
Abstract
In recent years, research into the role of complement in the immunopathogenesis of renal disease has broadened our understanding of the fragile balance between the protective and harmful functions of the complement system. Interventions into the complement system in various models of immune-mediated renal disease have resulted in both favourable and unfavourable effects and will allow us to precisely define the level of the complement cascade at which a therapeutic intervention will result in an optimal effect. The discovery of mutations of complement regulatory molecules has established a role of complement in the haemolytic uremic syndrome and membranoproliferative glomerulonephritis, and genotyping for mutations of the complement system are already leaving the research laboratory and have entered clinical practice. These clinical discoveries have resulted in the creation of relevant animal models which may provide crucial information for the development of highly specific therapeutic agents. Research into the role of complement in proteinuria has helped to understand pathways of inflammation which ultimately lead to renal failure irrespective of the underlying renal disease and is of major importance for the majority of renal patients. Complement science is a highly exciting area of translational research and hopefully will result in meaningful therapeutic advances in the near future.
Similar content being viewed by others

Introduction
The complement system involves approximately 30 plasma- and membrane-bound proteins (reviewed in [1, 2]). These proteins play an important role in anti-microbial defence and the clearance of immune complexes and apoptotic and necrotic cells. The role of complement is not restricted to the innate immune system but includes important functions in the regulation of the adaptive immune response. Although complement undoubtedly contributes to tissue damage in numerous forms of glomerulonephritis, we will also see that complement protects against immune-mediated tissue damage in a number of settings.
In the following, we will first give an introduction to the pathways of complement activation and their regulation. This will be followed by a review of glomerular renal diseases in which complement plays a prominent role. The role of complement in non-glomerular renal disease entities such as transplantation and ischaemia/reperfusion damage falls outside the scope of this study and is reviewed in references [3] and [4].
Overview of the complement pathways
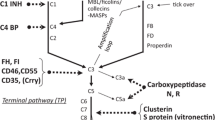
The complement system consists of three different pathways that all converge in the activation of the central complement molecule C3 (Fig. 1). Sufficient activation of C3 will then lead to the formation of the membrane attack complex.

Schematic overview of the three pathways of complement activation
The first component in the activation of the classical pathway is C1. Binding of at least two bindings sites of C1q to antigen-bound IgG or IgM, acute phase proteins such as CRP or dead cells leads to conformational changes that result in the activation of the associated serine proteases C1r and C1s. Activated C1s cleaves C4 into C4a and C4b. C4b then covalently binds to nearby structures. The bound C4b then binds C2 whereupon the smaller C2b fragment is cleaved off, resulting in the formation of the C4bC2a complex, which is the classical pathway C3 convertase.
The lectin pathway is related to the classical pathway and uses the same C3 convertase, C4bC2a. The initiation molecules of the lectin pathway, mannose-binding lectin (MBL) and the ficolins, recognise carbohydrate ligands present on a wide range of microorganisms in a pattern-like fashion. The interaction of MBL with its ligand leads to the activation of the MBL-associated serine proteases (MASP-1, MASP-2 and MASP-3). MASP-2 then cleaves C4 and subsequently C2 leading to the formation of the C3 convertase which is identical to the classical route C3 convertase, C4bC2a.
MBL consists of up to six trimeric subunits which are arranged in a structure similar to C1q. Frequently occurring single nucleotide polymorphisms within exon 1 of the MBL-2 gene and polymorphisms of the promoter region explain the up to 1,000-fold inter-individual variation in MBL plasma levels.
The activation of the alternative route of complement depends on spontaneous activation of the C3 molecule by hydrolysis of the internal thioester bond of C3. This results in the formation of the C3b-like molecule C3(H2O). Hydrolysed C3 then binds to factor B. This interaction renders factor B susceptible to cleavage by factor D resulting in the release of the Ba fragment and the formation of the C3 convertase C3(H2O)Bb. This initial convertase constantly cleaves C3 at a low rate generating C3b. This constant low rate generation of C3b is referred to as the “tick over” of the alternative pathway. The generated C3b can interact with factor B to form the more active alternative pathway C3 convertase C3bBb.
The majority of the generated C3b is rapidly inactivated by circulating factor I together with its co-factors, factor H (fH) and membrane co-factor protein (MCP). However, if C3b binds to an activator surface (e.g. a bacterial wall or damaged tissue), the molecule is protected against inactivation and further amplification of the alternative route will occur. Properdin is an important positive regulator of the alternative route. Binding of properdin leads to stabilisation of the labile C3 convertase and promotes the assembly of a complement-activating lattice by further binding factor B and C3b molecules [5–7].
