
Abstract
Neutrophils are the foot soldiers of the immune system. They home in to the site of infection and kill pathogens by phagocytosis, degranulation, and the release of web-like structures called neutrophil extracellular traps (NETs) that trap and kill a variety of microbes. NETs have been shown to play a multitude of additional roles in immunity but have also been implicated in inflammatory and autoimmune disease. Here, we discuss the role of NETs in these various contexts with a particular emphasis on the molecular mechanisms that regulate NET release and clearance. We highlight the comprehensive concepts and explore the important open questions in the field.
Similar content being viewed by others


Neutrophils are the major antimicrobial phagocytes of the innate immune system. Along with eosinophils, basophils, and mast cells, they comprise the granulocyte lineage. In the absence of infection, these short-lived, terminally differentiated cells will leave the bone marrow and die in the confines of the bloodstream. Upon infection, tissue-resident macrophages and other sentinel cells, secrete inflammatory cytokines and chemoattractants that are able to recruit and prime neutrophils. In response to these molecules, neutrophils leave the bloodstream and invade the infected tissues in a selectin- and integrin-mediated process known as extravasation. They are one of the first effector cells to arrive at the site of infection and play critical roles in pathogen clearance, recruitment, and activation of other immune cells and tissue repair. Neutrophils employ three major strategies to combat microbes: phagocytosis, degranulation, and the release of neutrophil extracellular traps (NETs) [1–3]. In this review, we focus on the mechanisms that regulate the release of NETs during infection and disease. We also cover various aspects of NET function in these contexts.
Neutrophil antimicrobial strategies
Neutrophils are equipped with cytokines and an arsenal of highly reactive, short-ranged antimicrobial proteins that exhibit broad specificity and can be equally toxic to microbes and host cells alike. Therefore, these antimicrobials are stored in specialized vesicles known as granules and are deployed in a rapid and spatiotemporally regulated manner [4]. Four different types of neutrophil granules are described to date: primary or azurophilic granules that among other factors contain myeloperoxidase (MPO), neutrophil elastase (NE), cathepsin G, lysozyme and defensins. The secondary or specific granules contain mainly antimicrobials such as lactoferrin and lysozyme, while the tertiary or gelatinase granules contain few antimicrobials but store metalloproteases such as gelatinase. Finally, secretory granules carry cytokines and important phagocytic receptors that are exposed to the neutrophil surface upon priming and fusion of these granules with the plasma membrane [5, 6]. During phagocytosis, neutrophils engulf and take up microbes into specialized compartments known as phagosomes in a receptor-mediated, clathrin-independent process. Fusion of neutrophil granules with the phagosome results in the formation of a phagolysosome, allowing for the assembly of the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase complex Nox2. The NADPH oxidase is a transmembrane multiprotein complex that transfers electrons to molecular oxygen, to generate superoxide anions into the lumen of the phagolysosome [7]. This highly oxidative environment in combination with the exposure to antimicrobial factors leads to the inactivation and killing of ingested microbes [8]. Degranulation involves the fusion of granules with the plasma membrane and the release of cytokines and antimicrobial contents into the extracellular space. These molecules help coordinate the immune response and control pathogens extracellularly [9–11]. Azurophilic granules lack the soluble N-ethylmaleimide-sensitive factor attachment protein receptors (SNAREs) that would direct them to fuse with the plasma membrane [12]. The contents of these granules are either deployed inside the phagosome or are released extracellularly via the third antimicrobial strategy in the neutrophil repertoire: the formation of NETs.
NETs are web-like structures that are composed of decondensed chromatin in complex with over 30 different neutrophil proteins that can capture, neutralize, and kill a variety of microbes. These large extracellular structures provide a physical barrier to prevent microbial dissemination and increase the local concentration of antimicrobial effectors [13–15]. Aside from infection, NETs have been recently found to regulate B cell function in the spleen [16] and to play a role in various sterile diseases, such as autoinflammation, autoimmune disease. An increasing number of bacteria, fungi, viruses, and protozoan parasites have been shown to induce NETs. Deficiencies leading to impaired NET formation result in high susceptibility to opportunistic infections in humans and mouse models and imply a significant contribution of NET formation in antimicrobial defense. In addition to NETosis, these deficiencies disrupt other neutrophil functions such as phagocytic killing, making it difficult to define the importance of NETs in the larger context of infection. This raises the necessity for dissecting the molecular mechanisms that regulate NETosis.
NETosis appears to be tightly regulated and dysregulation has been implicated in severe autoimmune and autoinflammatory disease. Below, we discuss the molecular mechanisms that lead to release of NETs, taking into consideration the differences between different physiological stimuli in infection and highlighting the importance of tight regulation of NET formation in autoimmunity and sterile inflammation.
NET formation and clearance
To date, two major NET release mechanisms have been described. In the first mechanism, neutrophils release NETs via a slow lytic cell death mechanism. This appears to be a major route for NET release. In addition, Pilsczek et al. have described that a small number of neutrophils rapidly expulse their nuclear content via vesicular secretion, yielding NETs and live intact cytoplasts that continue to crawl and digest microbes [17].
NETosis via cell death
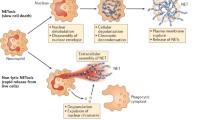
The first sign of NETosis via the cell death mechanism is a change in the morphology of the nucleus, which loses its characteristic lobulated architecture. Subsequently, nuclear membranes disassemble and chromatin decondenses into the cytoplasm while the plasma membrane remains intact. Finally, the plasma membrane bursts and the NETs are released [13] (Fig. 1b). This process is irreversible and is dependent on reactive oxygen species (ROS) such as superoxide that is generated by the NADPH oxidase Nox2. Neutrophils from chronic granulomatous disease (CGD) patients who bear mutations that disrupt the activity of the Nox2 enzyme, fail to form NETs [18, 19]. In addition, several enzymes that regulate Nox2 activity such as protein kinase C (PKC) isoforms and MAPK kinases have been implicated in NET formation [20, 21]. However, the receptors that trigger NET release and the molecular mechanisms by which ROS drives this process remain poorly understood. Superoxide spontaneously dismutates to hydrogen peroxide. MPO consumes H2O2 to generate hypochlorite and other halide ions [22]. MPO is also required for death-mediated NETosis but the mechanism of action has not yet been fully uncovered. The current evidence suggests that the downstream oxidant products of MPO are not involved in NETosis, as they fail to induce NET release and pharmacological inhibitors of the enzyme only slow down NETosis [23]. Moreover, NET formation is abrogated only in patients who are completely deficient in MPO. Experiments with neutrophils from patients with partial MPO deficiency show that even 5 % MPO activity is sufficient for NETosis [23]. MPO appears to drive NETosis independently from its enzymatic activity in a mechanism that involves synergy with NE. In response to ROS, the neutrophil-specific protease escapes from azurophilic granules into the cytoplasm and translocates to the nucleus where it partially degrades histones, the proteins that package DNA. This process drives the initial relaxation and decondensation of chromatin [24] (Fig. 2). Pharmacological inhibition of NE activity blocks NET formation and mice lacking NE fail to form NETs in a pulmonary model of Klebsiella pneumoniae infection. Subsequently, MPO associates with chromatin and promotes chromatin relaxation independent of the enzymatic activity. MPO and NE synergize to drive massive chromatin decondensation [24]. This two-step mechanism ensures that the antimicrobial components of NETs are not degraded by NE during the early phase of decondensation since binding to DNA downregulates the proteolytic activity. Furthermore, a strong ROS burst, and the expression of MPO and NE are hallmarks of neutrophils and help explain why extracellular trap (ET) release is confined to neutrophils and closely related granulocytes cells such as eosinophils and mast cells. The paradigm of ROS- and protease-dependent chromatin decondensation is employed for ET formation in macrophages, albeit using different factors.

Induction and release of NETs. a List of stimuli known to induce NETosis. b Two mechanisms of NET release have been discovered: an early rapid mechanism that takes 5–60 min after stimulation with S. aureus or LPS in the presence of platelets, undertaken by a small subset of neutrophils, yielding NETs and live cytoplasts that continue to phagocytose (left). First, the nuclear membranes separate and decondensed chromatin is seen in the intermembrane space. Chromatin-containing vesicles bud from the nucleus and accumulate below the plasma membrane. Finally, NETs are formed through the degranulation of vesicle content into the extracellular space and their assembly with decondensed chromatin. The majority of neutrophils undertake a cell death-mediated NETosis program that lasts from 2 to 4 h (right). First, the nucleus loses its characteristic lobulated shape. Subsequently, the nuclear membranes disintegrate and chromatin decondenses into the cytoplasm of the intact cell where it mixes with the granular contents. Finally, the plasma membrane ruptures and the NETs are released

Molecular mechanisms of NET release. To date, few receptors have been implicated in NET formation and little is known about the NET inducing ligands at a molecular level. Receptor engagement involves activation of phospholipase C (PLC), MAPK kinase (MAPKK) pathways and the activation of C (PKC). Assembly of the NADPH oxidase complex Nox2 on the membrane of the phagolysosome leads to production of reactive oxygen species (ROS) and acidification of the phagolysosome. Via an unknown mechanism, ROS drives the translocation of neutrophil elastase (NE) from the azurophilic (AZ) granules to the nucleus where it proteolytically processes histones and initiates the relaxation of chromatin. Subsequently, MPO associates with chromatin and synergizes with NE to promote chromatin decondensation. Together with other granule and cytoplasmic proteins, decondensed chromatin will eventually be released as NETs
Alternative rapid release of NETs from live cells during sepsis
NET release via cell death is a slow process and may leave an open time window for microbes to establish an infection. Interestingly, an alternative rapid mechanism for NET release has recently been described [17]. Neutrophils stimulated in vitro with Staphylococcus aureus, exhibit a unique form of rapid NET release that does not involve cell death. Reportedly, neutrophils release vesicles containing decondensed chromatin and granule antimicrobial proteins in the extracellular space where they assemble into NETs. This process occurs rapidly (5–60 min) in comparison to the cell death mechanism (120–240 min) (Fig. 1b). Release from live PMNs can also be triggered by Streptococcus pyogenes. Both NET formation strategies are dependent on TLR2 and complement factor 3 (C3). NET formation is deficient in mice lacking either of these molecules. NET release is restored by addition of wild-type mouse serum, highlighting the importance of opsonization for bacteria-triggered NETosis. However, TLR2 ligands and C3a are not sufficient to induce NET release in isolated neutrophils, suggesting that additional mediators or more complex mechanisms of activation are involved [25].
Rapid NET release was also observed by intravital microscopy in mice that were treated with MIP-2 (CXCL2) and intradermal infection of S. aureus or S. pyogenes. In vivo, the majority of neutrophils releasing NETs were undertaking a cell death-mediated pathway. These cells were highly motile and were phagocytosing bacteria. In addition, a minority of anuclear neutrophils had already released their nuclear material and were crawling slowly, still able to digest microbes [25]. Since neutrophils are terminally differentiated cells with low transcriptional activity, loss of the nucleus does not incapacitate these cells. On the contrary, this alternative mechanism of NET release provides a multitasking means for rapid extracellular antimicrobial action while maintaining the capacity for phagocytosis.
