [Re6(μ3-E)8(NCS)6]4-(E = S, Se). Polyhedron 19:437
Pietikainen J, Maaninen A, Laitinen RS, Oilunkaniemi R, Valkonen J (2002) Selective electrocatalytic hydroboration of aryl alkenes. Polyhedron 21:1089
Krebs B, Ahlers F-P (1990) Bis(tetraphenylphosphonium) di-μ-iodido-bis[diiodidotellurate(II)]. Adv Inorg Chem 35:235
Dammak M, Mhiri T, Jaud J, Savariault J (2001) Systematic prediction of new ferroelectrics in space group R3 II. Int J Inorg Mater 3:861873
Piecha-Bisiorek A, Mence K, Kinzhybalo V, Szota A, Jakubas R, Medyckl W, Zawrocki W (2018) Ferroelasticity and piezoelectricity of organic–inorganic hybrid materials with a one-dimensional anionic structure: so similar, yet so different. CrystEngComm. 20:2112
Sun T, Liang F, Zhang X, Tu H, Lin Z, Zhang G, Wu Y (2017) Multilayer stacked low-temperature-reduced graphene oxide films: preparation, characterization, and application in polymer memory devices. Polyhedron 127:8
Kchaou H, Ben Rhaiem A, Karoui K, Jomni F, Guidara K (2016) Electrical properties and phase transition of [(CH3)3NH]CdCl3 compound. Appl Phys A82:122
Brylev KA, Mironov YV, Naumov NG, Fedorov VE, Ibers JA (2004) New compounds from tellurocyanide rhenium cluster anions and 3d-transition metal cations coordinated with ethylenediamine. Inorg Chem 43:4833
Artemkina SB, Naumov NG, Virovets AV, Fedorov VE (2005) 3D-Coordination cluster polymers [Ln(H2O)3Re6Te8(CN)6]×nH2O (Ln: La3+, Nd3+): direct structural analogy with the mononuclear LnM(CN)6×nH2O family. Cheminform 36
Janiak C (2003) Engineering coordination polymers towards applications. Trans 2781
Dammak M, Mhiri T, Jaud J, Savariault JM (2001) Synthesis, structural study, thermal, optical properties and characterization of the new compound [C6H7N2O2]3TeCl5·2Cl. Int J Inorg Mater 3:861
Bochmann M, Coleman AP, Webb KJ, Hursthouse MB, Mazid M (1991) Synthesis and characterisation of manganese(II) chalcogenolato complexes Crystal and molecular structure of [{Mn(µ-SeC6H2Me3-2,4,6)2}∞]. Angew Chem Int Ed 30:97
Lee DJ, Freedman J, Melman H (1998) Trivalent lanthanide chalcogenolates: Ln(SePh)3, Ln2(EPh)6, Ln4(SPh)12, and [Ln(EPh)3]n (E = S, Se). How metal, chalcogen, and solvent influence structure. Inorg Chem 37:2512
Bird PH, Kumar V, Pant BC (1980) Syntheses and crystal structures of compounds containing short TeN bonds. Inorg. Chem. 19:2487
Alcock NW, Harrison WD (1982) Structure of catena-μ-bromo-dibromo(phenyl)-tellurium(IV). Acta Cryst. B38:2677
Maraii D, Dammak M (2020) Synthesis, structure, optical and thermal analysis of the new compound (C3N6H7)2Te(OH)6.2Cl. J Mol Struct 1217:128427
Bukvetskii BV, Sedakova TV, Mirochnik AG (2010) Russ J Coord Chem 36:651
Mhiri M, Kammoun O, Lhoste J, Auguste S, Ammar H, Boujelbene M (2021) Crystal structure and optical property of asupramolecular bromotellurate salt templated with 4 (Ethylammoniummethyl)pyridinium. J Mol Struct 2021:131994
Sheldrick GM (1986) SHELXS 2014. Crystal structure refinement with SHELXL. University of Gottingen, Germany
Sheldrick GM (2014) SHELXL 2014. University of Gottingen, Germany
Sheldrick GM (2008) Crystal structure refinement with SHELXL ShelXle: a Qt graphical user interface for SHELXL. Acta Crystallogr A 64:112
Farrugia LJ (1999) WinGX suite for small-molecule single-crystal crystallography. Appl J Crystallogr 32:837
Brandenburg K (1998) Diamond version 2.0 impact GbR, synthesis and crystal structure of a chiral aromatic amine chloride salt (C8H12N)Cl. Bonn, Germany
Wolf SK, Grimwood DJ, Mckinnon JJ, Turner MJ, Jayatilaka D, Spackman MA (2012) Crystal structure and Hirshfeld surface analysis of the new cyclodiphosphazane [EtNP(S)NMe2]2. In: Crystal Explorer3.1. University of Western Australia
Mckinnon JJ, Jayatilaka D, Spackman MA (2007) Towards quantitative analysis of intermolecular interactions with Hirshfeld surfaces. Chem Commun 37:3814
Wolff SK, Grimwood D, Mckinnon JJ, Turner MJ, Jayatilaka D, Spackman MA (2010) CrystalExplorer model energies and energy frameworks: extension to metal coordination compounds, organic salts, solvates and open-shell systems. In: 2010 crystal explorer 3.1. University of Western Australia
Seth SK, Sarkar D, Jana AD, Kar T (2011) Supramolecular self-assembly of M-IDA complexes involving lone-pair···π interactions: crystal structures, Hirshfeld surface analysis, and DFT calculations [H2IDA = iminodiacetic acid, M = Cu(II), Ni(II)]. J Cryst Growth Des 11:4837
Parsons S, Flack HD, Wagner T (2013) Use of intensity quotients and differences in absolute structure refinement. Acta Crystallogr. B 69:249
Gillespie RJ (1992) Atomic orbitals, molecular orbitals and related concepts: Conceptual difficulties among chemistry students. Chem Soc Rev 21:59