The early activation steps of the classical, lectin and alternative route of complement activation converge in a common terminal pathway. The addition of a further C3b molecule to the C3 convertase complex leads to the formation of C3bBbC3b in the case of the alternative pathway and to the formation of C4bC2aC3b in the case of both the classical and lectin pathways. These C5 convertases then initiate the assembly of the membrane attack complex by cleavage of C5 to C5a and C5b. C5a can then function as a potent anaphylotoxin. The newly formed C5b forms a tri-molecular complex by binding C6 and C7. After inserting into a cell membrane, this complex binds C8 and multiple C9 molecules. This results in the completion of the pore-forming membrane attack complex (C5b-9). This complex can lead to cell lysis and, in the absence of complete lysis, to cell activation.
Regulation of complement activation
The complement system consists of numerous regulatory molecules that protect the host from uncontrolled tissue destruction and activation by the complement system. Recently, defective complement regulation has been shown to play an important role in the pathogenesis of some forms of the haemolytic uremic syndrome (HUS) and membranoproliferative glomerulonephritis (MPGN). The role of complement in these diseases will be discussed in more detail below.
C1-inhibitor is a powerful inhibitor of the classical pathway of complement activation. It binds to activated C1r and C1s and causes dissociation of these inactivated enzymes from C1q. Recent data shows that C1 inhibitor also inhibits the activation of the lectin pathway by inactivation of MASP-2 and, at higher concentrations, fluid phase activation of the alternative pathway [8].
Factor I is a circulating serine protease that proteolytically degrades C3b and C4b in the presence of the co-factors fH and C4-binding protein (C4bp). Next to its function as a co-factor, fH also inhibits activation of the alternative pathway by binding to C3b and displacing Bb from the C3 convertase complex. Similarly, C4bp regulates activation of the classical and lectin pathway by displacing C2a from C4b. Both fH and C4bp promote the degradation of the C3 and C5 convertases of the respective pathways.
Cell-membrane-bound inhibitors of complement activation also contribute to the defence against inappropriate tissue damage by homologous complement. Decay-accelerating factor (CD55) exerts its effect early in the complement cascade by inhibiting the activation of C3 by preventing the formation and accelerating the decay of both the alternative and classical pathway C3 and C5 convertases. Membrane co-factor protein (MCP, CD46) serves as a co-factor for the cleavage of C3b and C4b by factor I. CD59 interacts with the final section of the complement activation pathway by inhibiting the formation of C5b-9.
Complement receptor 1 (CD35, CR1) also functions as a complement regulator by accelerating the decay of the C3 convertases. A functionally intact soluble form of CR1 can be detected in plasma [9]. Recently, a new complement receptor, the human complement receptor of the immunoglobulin superfamily (CRIg), has been described [10]. CRIg is present on macrophages in both humans and mice and plays a role in pathogen clearance. It binds to C3b and selectively inhibits the C3 and C5 convertases of the alternative pathway. A recombinant soluble form of CRIg suppressed inflammation in two murine models of arthritis [11].
Immune-complex-mediated glomerulonephritis
Immune complex glomerulonephritis is a good example for the dual role of the complement system. Immune complexes can either be deposited in the glomerulus by passive deposition from the circulation or by in situ formation via binding of antibody to local antigens. Alternatively, local formation of immune complexes may occur when a circulation antigen is recognised by antibodies after deposition in the glomerulus (planted antigen). Subepithelial complement deposition as found in membranous nephropathy leads to a non-inflammatory complement-mediated damage because the anaphylotoxins produced during the local activation do not reach circulating leucocytes. Subendothelial deposition of complement factors is associated with a brisk inflammatory response because the produced anaphylotoxins easily come into contact with circulating cells. Subendothelial immune complex deposition is typical of proliferative lupus nephritis.
Various studies have underscored the role of complement in immune-complex-mediated glomerulonephritis. Complement depletion by treating rats with aggregated human IgG resulted in a marked decrease of neutrophil influx and renal damage in a nephrotoxic serum model of acute glomerulonephritis [12]. Later studies demonstrating a beneficial effect of complement depletion in the non-inflammatory Heymann nephritis model of membranous nephropathy showed that complement-mediated damage is not dependent on the influx of inflammatory cells [13].
The contribution of terminal pathway of complement-mediated injury was established in various glomerulonephritis models in C6-depleted or C6-deficient rats. Renal damage is ameliorated in both the anti-Thy-1 and the passive Heymann nephritis models in the absence of C6 [14, 15]. As a follow-up to these findings, soluble CR1 was successfully used to treat the disease in both models [16]. A recent paper pointed towards an important role of the alternative pathway of complement activation in mouse models of type I and type II cryoglobulinemia [17]. The glomerular influx of neutrophils was significantly less in mice deficient for C3, factor B and C5, whereas C1q deficiency had no protective effect, suggesting involvement of the alternative pathway or lectin pathway.