NET formation in CGD immunodeficiency
ROS play a central role in cell death-mediated NET formation since neutrophils from CGD patients who carry mutations that inactivate the NADPH oxidase [26], do not form NETs [18]. The high susceptibility of CGD patients to severe life-threatening infections with opportunistic pathogens suggests that NETs may play an important role in containing these infections in healthy individuals. However, the central role of ROS in other essential neutrophil antimicrobial functions such as phagocytic killing makes it difficult to draw conclusions on the contribution of NETs in immune defense, from the severity of CGD immune deficiency alone. Rather, the role of NETs in the context of CGD has been inferred indirectly. Importantly, the restoration of NADPH activity by gene therapy was sufficient to completely eradicate systemic aspergillosis in a CGD patient [19]. Experiments exploring the mechanism of neutrophil clearance against the Aspergillus strain recovered from the patient showed that the genetically modified neutrophils isolated from the patient killed the pathogen mostly via NETosis. The importance of NETs was further highlighted by the observation that the restoration of NET formation in these CGD neutrophils enabled them to clear both the Aspergillus conidia and hyphae that due to their large size are resistant to killing via phagocytosis [19, 27].
MPO deficiency and the role of NETs in fungal infections
In contrast to CGD, MPO deficiencies are associated with more complex clinical symptoms. The majority of the patients that are classified as MPO deficient bear mutations that result only in a partial deficiency [28]. As MPO is among the most abundant neutrophil proteins, low levels of the protein in these healthy individuals are sufficient for proper neutrophil function. Only a small subset of patients is completely deficient in mature MPO protein and activity. These completely deficient MPO individuals are susceptible to chronic candidiasis and at rare occasions they present severe systemic infection with opportunistic, often fungal, pathogens that may render them clinically indistinguishable from CGD patients. In other cases, completely MPO-deficient individuals may present no symptoms [29, 30]. Only neutrophils derived from completely MPO-deficient patients are unable to form NETs [23]. The susceptibility of these patients to fungal infections is a more direct indication that NETs play an important role in antifungal defense. The difference in the degree of immune deficiency between CGD and MPO deficient individuals can be explained by comparing their ability to employ different antimicrobial strategies to contain infections.
CGD neutrophils are unable to kill via phagocytosis and NET formation, while completely MPO deficient neutrophils have diminished, yet adequate, phagocytic killing capacity but fail to form NETs. Many fungi are able to switch between a small yeast form, which is needed for systemic dissemination and a filamentous hyphal form that is adapted for tissue invasion [31]. Hyphae are too large to be phagocytosed. Thus, the ability of NETs to eradicate the large hyphae extracellularly renders them critical in host defense against fungal infections. Accordingly, the susceptibility of completely MPO-deficient patients to recurrent mild fungal infections and rare severe systemic episodes appears to be linked to the inability of their neutrophils to form NETs and is an indication that NETs are critical for antifungal defense.
The difference in the severity of the immune deficiency between CGD and MPO-deficient individuals illustrates that in order to provide protection against a broad variety of pathogens, the innate immune system has developed several layers of defense that under most circumstances can compensate for each other. This concept may help explain why completely MPO-deficient patients are mostly healthy but occasionally experience rare sporadic episodes of systemic fungal infection. While most localized fungal infections present low risk, systemic fungal infections represent serious threats and are difficult to eradicate. In the case of MPO-deficient neutrophils, phagocytosis appears to complement for the lack of NET formation in most infections [23]. This compensating mechanism may be overburdened when neutrophils are challenged with pathogens that are resistant to phagocytic killing such as fungal hyphae. Since completely MPO-deficient patients can clear the yeast form by phagocytosis, they are able to prevent dissemination and are at low risk for systemic fungal infection. However, the inability to clear hyphae via NETosis may lead to persistence of hyphae that may overwhelm the immune system and eventually allow for yeast dissemination and systemic infection.
Histone deimination
Several reports have implicated peptidylarginine deiminase 4 (PAD4) in NET formation [20, 32–34]. PAD4 drives citrullination of histones, which is readily observed during NET formation and is a useful marker for NETosis. However, despite several reports on the subject [34–36], there are still open questions over the requirement for PAD4 in NET formation. Wang et al. tested PAD4 against human leukemia 60 cells, which are very poor NET producers, but while they detected histone citrullination in neutrophil-derived NETs, they did not test the ability of PAD4 inhibitors to block NET release in neutrophils. Moreover, in studies involving PAD4 deficient mouse neutrophils stimulated with IL-8 and Shigella flexneri, WT neutrophils did not exhibit sufficient NET formation, to be able to make quantitative comparisons with PAD4-deficient neutrophils [35]. The requirement for PAD4 has been more convincingly demonstrated in the context of influenza infection (see below) [36], suggesting that the role of PAD4 in NETosis may be dependent on the stimulus. Consistently, Neeli et al. reported that PAD4 is regulated by specific isoforms of PKC. While many stimuli, including calcium ionophores, activate PKCζ, an inducer of PAD4, others such as phorbol myristate acetate (PMA), a strong inducer of NETosis, activate PKCα, an inhibitor of PAD4 [20]. Therefore, differential engagement of PKC isoforms may potentially regulate differential neutrophil fates. Evidently, histone citrullination may coincide with but not necessarily be required for NETosis. Still, citrullinated histones may serve as a diagnostic marker for the presence of NETs in tissues (see “Rheumatoid arthritis” for further discussion).
ETs by eosinophils, mast cells, and macrophages
Several studies have shown that extracellular chromatin traps are not exclusively released by neutrophils. Eosinophils [37] and mast cells [38] have also been reported to release ETs.
Yousefi et al. reported that release of ETs from eosinophils is dependent on ROS production and drives the release of mitochondrial DNA. Mitochondrial genes such as cytochrome b oxidase subunit 1 or NADH dehydrogenase subunit 1 have been found in eosinophil ETs. Furthermore, ET release is independent of cell death. Release of eosinophil ETs upon stimulation by LPS, eotaxin, and complement factor 5a has been shown to require priming with IL-5 or IFN-γ [37]. Another feature that clearly distinguishes eosinophil ETs from NETs is the timing of release, which is reportedly in a frame scale of seconds post stimulation, rather than hours. The requirement for ROS raises the question of whether the cell can generate an oxidative burst in time to drive rapid mitochondrial expulsion.
Since mitochondrial DNA does not contain histones, which are major antimicrobial proteins in NETs, the microbicidal activity of ETs is attributed to eosinophil antimicrobial proteins such as eosinophil cationic protein. The contribution of ETs to immunity in vivo has not been clearly demonstrated since experiments that specifically block ETosis in animal models have not been reported, aside from a transgenic model of eosinophilia, which exhibits a moderate survival advantage under cecal ligation [37].
Mast cells are important granulocytes that drive the allergic response. They have been shown to release ETs via cell death in response to stimulation in a ROS-dependent manner [38]. Mast cell-derived ETs (MCETs) contain mast cell-specific antimicrobial proteases such as tryptase and antimicrobial peptides such as cathelicidin (LL-37). The release of MCETs promotes the release of pro-inflammatory cytokines in psoriasis, in addition to T cell-derived cytokines [39]. Both NETing neutrophils and mast cells have been reported to release interleukin (IL)-17 in response to stimulation with IL-23 and IL-1β, signifying that inhibition of ET formation may be an important anti-inflammatory strategy.
To date, one recent report has provided significant evidence to suggest that macrophages may release ETs in response to Mycobacterium tuberculosis [40]. As in NET formation, a protease inhibitor (AAPV) blocked DNA release in macrophages. Macrophages do not express NE and AAPV does not inhibit chymotrypsin elastases [41] but may inhibit macrophage metaloelastase or other similar proteases [42]. Further genetic analysis is needed, accompanied by examination of histone processing, in order to address whether the reported MET release involves a mechanism that is analogous to the neutrophil cell death process.
NETs in infection
NETs display broad effectiveness against a variety of different species of gram-positive and gram-negative bacteria, fungi, parasites and viruses. On the other hand, experimental data suggest that NET formation appears to be restricted to specific microbes. The features that define potent NET stimuli are not known. In the following chapter we summarize the most common physiological NET stimuli and the implications for the role of NETs during infection.
Do NETs kill?
NETs contain a wide range of antimicrobial factors that are known to kill microbes [43, 44]. To date, NETs have been shown to kill or inhibit the growth of several bacterial, fungal, and parasite species. Some skepticism surrounding NET killing is founded in plating-based techniques to detect microbial killing, where NETs may have clumped microbes without killing them and thus reduce the number of plated colonies. One short study demonstrated that when these samples were treated with nucleases after the killing assay had been performed, the number of colonies in NET-treated samples was restored to levels of untreated bacteria [45]. However, a growth inhibition assay of Candida albicans and Aspergillus fumigatus by NETs that is based on fungal enzymatic activity confirms the killing capacity of NETs. This approach is insensitive to microbial clumping artifacts. Given that different groups employ different techniques to assess microbial killing and the fact that these assays are challenging, it is with little doubt that conflicting reports arise. However, there is a decent body of evidence to support the killing capacity of NETs against a broad range of microbes.
Bacterial infections
A variety of gram-negative as well as gram-positive bacteria have been shown to induce NET formation. S. flexneri is trapped and killed by NETs, which contain NE that degrades virulence factors such as IcsA and IpaB [13]. Furthermore, despite studies reporting that nonpathogenic Escherichia coli triggers NET release [46, 47] to date the only compelling data about E. coli-induced NETosis stems from studies where pathogenic E. coli strains were used [48]. The enteropathogenic strain WS2572 induces formation of NETs in neutrophils isolated from the bone marrow of WT mice. Interestingly NET formation is abrogated in neutrophils from glutathione reductase (GSR)-deficient mice. GSR maintains the reducing environment in the cytoplasm and these data suggest that NET release may be sensitive to the oxidative levels, where too high ROS may be inhibitory. Furthermore, the mammary pathogenic E. coli strain P4 induces NET release from primary bovine neutrophils ex vivo [49]. These observations suggest that microbial virulence may be an important stimulus in NET induction, as it may induce additional inflammatory cytokines that may act as co-activators.
Interestingly, the gram-negative bacterium K. pneumoniae is not sufficient to induce NETosis in isolated neutrophils ex vivo, but is a good inducer of NETosis in a mouse lung infection model [24]. NE-deficient mice, lacking the capacity to form NETs, are more susceptible to infection with K. pneumoniae [50], but this may be due to inefficient phagocytic killing as well as the absence of NETs. The role of NETs in combating K. pneumoniae is further corroborated by studies that dissect the antimicrobial capacity of NETs. NETs from myeloid related protein 14 (MRP14) deficient neutrophils fail to control K. pneumoniae in vitro and MRP14 knockout mice are more susceptible to K. pneumoniae, which disseminate more readily in the absence of MRP14 and support the role of NETs as effectors against bacterial dissemination [51]. Similarly, the adenosine A2B receptor (A2BR) is an anti-inflammatory receptor that is thought to suppress NET formation. A2BR deficient neutrophils show enhanced NET formation and increased K. pneumoniae killing and clearance [52]. Consistent with an increase in NET release in vivo, A2BR deficient mice exhibit increased survival rates in K. pneumoniae infection.
Streptococci are gram-positive bacteria that include non-pathogenic commensal strains, and highly virulent pathogenic strains. Pathogenicity depends on expression of virulence factors that allow the bacteria to establish an infection and evade the immune system. Recent studies have revealed a complex interplay between some of these virulence factors and NETs.