Ng SW, Zuckerman JJ (1985) The structures of the group 15 element(III) halides and halogenoanions. Adv Inorg Chem Radiochem 29:297
Stufkens DJ (1970) Dynamic Jahn–Teller effect in the excited state of TeCl62−. √T dependence of the splitting of the C absorption band. Recl Trav Chim 89:1185
Sheldrick GM (1986) SHELXS97, program for the refinement of crystal structures, synthesis and crystal structure of 3-ammonium-4-hydroxyphenyl sulfonate hemihydrate 1986. University of Gottingen, Germany
Sheldrick GM (1997) SHELXL97, program for the refinement of crystal structures, new synthesis of silver phosphonate complexes from polymeric silver phenylethynide as a structure-directing precursor 1997 University of Gottingen, Germany
Jagan R, Sivakumar K (2009) N-H…O and O-H…O hydrogen-bonded supramolecular networks in 4-chloroanilinium, 2-hydroxyanilinium and 3-hydroxyanilinium hydrogen phthalates. Acta Crystallogr. C 65:414
Jasinski JP, Golen JA, Praveen AS, Yathirajan HS, Narayana B (2011) 4-Chloro-anilinium 4-methyl-benzene-sulfonate. Acta Crystallogr E 67:3288
Chaabouni S, Hrizi C, Chaker H (2011) Synthesis, crystal structure, thermal and dielectric properties of bis(p-phenylenediammonium) chloride hexachlorobismuthate(III) monohydrate [C6H4(NH3)2]2ClBiCl6·H2O. Ionics 17:545
Spackman MA, McKinnon JJ (2002) Fingerprinting intermolecular interactions in molecular crystals. CrystEngComm 4:378
McKinnon JJ, Spackman MA, Mitchell AS (2004) Novel tools for visualizing and exploring intermolecular interactions in molecular crystals. Acta Crystallogr Sect B Struct Sci 60:627
McKinnon JJ, Jayatilaka D, Spackman MA (2007) Towards quantitative analysis of intermolecular interactions with Hirshfeld surfaces. Chem Commun 3814
Spackman MA, Jayatilaka D (2009) Hirshfeld surface analysis. CrystEngComm 11:19
Rodrigo Bitzer S, Lorenzo Visentin C, Vorner MH, Marco AC, Nascimento AL, Filgueiras C (2017) Synthesis, spectroscopic characterizations, crystal structures and DFT studies of nalidixic acid carbonyl hydrazones derivatives. J Mol Struct 1130:165
Thanigaimani K, Khlib NC, Temel E, Arshad S, Razak IA (2015) New supramolecular cocrystal of 2-amino-5-chloropyridine with 3-methylbenzoic acids: Syntheses, structural characterization, Hirshfeld surfaces and quantum chemical investigations. J Mol Struct 1099:246
Nataraj A, Balachandran V, Karthick T, Karabacak M, Atac A (2012) FT-Raman, FT-IR, UV spectra and DFT and ab initio calculations on monomeric and dimeric structures of 3,5-pyridinedicarboxylic acid. J Mol Struct 1027:1
Koczon P, Dobrowolski JCZ, Lewandowski W (2003) Experimental and theoretical IR and Raman spectra of picolinic, nicotinic and isonicotinic acids. J Mol Struct 655:89
Hamdouni M, Walha S, Kabadou A, Duhayon C (2013) Synthesis and crystal structures of various phases of the microporous three-dimensional coordination polymer [Zr(OH)2(C2O4)]n. J Pascal Growth Des 13:5100
Lyszczek R, Anal J (2009) Synthesis, crystal structure, spectroscopic and thermal investigations of neodymium(III) biphenyl-4,4′-dicarboxylate framework. Appl Pyrolsis 86:239
Stefanovich VA, Suslikov LM, Gad’mashi ZP, Yu Peresh E, Sidei VI, Zubak OV, Galagovets IV (2004) Synthesis, spectroscopic, structural and thermal characterizations of [(C7H6NO4)2TeBr6·4H2O]. Phys Solids State 46:1024
Suthan T, Rajesh NP, Mahadevan CK, Bhagavvannarayana G (2011) Mater Chem Phys 12:433
Krishnakumar V, Nagalakshmi R (2008) Studies on the first-order hyperpolarizability and terahertz generation in 3-nitroaniline. Phys B 403:1863
Karabacak M, Kose E, Atac A (2012) NMR, UV, FT-IR, FT-Raman spectra and molecular structure (monomeric and dimeric structures) investigation of nicotinic acid N-oxide: A combined experimental and theoretical study. Struct Acta Part A 91:83
Babu B, Chandrasekaran J, Mohanbabu B, Matsushita Y, Saravanakumar M (2016) Growth, physicochemical and quantum chemical investigations on 2-amino 5-chloropyridinium 4-carboxybutanoate—an organic crystal for biological and optoelectronic device applications. RSC Adv 6:110884
Acknowledgements
The authors greatly acknowledge the platforms of the “Diffusion et Diffraction des Rayons-X” and the “Microscopy” de l’IMMM, Le Mans Université. Miss A. Bali is gratefully acknowledged for her assistance in maintaining the grammatical structures of this work.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mhiri, M., Kammoun, O., Lhoste, J. et al. Growth, crystal structure, Hirshfeld surface and vibrational properties of a new supramolecular hybrid material: (C4H7N2)2TeBr6. Polym. Bull. 80, 829–846 (2023). https://doi.org/10.1007/s00289-021-04053-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00289-021-04053-6