It is interesting to note that many of the complement-deficient models of renal disease show spontaneous or worsened renal disease. This is compatible with the observation that the complement system plays an important role in the clearance of immune complexes from the circulation and in the solubilisation of deposited immune complexes. Immune complexes are rapidly opsonised with C4b and C3b. These complement components mediate the binding of the immune complexes to CR1 on erythrocytes. The complexes are then stripped off the erythrocytes when they pass through the liver or spleen. Thus, CR1-mediated clearance plays an important role in the handling of immune complexes and in keeping soluble immune complexes away from the endothelial surface thereby preventing vascular injury.
Numerous animal models of immune-complex-mediated renal disease demonstrate a protective role of complement. C3 deficiency did not protect mice from the formation of immune complexes and proteinuria in a planted antigen model of immune complex nephritis [18]. This can be explained by the role of Fc-receptor-mediated activation of either resident or infiltrating cells at the site of injury. In the complete absence of complement activation, deposition of immune complexes can still lead to renal damage via Fc-receptor-mediated cell activation [19, 20].
An interesting recent study demonstrated that factor-D-deficient mice spontaneously develop immune complex glomerulonephritis with mesangial deposition of IgM and C3 [21]. Apparently, amplification of C3 activation is necessary for the processing or dissociation of IgM-containing immune complexes in the kidney.
The important role of the complement system in immune complex clearance is underscored by the finding that humans with complement deficiency are prone to immune-complex-mediated disease. Systemic lupus erythematosus (SLE) is a highly relevant example for this dual role of the complement system as will be discussed in the next section.
Role of complement in lupus nephritis
The degree of peripheral complement consumption and the heavy glomerular deposition of complement in SLE nephritis point towards an important role of complement in lupus nephritis. Various approaches at inhibiting complement activation have been successful in treating experimental lupus nephritis. Wang et al. prevented the development of glomerulonephritis with an anti-C5 antibody in lupus-prone NZB/W mice. Both treatments with the soluble rodent complement inhibitor rCrry and transgenic expression of this CR1-like molecule limited renal damage in the MLR/lpr mouse model of SLE [22, 23].
However, in humans, deficiencies of the early complement proteins C1q, C2 and C4 are associated with an increased risk of developing SLE [24]. Similarly, mice with C1q or C4 deficiency develop an autoimmune disease which resembles SLE [25–27].
Next to the loss of clearance of immune complexes, complement deficiency may also lead to auto-immune phenomena due to defective clearance of apoptotic cells. C1q binds to apoptotic cells [28] and C1q-deficient mice are impaired in their capacity to clear these cells [29]. Defective clearance of this rich source of auto-antigens may contribute to the emergence of auto-immunity in patients with SLE.
Taken together, it seems that, in lupus, the early components of the classical pathway of complement activation are beneficial due to their role in the clearance of immune complexes and apoptotic cells. Probably, the damage caused by Fc-receptor-mediated mechanisms in the presence of an increased deposition of immune complexes overrides the benefit of complement inhibition in these models. However, the inhibition of complement activation downstream of C3 may be a promising therapeutic approach.
Lupus nephritis is strongly associated with the presence of anti-C1q antibodies. These antibodies are present in 30–40% of SLE patients [30] and correlate with active lupus nephritis with a sensitivity of 87 to 97% and a specificity of 92% [31, 32]. Antibodies against MBL are also present in SLE, but no association with disease activity was detected [33]. The strong association of anti-C1q antibodies with active lupus nephritis suggests a pathogenic role of these antibodies. To study this question, our group has generated homologous mouse anti-mouse C1q antibodies. The administration of these antibodies to healthy mice resulted in deposition of C1q in the glomeruli together with an influx of granulocytes. However, this was not accompanied by a reduction of renal function or significant proteinuria [34]. When mice were pre-treated with a sub-nephritogenic dose of rabbit anti-C1q antibodies, the subsequent administration of mouse anti-C1q antibodies resulted in an increased deposition of immunoglobulin. The use of mice deficient for C3, C4 or for all three Fc gamma receptors showed that both complement and Fc-gamma-receptor-mediated damage is involved in this model.
IgA nephropathy
Mesangial IgA deposition is the hallmark of IgA nephropathy. Early studies on complement deposition in IgA nephropathy reported co-deposition of C3 and properdin [35]. Because C1q and C4 are usually not detected in kidneys with IgA nephropathy, this complement deposition was thought to be caused by alternative pathway activation. Mesangial deposition of C5b-9 is also present and underscores the possible pathogenic importance of complement activation in this setting [36]. More recent studies suggest a role of the lectin pathway of complement activation in IgA nephropathy. Co-deposition of IgA and MBL has been described by several authors in both IgA nephropathy [37–39] and the related entity Henoch Schönlein purpura [40]. Hisano et al. found a relation between the presence of MBL deposition and the severity of the disease [41].