Infection with Streptococcus pneumoniae leads to community-acquired pneumonia and invasive diseases like meningitis and bacteraemia, whereas S. pyogenes is the major causative agent in group A Streptococcus (GAS) infections [53–55]. Likewise, S. pneumoniae and S. pyogenes induce NET formation but have evolved virulence mechanisms to evade NETs. Strikingly, it appears that neutrophils have also evolved to selectively release NETs in response to these virulence factors. The S. pyogenes virulence factor M1, modulates NET formation and confers resistance to NET killing. M1 induces NETosis by associating with fibrinogen and forming a complex that stimulates neutrophils. M1 mutant S. pyogenes exhibit a decreased ability to induce NET formation. Interestingly, although M1 increases NETosis, it also confers bacterial resistance to NET-mediated killing by sequestering and neutralizing the antimicrobial NET component cathelicidin, known as LL-37 in humans and mCRAMP in mice. Overexpression of M1 in susceptible S. pyogenes strains renders them resistant to extracellular killing, whereas the deletion of M1 in invasive serotypes leads to susceptibility towards killing by NETosis [56]. Importantly, these observations suggest that bacteria may have evolved to evade neutrophils by directing them to an antimicrobial program that they are able to resist, at the expense of other neutrophil strategies they are more susceptible to. Other such examples of pathogen specific molecules that are able to trigger NETosis are becoming apparent. α-enolase expression on the surface of S. pneumoniae is sufficient to trigger NET formation by binding to the neutrophil myoblast antigen 24.1D5. However, genetic ablation of the enolase does not disrupt NETosis [57].
Furthermore, S. pneumoniae has evolved strategies to escape NETs. In a passive manner, the polysaccharide capsule reduces NET binding [58]. In addition, streptococci also employ active strategies to counter NETs. The invasive pneumococcus type TIGR4 expresses the DNase endA, which enables escape from NETs, leading to increased virulence in vivo. Mice that were infected with endA-deficient TIGR4 bacteria showed lower mortality compared with the WT controls [59]. Similarly, the nuclease Sda1 in S. pyogenes degrades NETs and promotes high virulence in vivo [60, 61]. Mice that are infected with S. pyogenes that lack Sda1 cannot degrade NETs and exhibit low bacterial titers and reduced lesion size in vivo [61]. Furthermore GAS M1T1 express the protease SpeB that degrades Sda1 and eliminates the capacity to escape NETs. In mice infected with M1T1, bacteria that disseminated to internal organs accumulated mutations that inactivated the protease and allowed for an active Sda1 [62]. These results suggest that NETs act as physical barriers to prevent microbial dissemination and exert a selective pressure on the bacteria to degrade NETs in order to break away from the site of infection and spread systemically. However, recent work has also demonstrated that Sda1 degrades bacterial DNA to prevent alerting the immune system via the pattern recognition receptor Toll-like receptor 9 (TLR9), indicating that the virulence attributed to Sda1 cannot be attributed solely to NET evasion [63].
Likewise, S. aureus is a good inducer of NETosis [13, 18] and has also evolved a plethora of mechanisms to circumvent NET killing. One strategy involves the expression of pore-forming virulence factors that neutralize neutrophils and prevent NET formation by triggering necrosis. Moreover, catalase expression blocks the build-up of H2O2 and protects S. aureus from oxidation intracellularly and by blocking NETosis extracellularly [18]. Thus, neutrophils may have evolved their secondary rapid NET release mechanism that is independent of ROS in order to engage S. aureus and other catalase positive strains. NETs specifically degrade S. aureus virulence factors and thereby help to contain tissue damage, but methicillin-resistant S. aureus (MRSA) express extracellular nucleases for biofilm dispersal and degradation of NETs [64]. Mice infected with MRSA show increased mortality compared with controls infected with a nuclease deficient strain, which is more susceptible to extracellular killing by neutrophils. These findings suggest the potential of nuclease inhibitors for treatment of MRSA infections.
The facultative intracellular bacterium M. tuberculosis is capable to successfully evade the host immune system and to establish latent infection. Although macrophages play a key role during MTB infection, recent evidence has underlined the importance of neutrophils in containing the infection [65]. Interestingly, a recent study shows that the virulent M. tuberculosis H37Rv and the low virulent strain Mycobacterium canettii are sufficient to trigger ROS production and NET formation in vitro [66]. However, while NETs trapped M. tuberculosis, the bacteria were not killed, suggesting that NETs may be important for physically restricting M. tuberculosis. This hypothesis requires further testing in animal models and human studies.
Fungal infections
As mentioned above, neutrophils play a crucial role in containing fungal infections and NETs appear to be an important part of the neutrophil antifungal arsenal. However, to date, only a limited number of fungal species have been tested for their ability to induce NETs and to be efficiently killed by them. C. albicans is the most common fungal commensal microorganism with asymptomatic colonization of the skin and mucosal surfaces in 30–50 % of the human population. Moreover, Candida species are also amongst the most prevalent opportunistic human pathogens in immunocompromised individuals with a mortality of up to 40 % [67, 68]. C. albicans virulence is strongly depended on its dimorphic ability: The budding yeast stage is crucial for systemic dissemination, whereas hyphae are needed for persistence and tissue invasion [31]. Since hyphae are too large to be phagocytosed, extracellular killing by release of NETs is an ideal strategy to contain the hyphal form and a number of studies have demonstrated that NETs are sufficient to kill C. albicans yeast and hyphae [19, 69].
Efforts to characterize the antifungal components of NETs led to the identification of calprotectin as major antifungal agent in the response against C. albicans [43]. Calprotectin is a cytoplasmic protein that is released via NETosis both as a soluble molecule and bound to NETs, highlighting the importance of NETosis as a major route for secretion of soluble factors alongside NET-bound antimicrobials. While calprotectin is found associated with C. albicans in NETs, direct contact of the protein with the microbe is not crucial since calprotectin chelates Mn2+ and Zn2+ which are required for C. albicans growth. The importance of calprotectin in antifungal defense is highlighted by the high susceptibility of calprotectin-deficient mice towards subcutaneous and pulmonary candidiasis [43]. Calprotectin has also been shown to be an important NET component in the defense against Aspergillus nidulans as calprotectin deficient mice are more susceptible to aspergillosis.
Interestingly, the protective effect of calprotectin is dependent on the presence of both subunits of the heteroduplex between the S1008A and S1009A subunits that provides anti-A. nidulans protection in a dose-dependent manner. At low doses calprotectin acts reversibly as a fungistatic, whereas high doses, it leads to starvation and subsequent killing of the fungus [27]. CGD patients suffer from frequent Aspergillus infections due to abrogated ROS production and insufficient extracellular killing via NETosis. Patients that received gene therapy to restore NADPH oxidase function and therefore NET release have been shown to regain capacity to clear A. nidulans infections in a NET- and calprotectin-dependent manner [19, 27].
Little is known about the upstream recognition events that regulate NETosis in response to fungi. Induction of NETosis by A. fumigatus is independent of viable fungus; inactivated conidia and hyphae are sufficient to trigger NET release [70]. Interestingly, the ability of fungal morphotypes to induce NETs appears to be influenced by different fungal surface molecules. Bruns et al. proposed that conidia induce slightly fewer NETs than hyphae because they express hydrophobin RodA, a surface protein that renders them immunologically inert. Thus, conidia may be better protected against neutrophils than hyphae, which do not bear RodA on the surface. RodA-deficient A. fumigatus conidia induce NETs better than wild-type hyphae suggesting that RodA exhibits an inhibitory effect on NET formation by shielding yet unidentified NET inducing elements in conidia. Consistently, A. fumigatus conidia were shown to be killed primarily by phagocytosis and not NETosis [71]. In contrast, another report has indicated that A. nidulans conidia can be killed via NETs, albeit less efficiently than hyphae [19]. One note of caution with regards to the RodA studies is that NET formation was not assessed by microscopy but by a total fluorescence assay, which may not be able to distinguish between NETs and necrotic neutrophils with condensed nuclei. Furthermore, the effect of RodA may be indirect since RodA deficient conidia may be more fragile and more likely to induce NETosis, although Bruns et al. show that these mutants are more resistant to phagocytic killing.
While further studies are needed to fully understand the contribution of NETs in the immune defense against fungal infection, there is strong evidence that NETs constitute an important antifungal innate immune strategy.
Viral infections
Neutrophils are not regarded as important effector cells against viruses and few studies have examined the role of NET formation in response to viral infection. Recently, human immunodeficiency virus (HIV)-1 was shown to induce NETosis via the cell death pathway, although the requirement for ROS, MPO and NE was not addressed [72]. NETs are able to capture and neutralize the negatively charged HIV virions, significantly decreasing HIV infectivity. Thus, neutrophils and NETs may play important roles in combating HIV. Interestingly, HIV is capable of manipulating neutrophil activation in order to suppress NET formation. Saitoh et al. demonstrated that HIV engages DC-SIGN (CD209) on dendritic cells (DCs) with its envelope glycoprotein gp120. Engagement of DC-SIGN leads to production of IL-10 by DCs, which suppresses NET formation [72]. Therefore, HIV not only takes advantage of DC-SIGN on DCs for efficient infection of CD4+ T-cells via the DC-T cell synapse, but also to evade NET killing. This study impressively demonstrates a surprising feature of NETs as antiviral effectors and the capacity of HIV to coevolve and adapt to the innate immune response.
Other viruses modulate host responses in order to suppress NET formation. Feline leukemia virus (FeLV) inhibits neutrophil activation by inhibiting the activation of PKC to reduce ROS production. Progressive chronic FeLV infection reduces neutrophil responsiveness to secondary stimulation with Leishmania promastigotes due to exhaustive neutrophil activation by FeLV, demonstrating that neutrophil activation and NET induction can be modulated by viral infection [73].
Finally, the role of NETs in influenza infection has also been explored. NETs are induced secondarily by influenza-activated lung epithelium producing superoxide and H2O2 [74]. Moreover, while influenza has been shown to induce NETs in the mouse lung, NET deficiency in PAD4 knockout mice was not associated with an increase in viral titers and susceptibility to infection [36].
Therefore, while few studies have addressed the role of NETs in viral infection, given that many viruses elicit strong neutrophil recruitment, there is enough evidence to suggest that NETs may be implicated in antiviral defense.
Parasitic infections
To date, several studies have explored the potential role of NETs in the immune response against protozoan parasites. Circulating NET structures have been detected in the blood of Plasmodium falciparum-infected children with uncomplicated malaria. NETs entangled parasitized erythrocytes and throphozoites, while titers of antinuclear antibodies (ANAs) were significantly above levels associated with auto-immunity [75]. Thus, the presence of NETs and the formation of ANAs may contribute to disease in children and development of autoimmune phenotypes, but may also induce immune protection in adults over time. Furthermore, it is speculated that the presence of ANAs may lead to hyporesponsiveness towards CpG oligonucleotide-based malaria vaccines [75].
The role of NETs has also been explored in parasites causing leishmaniasis. Leishmania parasites are the inducing agents in a variety of leishmaniasis infections where, depending on the species, the phenotype can range from cutaneous and subcutaneous lesions to potentially lethal visceral leishmaniasis (kala-azar). The two major parasite stages, amastigotes that reside primarily in macrophages and promastigotes found primarily in the sandfly vector, induce formation of NETs ex vivo. This effect is observed in promastigotes of Leishmania amozonensis, Leishmania major, Leishmania chagasi, Leishmania major, and Leishmania donovani as well as in amastigotes of L. amozonensis and seems therefore to be an important defense-mechanism against Leishmania spp. Induction of NETs by Leishmania spp has been reported to be independent of NADPH oxidase activity and ROS production. Entrapment in NETs leads to decreased viability of the parasites, although authors of different studies conclude that the main function of NETs in Leishmania infection is the immobilization of the parasite and containment of the infection [76, 77].
With regards to the molecular recognition of these parasites, different groups report a conflicting role for the promastigote surface glycolipid lipophosphoglycan (LPG) in NETosis. Guimaraes-Costa et al. describe NET induction by LPG and provide evidence that purified LPG triggers NET-formation in a dose-dependent manner [77]. In contrast, Gabriel et al. report that NET formation is independent of LPG and the surface metalloprotease leishmanolysin (GP63), showing that LPG-deficient L. donovani promastigotes induce NET sufficiently. Furthermore, LPG protects the parasite against killing by NETs since an LPG-mutant strain is more susceptible NET-mediated killing [76]. Anti-histone antibodies abrogate the killing capacity of NETs towards Leishmania and addition of purified histone H2B kills the parasite efficiently, highlighting the important role of histones as NET antimicrobial effector proteins [77, 78]. Therefore, LPG may shelter the parasite from NET antimicrobial histones.
NETs are also implicated in Toxoplasma gondii infection. T. gondii is an obligate intracellular parasite that infects many warm-blooded animals. It leads to asymptomatic infections in immunocompetent hosts, but causes severe disease in immunocompromised individuals and is a significant risk factor in pregnant women. T. gondii elicits NET formation in a strain-independent manner. Invasion of the parasite is not needed to trigger NET release, since treatment with cytochalasin D inhibits entry of the parasite into the host cell, but has no effect on NET formation [79]. NETs kill approximately 25 % of the entangled parasites, which indicates that the primary function of NETs may be to physically contain the infection. In vivo, pulmonary infection with Toxoplasma in mice leads to recruitment of neutrophils and high levels of extracellular dsDNA in the broncho-alveolar fluid, which are abrogated by neutrophil depletion. In addition to mouse, human blood-derived neutrophils have also been shown to produce NETs in response to T. gondii. The Raf-MEK-ERK pathway is implicated in the induction of NETs by T. gondii, which elicit strong phosphorylation of ERK1/2. However, inhibition of ERK1/2 leads to a decrease in NET formation, but not complete abrogation. Therefore ERK1/2 activation augments NET formation, but is not absolutely required [79].
Finally, Eimeria bovis that induces enteritis with intestinal lesions in calves and young cattle is a strong inducer of NETosis ex vivo. In fact, NET formation, 30 min postinfection with E. bovis is more efficient as compared to induction with PMA (3–4 h), which is considered to be a strong pharmacological NET inducer [80]. Interestingly, the induction of NETs is dependent on viable parasite particles. Inactivated and homogenized E. bovis still induce NETs, but at levels below PMA induction. The induction is dependent on the NADPH oxidase since inhibition with diphenylene iodoniumn reduced NET formation to background levels.
These studies suggest that NETs play an important role in the defense against protozoan parasites. However, the available data indicate that NETs may be important primarily to physically restrict the entrapped pathogens and secondarily to kill them.
NET formation and clearance in disease
NETs are a potent neutrophil defense mechanism and complement the immune system’s capacity to fight infectious diseases. However, defects and dysregulation of this sensible process can lead to severe pathologies and autoimmune conditions. Excessive NET formation and impaired clearance of NETs have been shown to contribute to inflammation and organ damage in autoimmune disorders. Furthermore, NETs display a range of self-antigens and may promote production of autoantibodies.
Autoimmunity
Systemic lupus erythematosus and ANCA vasculitis
Systemic lupus erythematosus (SLE) is a diverse autoimmune disease characterized by skin rashes, and internal organ pathology. SLE patients exhibit elevated levels of anti-neutrophil cytoplasmic antibodies (ANCAs), antibodies against histones, DNA (ANAs) and ribonucleoproteins (RNP) that target host tissues [81]. The most severe symptoms of SLE include renal pathology. Long-term immunosuppressive therapy increases the risk for infection and sepsis in SLE patients and more targeted treatments are needed [82]. The recent implication of NETosis in SLE pathology helps explain the high prevalence of neutrophil specific autoantigens in these diseases [83]. During infection, NETs are a major source of extracellular chromatin, neutrophil proteins and microbial co-stimulatory adjuvants, and are therefore a primary suspect as an antigenic source in SLE (Fig. 3).

NETs are implicated in the onset of inflammatory and autoimmune diseases. (clockwise) Gouty arthritis: deposition of MSU needles in the SF leads to release of IL-1β by monocytes and induction of IL-8, with subsequent neutrophil recruitment. MSU needles induce release of NETs that stimulate a feedback loop of IL-1β production by monocytes. Deep vein thrombosis: fibrin and von Willebrand factor (vWF) promote blood clotting and deposition of thrombi in the blood vessels. Activated endothelial cells release IL-8 and ROS, thereby recruiting neutrophils and triggering NET formation. NETs act as scaffolds for thrombi in the circulation by interacting with coagulation factors, platelets, and red blood cells (RBC). Systemic lupus erythematosus (SLE): a subset of low-density granulocytes has been shown to spontaneously release NETs in SLE patients. The NET component LL-37 enhances the ability of naked DNA to activate plasmacytoid dendritic cells (pDC) via toll-like receptor 9 (TLR9). pDCs subsequently secrete IFN-α that primes neutrophils to recognize autoimmune complexes of antinuclear antibodies (ANA) and DNA and release NETs. This feedback loop further activates pDCs and may contribute to SLE progression. Cystic fibrosis (CF): NETs form into the sputum in an NE-dependent manner. i Inhibiting NE in intact neutrophils would prevent NET release, but NE is also important in decondensing chromatin extracellularly, which enhances sputum solubilization by therapeutic DNases. ii NE inhibition extracellular would prevent tissue damage iii but would also interfere with sputum solubilization
Furthermore, the antimicrobial peptide LL-37, an important NET component, greatly enhances the ability of DNA to activate plasmacytoid DCs via TLR9 [84, 85]. Subsequently, a low-density granulocyte subset in the blood of SLE patients was found to undergo spontaneous NET formation ex vivo [86, 87]. Autoimmune complexes targeting NET components were detected in sera from SLE patients and induce NET formation in neutrophils from healthy control donors [85]. In turn, NETs induce pDCs to secrete IFN-α, which promotes autoreactivity of antigen-presenting and antibody-producing cells and SLE progression [85, 87]. IFN-α has been shown to prime neutrophils to respond to autoimmune complexes by releasing NETs, which in turn activate pDCs further [84, 85]. Low-density granulocytes in SLE patients may be neutrophils primed by IFN. The origins of such a priming event and why it afflicts only a subset of granulocytes remain unclear. High density SLE neutrophils do not form NETs spontaneously and require IFN priming to respond to challenge with autoimmune complexes [85].
This positive feedback loop may play an important role in autoimmune disease development in SLE and ANCA vasculitis. Several clinical trials involving anti-IFN-I strategies are underway [88]. Additionally, targeting NETs directly may provide a beneficial therapeutic strategy since studies have demonstrated that SLE patients who appear defective in NET clearance mechanisms exhibit higher autoantibody titers and are more likely to develop lupus nephritis. Sera from these patients do not degrade NETs either due to low DNase I activity, expression of DNase I inhibitors or presence of autoantibodies protecting NETs against degradation by DNase I [83, 89]. Moreover, in DNase I-deficient mice, production of ANCAs and immune complex formation is detected in the kidneys and associates with severe glomerulonephritis [90].
Interestingly, Campbell et al. have recently suggested that the NADPH oxidase acts to inhibit disease pathogenesis. NADPH oxidase deficient mice, incapable to form NETs that were crossed to a strain of lupus-prone mice, developed SLE symptoms, suggesting that NET formation is not implicated in SLE pathogenesis [91]. However, the mouse MRL/ltr model is in a not well-defined genetic background, making it difficult to compare to NADPH oxidase deficient mice. In addition, these mice do not generate neutrophil-specific ANCA antibodies and thus pathogenesis does not seem to implicate NETs. Mutations in the human LTR Fas receptor gene result in the absence of DCs in humans. These patients are very rare and show no symptoms of autoimmune disease. Furthermore, there are no reports of CGD patients exhibiting autoimmune disease, but a case report of one CGD patient and a few mothers of CGD patients suffering from autoinflammatory disease without any autoantibody titers.
Rheumatoid arthritis
Rheumatoid arthritis (RA) is a systemic autoimmune disease associated with chronic inflammation, primarily in the synovial joints. NETs are abundant in synovial fluids (SF), rheumatoid nodules, and skin of RA patients. High levels of citrullinated proteins [92] and elevated titers of anticitrullinated peptide antibodies (ACPAs) in the SF are RA diagnostic markers. ACPA containing serum and SF induce NETosis in neutrophils from RA patients and control neutrophils ex vivo [93]. Furthermore, citrullinated vimentin externalized through NET formation and anticitrullinated vimentin autoantibodies from RA patients induce NETosis. In this vicious circle, NETs present autoantigens in the context of immunostimulatory molecules thereby enhancing further NET release.
RA patients show elevated levels of proinflammatory cytokines such as IL-17A and TNF-α [94]. Whereas IL-17A or TNF-α alone are not sufficient to induce NETosis in control neutrophils, stimulation with LPS, IL-17A, or TNF-α was sufficient to trigger NET release in RA neutrophils without further priming in a ROS-dependent manner. Furthermore, the presence of anti-IL-17A or anti-TNF-α neutralizing antibodies decreased NET formation in RA neutrophils incubated with RA serum and the presence of both antibodies together abrogated NET formation completely [93]. It is unclear how IL-17 activates neutrophils since reports suggest that these cells do not express the IL-17 receptor C which forms a multimeric complex with IL-17 receptor A and is required for IL-17 receptor signal transduction [95, 96].
Interestingly, Khandpur et al. describe a different NET protein composition depending on the stimulus [93]. NETs induced with RA autoantibodies contain matrix-metalloprotease-8 (MMP-8), histone H3 and vasodilator-stimulated phosphoprotein, which are not found in TNF-α-induced NETs. NETs triggered by PMA contain histone H3 but not MMP-8 [43]. MMP8 plays critical roles in tissue destruction in arthritis and cancer metastasis [97], suggesting that NETs may be an important source of this metalloprotease in these diseases. Other proteins have been shown to be present in NETs independent of the specific stimulus [93]. The concept that different stimuli may lead to NETs with different components and different attributes is an interesting one, and should be explored further.
RA patients show elevated PAD4 expression and high levels of anti-PAD4 autoantibodies [92, 98, 99]. Khandpur et al. report that NET formation is decreased in the presence of anti-PAD4 neutralizing antibodies in human neutrophils ex vivo [93]. However, a recent study showed that PAD4 is dispensable for development of RA in a mouse model of serum transfer disease [100]. Therefore, while hypercitrullinated NET histones may represent an important source of citrullinated proteins in the joint of RA patients, the role PAD4 plays in the regulation of NET formation and disease progression remains unclear. Accordingly, although the presence of citrullinated proteins may be a good diagnostic marker for monitoring NET release in RA joints, PAD4 may not prove to be a good target for therapy.
Gouty arthritis
Gouty arthritis is the most common form of arthritis and is caused by precipitation of uric acid in sodium containing fluids as monosodium urate (MSU) needles. Deposition of MSU needles in the SF leads to activation of the NALP3 inflammasome and release of IL-1β by monocytes. IL-1β induces the neutrophil chemoattractant IL-8, which recruits neutrophils (Fig. 3). Importantly, MSU needles trigger NET release in gouty arthritis [101]. NETs are spared from opsonization with C-reactive protein and complement. Thus, gouty-associated NETs are not targeted for clearance through scavenger cells and may persist long enough to contribute to chronic inflammation [99].
NETs in inflammation
Cystic fibrosis
Cystic fibrosis (CF) is a debilitating hereditary disease, which is caused by a mutation in the CF transmembrane conductance regulator ion channel which leads to high sputum viscosity and sputum that is difficult to clear from the airways. The lack of sputum clearance promotes bacterial colonization leading to chronic airway inflammation and low life expectancy of the affected individuals [102]. Sputum viscosity is caused by high levels of decondensed extracellular DNA since treatment with recombinant DNase promotes sputum solubilization and is a palliative treatment for CF [103, 104]. Neutrophil antimicrobial proteins such as NE and MPO are also abundant in CF sputum [105]. The above observations hinted that NETs may be an important source of extracellular DNA in CF sputum. Indeed several studies have now confirmed the abundance of NETs in CF sputum microscopically and biochemically [106, 107]. Notably, a study claiming that NETosis in CF is triggered by IL-8 in a ROS-independent manner has since been retracted [107]. In our hands IL-8 alone is not sufficient to induce NETs. (Metzler and Zychlinsky, unpublished data).
Neutrophil infiltration in the lung and release of NE is thought to have detrimental effects on lung epithelium. NE directly injures epithelial cells, alters mucus secretion and upregulates expression of pro-inflammatory cytokines [108]. Therefore, extracellular NE release via NETosis may be an important cause of lung tissue damage and disease progression in CF. NE inhibitors have been explored as therapeutics but recent studies suggest that the potential of this strategy should be viewed with caution. Recent findings show that although NE contributes to airway tissue damage, most of it is bound to chromatin [106]. DNA binding has been shown to dramatically downregulate NE proteolytic activity. Moreover, as it is anchored to NETs the residual NE activity is more likely to be directed against histones rather than host tissues as NE continues to degrade histones after NET release. Similarly to its role in NET formation, NE-mediated histone degradation promotes chromatin relaxation and decondensation. Ex vivo experiments with CF sputum suggest that the ability of NE to decondense extracellular chromatin is important for the effectiveness of DNase therapy [106]. Treatment of CF sputum from patients receiving DNase therapy with NE inhibitors blocks the sputum solubilization. On the other hand, NE activity is required for cell death-mediated NET release and blocking NE activity early may reduce the levels of extracellular DNA and sputum viscosity. Considering these findings, the nature and timing of administration of these molecules would have to be carefully examined. Soluble NE inhibitors would be ideal for blocking NETosis in CF patients who are not receiving DNase therapy. However, since these patients suffer from chronic infection, blocking NET formation may have adverse effects on their ability to contain the infection. Therefore, although promising, this field requires further study.
Deep vein thrombosis
Deep vein thrombosis (DVT) is caused by disturbances and stagnation in venous blood flow. A wide range of conditions is known to increase the risk for DVT such as pregnancy, obesity, trauma and certain cancers. Importantly, NETs have been shown to form scaffolds in circulation that promote thrombus formation by interacting with the endothelium, platelets, coagulation factors and red blood cells. Consistently, depletion of neutrophils [109] or injection of exogenous DNase I have been shown to prevent thrombus formation in mouse models and humans [109, 110]. IL-8 and ROS released from endothelial cells can recruit and trigger neutrophils to form NETs, which in return activate and damage the endothelium by binding of histones to endothelial membranes [111] (Fig. 3). The release of Weibel–Palade bodies from the endothelium and deposition of fibrin and von Willebrand factor (vWF) promotes blood coagulation by formation of thrombus scaffolds. vWF and fibrin have a high affinity for histones and therefore readily bind to NETs [112, 113]. Furthermore, histones have been shown to inhibit anticoagulants in the plasma, thereby further promoting thrombus formation [114].
Additionally, the interaction of NETs with platelets plays an important role in NET-mediated thrombus formation. Platelets aggregate on the NETs due to proteoloytic activation of platelet receptors via NE and cathepsin G [115, 116]. Notably, a subset of SLE patients with specific antiplatelet autoantibodies is in high risk of DVT in what is known as antiphospholipid (or Hughes) syndrome [117]. Deficiency in NET clearance in lupus patients may lead to a prolonged persistence of associated immune complexes [83].
These studies suggest that targeting NET release and driving NET clearance may constitute important therapeutic strategies to treat DVT.
Preeclampsia
Preeclampsia is a severe condition during pregnancy that affects 1–2 % of pregnant women and is characterized by proteinuria, oedema and drastic elevation of blood pressure. Untreated preeclampsia can develop into eclampsia accompanied by seizures, which can be life threatening for the mother and the fetus [118]. Currently, the only treatment for preeclampsia is the premature delivery of the baby. Preeclampsia is associated with abundance of placental microdebris in the maternal blood circulation. Interestingly, micro-debris has been shown to activate neutrophils and stimulate the release of NETs in a dose-dependent manner [119–121]. However, although IL-8 is not sufficient to trigger NET release in isolated neutrophils (Zychlinsky, unpublished data) the elevated levels of this cytokine in preeclampsia recruit neutrophils and may be important for priming them to respond to micro-debris [119]. The interplay between microdebris and IL-8 in neutrophil activation and deciphering the role of NETs in preeclampsia remain to be investigated.
Periodontitis
Periodontitis is a chronic inflammation of the periodontium caused by bacteria in the gingival crevice. Neutrophil influx leads to formation of a purulent crevicular exudate. This exudate has been shown to contain NETs that are thought to prevent bacterial spread to the gingival surface [122]. NETs may have evolved as an alternative defense mechanism to the host cells’ inability to phagocytose bacterial biofilms in the gingival crevice. However, the concomitant presence of phagocytosing neutrophils indicates that both strategies may be complementary.
The role of NETs in cancer
The role of tumor-associated neutrophils (TANs) in cancer is still unclear. A recent study suggests that neutrophils can be programmed to promote (N2) or suppress tumor growth (N1) according to the tumor environment [123]. The transforming growth factor-β (TGF-β) plays a central role in determining this neutrophil phenotype. TGF-β appears to polarize a pro-tumor N2 neutrophil state that may upregulate tumor cell proliferation. In contrast, inhibiting TGF-β leads to antitumor N1 TANs that may target tumor cells and prevent tumor growth. It is unclear how TGF-β affects neutrophil function and in particular NET release, but TGF-β inhibition resulted in upregulation of neutrophil activation and ROS suggesting that NETosis would be favored.
Indeed, lung adenocarcinomas elicited in NE knockout mice grow significantly slower than in WT mice suggesting that NE promotes tumor cell proliferation [124]. This study provides evidence that NE is taken up by tumor cells and upregulates the PI3K pathway. However, the proliferation effects of NE on these cells in vitro appear modest and alternative mechanisms, such as changes in NET release, may also play a role. Interestingly, NETs have been recently visualized in pediatric Ewing sarcoma (ES) tumors [125]. NETs may kill tumor cells but they may also release NE that is mostly bound to NETs. By sequestering NE, NETs may prevent the uptake of NE by tumor cells and create a cytotoxic antitumor environment. Given tumor variability, the role of neutrophil and NETs in cancer will likely be complex and specific to different tumor types and stages.
NETs as modulators of the adaptive immune system
Interestingly, in addition to activating pDCs, NETs appear to play wider immunomodulatory roles as they were recently shown to regulate B cell function in humans. Upon colonization of the gut with microbiota, neutrophils infiltrate the spleen marginal zone (MZ) and form NETs, in a process that depends on IL-10 release by splenic sinusoidal endothelial cells [16]. NETs interact with and activate B cells via the release of B cell regulatory proteins: a proliferation-inducing ligand, B cell-activating factor BAFF and IL-21. NET-mediated B cell activation triggers class switching, somatic hypermutation and antibody production. The authors examined human neutropenic patients and found that these patients express low IgG and IgM levels, indicating a requirement of neutrophils in class switching. Close examination of the list of patients provided reveals that although they are classified as neutropenic, they are in their majority patients who are deficient in NE. In fact, most severe cyclic neutropenias are caused by mutations in NE that promote misfolding of the protein and premature neutrophil apoptosis due to the activation of the unfolded protein response [126]. SCN patients are treated with GM-CSF, which restores the number of mature circulating neutrophils. However, these neutrophils are still deficient in NE. The MZ of SCN patients was still infiltrated by neutrophils but NETosis was severely diminished. These data further confirm the requirement of NE for NET formation [24] and highlight the role of NETosis as a route for the release of immunomodulatory molecules that affect innate and adaptive responses.
Conclusions
In summary, NETs are effective against a variety of microbes but may be critical against pathogens that are resilient against phagocytosis. NETs appear to play a key role in physically trapping microbes to prevent microbial dissemination. However, dysregulation of NET formation and clearance appears to have detrimental effects. One common underlying theme in the work we have covered is that spatiotemporal regulation is key to ensure proper neutrophil function and health: NETs are beneficial in some places and have detrimental effects in others. Timing is also important since NET persistence causes adverse effects. Important mechanisms must be in place at the subcellular and organismal level to ensure that NETs are made and cleared at the right place and time. Understanding of the molecular mechanisms and the spatiotemporal dynamics that regulate NET release and clearance in these various contexts, may pave the way for the development of new therapeutics for a number of human diseases
References
Amulic B, Cazalet C, Hayes GL, Metzler KD, Zychlinsky A (2012) Neutrophil function: from mechanisms to disease. Annu Rev Immunol 30:459–489. doi:10.1146/annurev-immunol-020711-074942
Mantovani A, Cassatella MA, Costantini C, Jaillon S (2011) Neutrophils in the activation and regulation of innate and adaptive immunity. Nat Rev Immunol 11(8):519–531. doi:10.1038/nri3024
Borregaard N (2010) Neutrophils, from marrow to microbes. Immunity 33(5):657–670. doi:10.1016/j.immuni.2010.11.011
Sengelov H, Kjeldsen L, Borregaard N (1993) Control of exocytosis in early neutrophil activation. J Immunol 150(4):1535–1543
Faurschou M, Borregaard N (2003) Neutrophil granules and secretory vesicles in inflammation. Microbes Infect 5(14):1317–1327. doi:10.1016/j.micinf.2003.09.008
Borregaard N, Sorensen OE, Theilgaard-Monch K (2007) Neutrophil granules: a library of innate immunity proteins. Trends Immunol 28(8):340–345. doi:10.1016/j.it.2007.06.002
El-Benna J, Dang PM-C, Gougerot-Pocidalo M-A (2008) Priming of the neutrophil NADPH oxidase activation: role of p47phox phosphorylation and NOX2 mobilization to the plasma membrane. Semin Immunopathol 30(3):279–289. doi:10.1007/s00281-008-0118-3
Hampton MB, Kettle AJ, Winterbourn CC (1998) Inside the neutrophil phagosome: oxidants, myeloperoxidase, and bacterial killing. Blood 92(9):3007–3017
Soehnlein O, Zernecke A, Weber C (2009) Neutrophils launch monocyte extravasation by release of granule proteins. Thromb Haemost 102(2):198–205. doi:10.1160/TH08-11-0720
Scapini P, Lapinet-Vera JA, Gasperini S, Calzetti F, Bazzoni F, Cassatella MA (2000) The neutrophil as a cellular source of chemokines. Immunol Rev 177:195–203
Soehnlein O, Weber C, Lindbom L (2009) Neutrophil granule proteins tune monocytic cell function. Trends Immunol 30(11):538–546. doi:10.1016/j.it.2009.06.006
Lacy P (2005) The role of rho GTPases and SNAREs in mediator release from granulocytes. Pharmacol Ther 107(3):358–376. doi:10.1016/j.pharmthera.2005.03.008
Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A (2004) Neutrophil extracellular traps kill bacteria. Science 303(5663):1532–1535
Brinkmann V, Zychlinsky A (2007) Beneficial suicide: why neutrophils die to make NETs. Nat Rev Microbiol 5(8):577–582
Papayannopoulos V, Zychlinsky A (2009) NETs: a new strategy for using old weapons. Trends Immunol 30(11):513–521. doi:10.1016/j.it.2009.07.011
Puga I, Cols M, Barra CM, He B, Cassis L, Gentile M, Comerma L, Chorny A, Shan M, Xu W, Magri G, Knowles DM, Tam W, Chiu A, Bussel JB, Serrano S, Lorente JA, Bellosillo B, Lloreta J, Juanpere N, Alameda F, Baro T, de Heredia CD, Toran N, Catala A, Torrebadell M, Fortuny C, Cusi V, Carreras C, Diaz GA, Blander JM, Farber CM, Silvestri G, Cunningham-Rundles C, Calvillo M, Dufour C, Notarangelo LD, Lougaris V, Plebani A, Casanova JL, Ganal SC, Diefenbach A, Arostegui JI, Juan M, Yague J, Mahlaoui N, Donadieu J, Chen K, Cerutti A (2012) B cell-helper neutrophils stimulate the diversification and production of immunoglobulin in the marginal zone of the spleen. Nat Immunol 13(2):170–180. doi:10.1038/ni.2194
Pilsczek FH, Salina D, Poon KK, Fahey C, Yipp BG, Sibley CD, Robbins SM, Green FH, Surette MG, Sugai M, Bowden MG, Hussain M, Zhang K, Kubes P (2010) A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus. J Immunol 185(12):7413–7425. doi:10.4049/jimmunol.1000675
Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, Weinrauch Y, Brinkmann V, Zychlinsky A (2007) Novel cell death program leads to neutrophil extracellular traps. J Cell Biol 176(2):231–241
Bianchi M, Hakkim A, Brinkmann V, Siler U, Seger RA, Zychlinsky A, Reichenbach J (2009) Restoration of NET formation by gene therapy in CGD controls aspergillosis. Blood 114(13):2619–2622. doi:10.1182/blood-2009-05-221606
Neeli I, Radic M (2013) Opposition between PKC isoforms regulates histone deimination and neutrophil extracellular chromatin release. Front Immunol 4:38. doi:10.3389/fimmu.2013.00038
Keshari RS, Verma A, Barthwal MK, Dikshit M (2012) Reactive oxygen species-induced activation of ERK and p38 MAPK mediates PMA-induced NETs release from human neutrophils. J Cell Biochem. doi:10.1002/jcb.24391
Akong-Moore K, Chow OA, von Kockritz-Blickwede M, Nizet V (2012) Influences of chloride and hypochlorite on neutrophil extracellular trap formation. PLoS One 7(8):e42984. doi:10.1371/journal.pone.0042984
Metzler KD, Fuchs TA, Nauseef WM, Reumaux D, Roesler J, Schulze I, Wahn V, Papayannopoulos V, Zychlinsky A (2011) Myeloperoxidase is required for neutrophil extracellular trap formation: implications for innate immunity. Blood 117(3):953–959. doi:10.1182/blood-2010-06-290171
Papayannopoulos V, Metzler KD, Hakkim A, Zychlinsky A (2010) Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J Cell Biol 191(3):677–691. doi:10.1083/jcb.201006052
Yipp BG, Petri B, Salina D, Jenne CN, Scott BN, Zbytnuik LD, Pittman K, Asaduzzaman M, Wu K, Meijndert HC, Malawista SE, de Boisfleury CA, Zhang K, Conly J, Kubes P (2012) Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat Med 18(9):1386–1393. doi:10.1038/nm.2847
Seger RA (2008) Modern management of chronic granulomatous disease. Br J Haematol 140(3):255–266. doi:10.1111/j.1365-2141.2007.06880.x
Bianchi M, Niemiec MJ, Siler U, Urban CF, Reichenbach J (2011) Restoration of anti-Aspergillus defense by neutrophil extracellular traps in human chronic granulomatous disease after gene therapy is calprotectin-dependent. J Allergy Clin Immunol 127(5):1243–1252. doi:10.1016/j.jaci.2011.01.021
Kameoka Y, Persad AS, Suzuki K (2004) Genomic variations in myeloperoxidase gene in the Japanese population. Jpn J Infect Dis 57(5):S12–13
Kutter D (1998) Prevalence of myeloperoxidase deficiency: population studies using Bayer-Technicon automated hematology. J Mol Med-Jmm 76(10):669–675
Prokopowicz Z, Marcinkiewicz J, Katz DR, Chain BM (2012) Neutrophil myeloperoxidase: soldier and statesman. Arch Immunol Ther Exp 60(1):43–54. doi:10.1007/s00005-011-0156-8
Gow NA, van de Veerdonk FL, Brown AJ, Netea MG (2012) Candida albicans morphogenesis and host defence: discriminating invasion from colonization. Nat Rev Microbiol 10(2):112–122. doi:10.1038/nrmicro2711
Neeli I, Khan SN, Radic M (2008) Histone deimination as a response to inflammatory stimuli in neutrophils. J Immunol 180(3):1895–1902
Leshner M, Wang S, Lewis C, Zheng H, Chen XA, Santy L, Wang Y (2012) PAD4 mediated histone hypercitrullination induces heterochromatin decondensation and chromatin unfolding to form neutrophil extracellular trap-like structure. Front Immunol 3:307. doi:10.3389/fimmu.2012.00307
Wang YM, Li M, Stadler S, Correll S, Li PX, Wang DC, Hayama R, Leonelli L, Han H, Grigoryev SA, Allis CD, Coonrod SA (2009) Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J Cell Biol 184(2):205–213. doi:10.1083/jcb.200806072
Li P, Li M, Lindberg MR, Kennett MJ, Xiong N, Wang Y (2010) PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J Exp Med 207(9):1853–1862. doi:10.1084/jem.20100239
Hemmers S, Teijaro JR, Arandjelovic S, Mowen KA (2011) PAD4-mediated neutrophil extracellular trap formation is not required for immunity against influenza infection. PLoS One 6(7):e22043. doi:10.1371/journal.pone.0022043
Yousefi S, Gold JA, Andina N, Lee JJ, Kelly AM, Kozlowski E, Schmid I, Straumann A, Reichenbach J, Gleich GJ, Simon HU (2008) Catapult-like release of mitochondrial DNA by eosinophils contributes to antibacterial defense. Nat Med 14(9):949–953. doi:10.1038/nm.1855
von Kockritz-Blickwede M, Goldmann O, Thulin P, Heinemann K, Norrby-Teglund A, Rohde M, Medina E (2008) Phagocytosis-independent antimicrobial activity of mast cells by means of extracellular trap formation. Blood 111(6):3070–3080. doi:10.1182/blood-2007-07-104018
Lin AM, Rubin CJ, Khandpur R, Wang JY, Riblett M, Yalavarthi S, Villanueva EC, Shah P, Kaplan MJ, Bruce AT (2011) Mast cells and neutrophils release IL-17 through extracellular trap formation in psoriasis. J Immunol 187(1):490–500. doi:10.4049/jimmunol.1100123
Wong KW, Jacobs WR, Jr. (2013) Mycobacterium tuberculosis exploits human interferon gamma to stimulate macrophage extracellular trap formation and necrosis. J Infect Dis. doi:10.1093/infdis/jit097
Nakajima K, Powers JC, Ashe BM, Zimmerman M (1979) Mapping the extended substrate binding site of cathepsin G and human leukocyte elastase. Studies with peptide substrates related to the alpha 1-protease inhibitor reactive site. J Biol Chem 254(10):4027–4032
Lavie G, Zucker-Franklin D, Franklin EC (1980) Elastase-type proteases on the surface of human blood monocytes: possible role in amyloid formation. J Immunol 125(1):175–180
Urban CF, Ermert D, Schmid M, Abu-Abed U, Goosmann C, Nacken W, Brinkmann V, Jungblut PR, Zychlinsky A (2009) Neutrophil Extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against Candida albicans. PLoS Pathog 5(10):e1000639. doi:10.1371/journal.ppat.1000639
Urban CF, Lourido S, Zychlinsky A (2006) How do microbes evade neutrophil killing? Cell Microbiol 8(11):1687–1696
Menegazzi R, Decleva E, Dri P (2012) Killing by neutrophil extracellular traps: fact or folklore? Blood 119(5):1214–1216. doi:10.1182/blood-2011-07-364604
Marin-Esteban V, Turbica I, Dufour G, Semiramoth N, Gleizes A, Gorges R, Beau I, Servin AL, Lievin-Le Moal V, Sandre C, Chollet-Martin S (2012) Afa/Dr diffusely adhering Escherichia coli strain C1845 induces neutrophil extracellular traps that kill bacteria and damage human enterocyte-like cells. Infect Immun 80(5):1891–1899. doi:10.1128/IAI.00050-12
Parker H, Dragunow M, Hampton MB, Kettle AJ, Winterbourn CC (2012) Requirements for NADPH oxidase and myeloperoxidase in neutrophil extracellular trap formation differ depending on the stimulus. J Leukoc Biol. doi:10.1189/jlb.1211601
Yan J, Meng X, Wancket LM, Lintner K, Nelin LD, Chen B, Francis KP, Smith CV, Rogers LK, Liu Y (2012) Glutathione reductase facilitates host defense by sustaining phagocytic oxidative burst and promoting the development of neutrophil extracellular traps. J Immunol 188(5):2316–2327. doi:10.4049/jimmunol.1102683
Grinberg N, Elazar S, Rosenshine I, Shpigel NY (2008) Beta-hydroxybutyrate abrogates formation of bovine neutrophil extracellular traps and bactericidal activity against mammary pathogenic Escherichia coli. Infect Immun 76(6):2802–2807. doi:10.1128/IAI.00051-08
Belaaouaj A, McCarthy R, Baumann M, Gao ZM, Ley TJ, Abraham SN, Shapiro SD (1998) Mice lacking neutrophil elastase reveal impaired host defense against gram negative bacterial sepsis. Nat Med 4(5):615–618. doi:10.1038/nm0598-615
Achouiti A, Vogl T, Urban CF, Rohm M, Hommes TJ, van Zoelen MA, Florquin S, Roth J, van ’t Veer C, de Vos AF, van der Poll T (2012) Myeloid-related protein-14 contributes to protective immunity in gram-negative pneumonia derived sepsis. PLoS Pathog 8(10):e1002987. doi:10.1371/journal.ppat.1002987
Barletta KE, Cagnina RE, Burdick MD, Linden J, Mehrad B (2012) Adenosine A(2B) receptor deficiency promotes host defenses against gram-negative bacterial pneumonia. Am J Respir Crit Care Med 186(10):1044–1050. doi:10.1164/rccm.201204-0622OC
Bogaert D, de Groot R, Hermans PWM (2004) Streptococcus pneumoniae colonisation: the key to pneumococcal disease. Lancet Infect Dis 4(3):144–154. doi:10.1016/s1473-3099(04)00938-7
O’Loughlin RE, Roberson A, Cieslak PR, Lynfield R, Gershman K, Craig A, Albanese BA, Farley MM, Barrett NL, Spina NL, Beall B, Harrison LH, Reingold A, Van Beneden C (2007) The epidemiology of invasive group A streptococcal infection and potential vaccine implications: United States, 2000–2004. Clin Infect Dis : Off Publ Infect Dis Soc Am 45(7):853–862. doi:10.1086/521264
Aziz RK, Kotb M (2008) Rise and persistence of global M1T1 clone of Streptococcus pyogenes. Emerg Infect Dis 14(10):1511–1517. doi:10.3201/eid1410.071660
Lauth X, von Kockritz-Blickwede M, McNamara CW, Myskowski S, Zinkernagel AS, Beall B, Ghosh P, Gallo RL, Nizet V (2009) M1 protein allows group A streptococcal survival in phagocyte extracellular traps through cathelicidin inhibition. J Innate Immun 1(3):202–214. doi:10.1159/000203645
Mori Y, Yamaguchi M, Terao Y, Hamada S, Ooshima T, Kawabata S (2012) Alpha-enolase of Streptococcus pneumoniae induces formation of neutrophil extracellular traps. J Biol Chem 287(13):10472–10481. doi:10.1074/jbc.M111.280321
Wartha F, Beiter K, Albiger B, Fernebro J, Zychlinsky A, Normark S, Henriques-Normark B (2007) Capsule and d-alanylated lipoteichoic acids protect Streptococcus pneumoniae against neutrophil extracellular traps. Cell Microbiol 9(5):1162–1171. doi:10.1111/j.1462-5822.2006.00857.x
Beiter K, Wartha F, Albiger B, Normark S, Zychlinsky A, Henriques-Normark B (2006) An endonuclease allows Streptococcus pneumoniae to escape from neutrophil extracellular traps. Curr Biol 16(4):401–407. doi:10.1016/j.cub.2006.01.056
Sumby P, Barbian KD, Gardner DJ, Whitney AR, Welty DM, Long RD, Bailey JR, Parnell MJ, Hoe NP, Adams GG, Deleo FR, Musser JM (2005) Extracellular deoxyribonuclease made by group A Streptococcus assists pathogenesis by enhancing evasion of the innate immune response. Proc Natl Acad Sci U S A 102(5):1679–1684. doi:10.1073/pnas.0406641102
Buchanan JT, Simpson AJ, Aziz RK, Liu GY, Kristian SA, Kotb M, Feramisco J, Nizet V (2006) DNase expression allows the pathogen group A Streptococcus to escape killing in neutrophil extracellular traps. Curr Biol 16(4):396–400. doi:10.1016/j.cub.2005.12.039
Walker MJ, Hollands A, Sanderson-Smith ML, Cole JN, Kirk JK, Henningham A, McArthur JD, Dinkla K, Aziz RK, Kansal RG, Simpson AJ, Buchanan JT, Chhatwal GS, Kotb M, Nizet V (2007) DNase Sda1 provides selection pressure for a switch to invasive group A streptococcal infection. Nat Med 13(8):981–985. doi:10.1038/nm1612
Uchiyama S, Andreoni F, Schuepbach RA, Nizet V, Zinkernagel AS (2012) DNase Sda1 allows invasive M1T1 group A Streptococcus to prevent TLR9-dependent recognition. PLoS Pathog 8(6):e1002736. doi:10.1371/journal.ppat.1002736
Berends ET, Horswill AR, Haste NM, Monestier M, Nizet V, von Kockritz-Blickwede M (2010) Nuclease expression by Staphylococcus aureus facilitates escape from neutrophil extracellular traps. J Innate Immun 2(6):576–586. doi:10.1159/000319909
Nandi B, Behar SM (2011) Regulation of neutrophils by interferon-gamma limits lung inflammation during tuberculosis infection. J Exp Med 208(11):2251–2262. doi:10.1084/jem.20110919
Ramos-Kichik V, Mondragon-Flores R, Mondragon-Castelan M, Gonzalez-Pozos S, Muniz-Hernandez S, Rojas-Espinosa O, Chacon-Salinas R, Estrada-Parra S, Estrada-Garcia I (2009) Neutrophil extracellular traps are induced by Mycobacterium tuberculosis. Tuberc (Edinb) 89(1):29–37. doi:10.1016/j.tube.2008.09.009
Netea MG, Brown GD, Kullberg BJ, Gow NA (2008) An integrated model of the recognition of Candida albicans by the innate immune system. Nat Rev Microbiol 6(1):67–78. doi:10.1038/nrmicro1815
Cheng SC, Joosten LA, Kullberg BJ, Netea MG (2012) Interplay between Candida albicans and the mammalian innate host defense. Infect Immun 80(4):1304–1313. doi:10.1128/IAI.06146-11
Urban CF, Reichard U, Brinkmann V, Zychlinsky A (2006) Neutrophil extracellular traps capture and kill Candida albicans yeast and hyphal forms. Cell Microbiol 8(4):668–676. doi:10.1111/j.1462-5822.2005.00659.x
McCormick A, Heesemann L, Wagener J, Marcos V, Hartl D, Loeffler J, Heesemann J, Ebel F (2010) NETs formed by human neutrophils inhibit growth of the pathogenic mold Aspergillus fumigatus. Microbes Infect/Inst Pasteur 12(12–13):928–936. doi:10.1016/j.micinf.2010.06.009
Bruns S, Kniemeyer O, Hasenberg M, Aimanianda V, Nietzsche S, Thywissen A, Jeron A, Latge JP, Brakhage AA, Gunzer M (2010) Production of extracellular traps against Aspergillus fumigatus in vitro and in infected lung tissue is dependent on invading neutrophils and influenced by hydrophobin RodA. PLoS Pathog 6(4):e1000873. doi:10.1371/journal.ppat.1000873
Saitoh T, Komano J, Saitoh Y, Misawa T, Takahama M, Kozaki T, Uehata T, Iwasaki H, Omori H, Yamaoka S, Yamamoto N, Akira S (2012) Neutrophil extracellular traps mediate a host defense response to human immunodeficiency virus-1. Cell Host Microbe 12(1):109–116
Wardini AB, Guimaraes-Costa AB, Nascimento MT, Nadaes NR, Danelli MG, Mazur C, Benjamim CF, Saraiva EM, Pinto-da-Silva LH (2010) Characterization of neutrophil extracellular traps in cats naturally infected with feline leukemia virus. J Gen Virol 91(Pt 1):259–264. doi:10.1099/vir.0.014613-0
Narasaraju T, Yang E, Samy RP, Ng HH, Poh WP, Liew AA, Phoon MC, van Rooijen N, Chow VT (2011) Excessive neutrophils and neutrophil extracellular traps contribute to acute lung injury of influenza pneumonitis. Am J Pathol 179(1):199–210. doi:10.1016/j.ajpath.2011.03.013
Baker VS, Imade GE, Molta NB, Tawde P, Pam SD, Obadofin MO, Sagay SA, Egah DZ, Iya D, Afolabi BB, Baker M, Ford K, Ford R, Roux KH, Keller TC 3rd (2008) Cytokine-associated neutrophil extracellular traps and antinuclear antibodies in Plasmodium falciparum infected children under six years of age. Malar J 7:41. doi:10.1186/1475-2875-7-41
Gabriel C, McMaster WR, Girard D, Descoteaux A (2010) Leishmania donovani promastigotes evade the antimicrobial activity of neutrophil extracellular traps. J Immunol 185(7):4319–4327. doi:10.4049/jimmunol.1000893
Guimaraes-Costa AB, Nascimento MT, Froment GS, Soares RP, Morgado FN, Conceicao-Silva F, Saraiva EM (2009) Leishmania amazonensis promastigotes induce and are killed by neutrophil extracellular traps. Proc Natl Acad Sci U S A 106(16):6748–6753. doi:10.1073/pnas.0900226106
Wang Y, Chen Y, Xin L, Beverley SM, Carlsen ED, Popov V, Chang KP, Wang M, Soong L (2011) Differential microbicidal effects of human histone proteins H2A and H2B on Leishmania promastigotes and amastigotes. Infect Immun 79(3):1124–1133. doi:10.1128/IAI.00658-10
Abi Abdallah DS, Lin C, Ball CJ, King MR, Duhamel GE, Denkers EY (2012) Toxoplasma gondii triggers release of human and mouse neutrophil extracellular traps. Infect Immun 80(2):768–777. doi:10.1128/IAI.05730-11
Behrendt JH, Ruiz A, Zahner H, Taubert A, Hermosilla C (2010) Neutrophil extracellular trap formation as innate immune reactions against the apicomplexan parasite Eimeria bovis. Vet Immunol Immunopathol 133(1):1–8. doi:10.1016/j.vetimm.2009.06.012
Tsokos GC (2011) Systemic lupus erythematosus. N Engl J Med 365(22):2110–2121. doi:10.1056/NEJMra1100359
Cuchacovich R, Gedalia A (2009) Pathophysiology and clinical spectrum of infections in systemic lupus erythematosus. Rheum Dis Clin N Am 35(1):75–93. doi:10.1016/j.rdc.2009.03.003
Hakkim A, Furnrohr BG, Amann K, Laube B, Abed UA, Brinkmann V, Herrmann M, Voll RE, Zychlinsky A (2010) Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc Natl Acad Sci U S A 107(21):9813–9818. doi:10.1073/pnas.0909927107
Lande R, Ganguly D, Facchinetti V, Frasca L, Conrad C, Gregorio J, Meller S, Chamilos G, Sebasigari R, Riccieri V, Bassett R, Amuro H, Fukuhara S, Ito T, Liu YJ, Gilliet M (2011) Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA-peptide complexes in systemic lupus erythematosus. Sci Transl Med 3(73):73. doi:10.1126/scitranslmed.3001180
Garcia-Romo GS, Caielli S, Vega B, Connolly J, Allantaz F, Xu Z, Punaro M, Baisch J, Guiducci C, Coffman RL, Barrat FJ, Banchereau J, Pascual V (2011) Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci Transl Med 3(73):73. doi:10.1126/scitranslmed.3001201
Denny MF, Yalavarthi S, Zhao W, Thacker SG, Anderson M, Sandy AR, McCune WJ, Kaplan MJ (2010) A distinct subset of proinflammatory neutrophils isolated from patients with systemic lupus erythematosus induces vascular damage and synthesizes type I IFNs. J Immunol 184(6):3284–3297. doi:10.4049/jimmunol.0902199
Villanueva E, Yalavarthi S, Berthier CC, Hodgin JB, Khandpur R, Lin AM, Rubin CJ, Zhao W, Olsen SH, Klinker M, Shealy D, Denny MF, Plumas J, Chaperot L, Kretzler M, Bruce AT, Kaplan MJ (2011) Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus. J Immunol 187(1):538–552. doi:10.4049/jimmunol.1100450
Alliance for Lupus Research (2012) The latest lupus research and drug developments. http://www.lupusresearch.org/about-lupus/newtherapiesforlupusintrial.html
Leffler J, Martin M, Gullstrand B, Tyden H, Lood C, Truedsson L, Bengtsson AA, Blom AM (2012) Neutrophil extracellular traps that are not degraded in systemic lupus erythematosus activate complement exacerbating the disease. J Immunol 188(7):3522–3531. doi:10.4049/jimmunol.1102404
Napirei M, Karsunky H, Zevnik B, Stephan H, Mannherz HG, Moroy T (2000) Features of systemic lupus erythematosus in Dnase1-deficient mice. Nat Genet 25(2):177–181. doi:10.1038/76032
Campbell AM, Kashgarian M, Shlomchik MJ (2012) NADPH oxidase inhibits the pathogenesis of systemic lupus erythematosus. Sci Transl Med 4(157):157. doi:10.1126/scitranslmed.3004801
Chang X, Yamada R, Suzuki A, Sawada T, Yoshino S, Tokuhiro S, Yamamoto K (2005) Localization of peptidylarginine deiminase 4 (PADI4) and citrullinated protein in synovial tissue of rheumatoid arthritis. Rheumatol (Oxford) 44(1):40–50. doi:10.1093/rheumatology/keh414
Khandpur R, Carmona-Rivera C, Vivekanandan-Giri A, Gizinski A, Yalavarthi S, Knight JS, Friday S, Li S, Patel RM, Subramanian V, Thompson P, Chen P, Fox DA, Pennathur S, Kaplan MJ (2013) NETs are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Sci Transl Med 5(178):178. doi:10.1126/scitranslmed.3005580
Tarner IH, Muller-Ladner U, Gay S (2007) Emerging targets of biologic therapies for rheumatoid arthritis. Nat Clin Pract Rheumatol 3(6):336–345. doi:10.1038/ncprheum0506
Pelletier M, Maggi L, Micheletti A, Lazzeri E, Tamassia N, Costantini C, Cosmi L, Lunardi C, Annunziato F, Romagnani S, Cassatella MA (2010) Evidence for a cross-talk between human neutrophils and Th17 cells. Blood 115(2):335–343. doi:10.1182/blood-2009-04-216085
Shen F, Gaffen SL (2008) Structure-function relationships in the IL-17 receptor: implications for signal transduction and therapy. Cytokine 41(2):92–104. doi:10.1016/j.cyto.2007.11.013
Malemud CJ (2006) Matrix metalloproteinases (MMPs) in health and disease: an overview. Front Biosci: J Virtual Library 11:1696–1701
Suzuki A, Yamada R, Yamamoto K (2007) Citrullination by peptidylarginine deiminase in rheumatoid arthritis. Ann N Y Acad Sci 1108(1):323–339. doi:10.1196/annals.1422.034
Schorn C, Janko C, Krenn V, Zhao Y, Munoz LE, Schett G, Herrmann M (2012) Bonding the foe—NETting neutrophils immobilize the pro-inflammatory monosodium urate crystals. Front Immunol 3:376. doi:10.3389/fimmu.2012.00376
Rohrbach AS, Hemmers S, Arandjelovic S, Corr M, Mowen KA (2012) PAD4 is not essential for disease in the K/BxN murine autoantibody-mediated model of arthritis. Arthr Res Ther 14(3):R104. doi:10.1186/ar3829
Mitroulis I, Kambas K, Chrysanthopoulou A, Skendros P, Apostolidou E, Kourtzelis I, Drosos GI, Boumpas DT, Ritis K (2011) Neutrophil extracellular trap formation is associated with IL-1beta and autophagy-related signaling in gout. PLoS One 6(12):e29318. doi:10.1371/journal.pone.0029318
McIntosh I, Cutting GR (1992) Cystic fibrosis transmembrane conductance regulator and the etiology and pathogenesis of cystic fibrosis. FASEB J 6(10):2775–2782
Cantin AM (1998) DNase I acutely increases cystic fibrosis sputum elastase activity and its potential to induce lung hemorrhage in mice. Am J Respir Crit Care Med 157(2):464–469. doi:10.1164/ajrccm.157.2.9608033
Vogelmeier C, Döring G (1996) Neutrophil proteinases and rhDNase therapy in cystic fibrosis. Eur Respir J 9(11):2193–2195. doi:10.1183/09031936.96.09112193
Ratjen F (2008) Recent advances in cystic fibrosis. Paediatr Respir Rev 9(2):144–148. doi:10.1016/j.prrv.2008.01.004
Papayannopoulos V, Staab D, Zychlinsky A (2011) Neutrophil elastase enhances sputum solubilization in cystic fibrosis patients receiving DNase therapy. PLoS One 6(12):e28526. doi:10.1371/journal.pone.0028526
Marcos V, Zhou Z, Yildirim AO, Bohla A, Hector A, Vitkov L, Wiedenbauer EM, Krautgartner WD, Stoiber W, Belohradsky BH, Rieber N, Kormann M, Koller B, Roscher A, Roos D, Griese M, Eickelberg O, Doring G, Mall MA, Hartl D (2010) CXCR2 mediates NADPH oxidase-independent neutrophil extracellular trap formation in cystic fibrosis airway inflammation. Nat Med 16(9):1018–U1114. doi:10.1038/nm.2209
Roghanian A, Sallenave J-M (2008) Neutrophil elastase (NE) and NE inhibitors: Canonical and noncanonical functions in lung chronic, inflammatory diseases (cystic fibrosis and chronic obstructive pulmonary disease). J Aerosol Med Pulm Drug Deliv 21(1):125–144. doi:10.1089/jamp.2007.0653
von Bruhl ML, Stark K, Steinhart A, Chandraratne S, Konrad I, Lorenz M, Khandoga A, Tirniceriu A, Coletti R, Kollnberger M, Byrne RA, Laitinen I, Walch A, Brill A, Pfeiler S, Manukyan D, Braun S, Lange P, Riegger J, Ware J, Eckart A, Haidari S, Rudelius M, Schulz C, Echtler K, Brinkmann V, Schwaiger M, Preissner KT, Wagner DD, Mackman N, Engelmann B, Massberg S (2012) Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med 209(4):819–835. doi:10.1084/jem.20112322
Brill A, Fuchs TA, Savchenko AS, Thomas GM, Martinod K, De Meyer SF, Bhandari AA, Wagner DD (2012) Neutrophil extracellular traps promote deep vein thrombosis in mice. J Thromb Haemost 10(1):136–144. doi:10.1111/j.1538-7836.2011.04544.x
Gupta AK, Joshi MB, Philippova M, Erne P, Hasler P, Hahn S, Resink TJ (2010) Activated endothelial cells induce neutrophil extracellular traps and are susceptible to NETosis-mediated cell death. FEBS Lett 584(14):3193–3197. doi:10.1016/j.febslet.2010.06.006
Gonias SL, Pasqua JJ, Greenberg C, Pizzo SV (1985) Precipitation of fibrinogen, fibrinogen degradation products and fibrin monomer by histone H3. Thromb Res 39(1):97–116
Ward CM, Tetaz TJ, Andrews RK, Berndt MC (1997) Binding of the von Willebrand factor A1 domain to histone. Thromb Res 86(6):469–477
Ammollo CT, Semeraro F, Xu J, Esmon NL, Esmon CT (2011) Extracellular histones increase plasma thrombin generation by impairing thrombomodulin-dependent protein C activation. J Thromb Haemost 9(9):1795–1803. doi:10.1111/j.1538-7836.2011.04422.x
Renesto P, Chignard M (1993) Enhancement of cathepsin G-induced platelet activation by leukocyte elastase: consequence for the neutrophil-mediated platelet activation. Blood 82(1):139–144
Si-Tahar M, Pidard D, Balloy V, Moniatte M, Kieffer N, Van Dorsselaer A, Chignard M (1997) Human neutrophil elastase proteolytically activates the platelet integrin alphaIIbbeta3 through cleavage of the carboxyl terminus of the alphaIIb subunit heavy chain. Involvement in the potentiation of platelet aggregation. J Biol Chem 272(17):11636–11647
Al-Homood IA (2012) Thrombosis in systemic lupus erythematosus: a review article. ISRN Rheumatol 2012:428269–10.5402/2012/428269
Roberts JM, Redman CW (1993) Pre-eclampsia: more than pregnancy-induced hypertension. Lancet 341(8858):1447–1451
Gupta AK, Hasler P, Holzgreve W, Gebhardt S, Hahn S (2005) Induction of neutrophil extracellular DNA lattices by placental microparticles and IL-8 and their presence in preeclampsia. Hum Immunol 66(11):1146–1154. doi:10.1016/j.humimm.2005.11.003
Gupta A, Hasler P, Gebhardt S, Holzgreve W, Hahn S (2006) Occurrence of neutrophil extracellular DNA traps (NETs) in pre-eclampsia: a link with elevated levels of cell-free DNA? Ann N Y Acad Sci 1075:118–122. doi:10.1196/annals.1368.015
Gupta AK, Hasler P, Holzgreve W, Hahn S (2007) Neutrophil NETs: a novel contributor to preeclampsia-associated placental hypoxia? Semin Immunopathol 29(2):163–167. doi:10.1007/s00281-007-0073-4
Vitkov L, Klappacher M, Hannig M, Krautgartner WD (2009) Extracellular neutrophil traps in periodontitis. J Periodontal Res 44(5):664–672. doi:10.1111/j.1600-0765.2008.01175.x
Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, Worthen GS, Albelda SM (2009) Polarization of tumor-associated neutrophil phenotype by TGF-beta: "N1" versus "N2" TAN. Cancer Cell 16(3):183–194. doi:10.1016/j.ccr.2009.06.017
Houghton AM, Rzymkiewicz DM, Ji H, Gregory AD, Egea EE, Metz HE, Stolz DB, Land SR, Marconcini LA, Kliment CR, Jenkins KM, Beaulieu KA, Mouded M, Frank SJ, Wong KK, Shapiro SD (2010) Neutrophil elastase-mediated degradation of IRS-1 accelerates lung tumor growth. Nat Med 16(2):219–223. doi:10.1038/nm.2084
Berger-Achituv S, Brinkmann V, Abed UA, Kuhn LI, Ben-Ezra J, Elhasid R, Zychlinsky A (2013) A proposed role for neutrophil extracellular traps in cancer immunoediting. Front Immunol 4:48. doi:10.3389/fimmu.2013.00048
Ward AC, Dale DC (2009) Genetic and molecular diagnosis of severe congenital neutropenia. Curr Opin Hematol 16(1):9–13. doi:10.1097/MOH.0b013e32831952de
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is a contribution to the special issue on Neutrophils - Guest Editors: Paul Hasler and Sinuhe Hahn
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Branzk, N., Papayannopoulos, V. Molecular mechanisms regulating NETosis in infection and disease. Semin Immunopathol 35, 513–530 (2013). https://doi.org/10.1007/s00281-013-0384-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00281-013-0384-6