The discovery of MBL-binding properties of IgA has provided a mechanistic explanation for the link between IgA and MBL deposition [42]. MBL binds to polymeric IgA in a calcium-dependent fashion via its lectin domain, suggesting an interaction with carbohydrates exposed on the IgA molecule. The binding of MBL to IgA results in complement deposition and offers an explanation for the complement deposition found in IgA nephropathy.
A recent publication underscored the potential clinical importance of MBL deposition in IgA nephropathy [43]. About 75% of the 60 biopsies in this study were negative for MBL and C4d, indicating that the C3 and C5b-9 deposition in these patients is most probably caused by the alternative pathway. The 25% of biopsies in which glomerular MBL deposition was found were positive for C4d in the absence of Clq, indicating complement activation via the lectin pathway. MBL deposition in the glomerulus was associated with both clinical and histological markers of more severe renal damage including more severe proteinuria, renal failure, extracapillary proliferation, glomerular sclerosis and interstitial fibrosis. The MASP-associated lectin, L-ficolin, was also present in these biopsies and may contribute to the activation of the lectin pathway next to MBL.
Membranoproliferative glomerulonephritis
Complement research has greatly contributed to the understanding of the pathophysiology of type II membranoproliferative glomerulonephritis (dense deposit disease) [44]. MPGN type II is characterised by deposits within the glomerular basement membrane together with staining for C3 along the glomerular basement membrane. In contrast to MPGN type I, deposition of immunoglobulins is usually not detected. The glomerular complement deposition is usually accompanied by decreased circulating C3 levels and alternative route activity. More than 80% of patients with MPGN type II are positive for serum C3-nephritic factor (C3NeF) [45].
C3NeF is an antibody directed against the alternative pathway C3 convertase. The binding of C3NeF to C3bBb prolongs the half-life of the C3 convertase by slowing down the dissociation of Bb form C3b [46]. One of the mechanisms by which C3NeF increases the half-life of C3bBb is by inhibition of fH-mediated inactivation of the convertase [47]. Consistent with complement activation by stabilisation of alternative pathway convertase activity, serum complement profiles of patients with MPGN II show predominant depletion of C3 with no consumption of C1q and C4. The glomeruli of affected kidneys show marked deposition of C3 along the glomerular capillary walls without deposition of C1q, C4 or immunoglobulins. MPGN II is also associated with acquired partial lipodystrophy. This entity is also associated with the presence of C3NeF and marked C3 depletion. In vitro data shows that C3NeF is capable of inducing alternative-pathway-mediated damage on adipocytes [48]
The role of a deregulated alternative pathway in MPGN type II was highlighted by the discovery of FH mutations in both humans [49] and pigs with MPGN type II. Mice with a targeted deletion of FH have significantly reduced levels of C3 and consistently develop MPGN with deposition of C3 in the capillary walls [50]. If fH-deficient mice are also deficient for factor B, they cannot activate the alternative pathway and no renal disease develops.
A chronic serum sickness model of immune complex disease demonstrated increased deposition if IgG immune complexes with increased C3 deposition in fH-deficient mice compared to wild-type mice. The fH-deficient mice developed diffuse proliferative glomerulonephritis, while the wild-type mice were protected against glomerular pathology. These findings indicate a role of fH in processing immune complexes and protecting the glomerulus against immune-complex-mediated disease.
Patients with MPGN type II develop ocular lesions which are similar to the drusen that are found in patients with age-related macular degeneration (AMD). The finding of a close association of AMD with factor H mutations suggests that complement is also involved in the pathogenesis of this visually disabling disease [51–54].
A recent study into the role of the terminal complement pathway in MPGN has pointed towards the potential of therapeutic inhibition of the complement system in MPGN [55]. Mice deficient for both fH and C5 developed less severe glomerulonephritis with better renal function, lower mortality and reduced glomerular cellularity in comparison to fH-deficient mice with normal C5. It is interesting to note that C5-deficient mice were not protected against proteinuria, suggesting that the glomerular C3 activation is sufficient to disrupt the glomerular permselectivity. The induction of heterologous nephrotoxic serum nephritis in fH-deficient mice resulted in markedly increased renal damage when compared to wild-type mice. The renal damage was clearly reduced in fH-deficient mice lacking C5 whereas C6-deficient mice were not protected. These findings demonstrated that the complement-mediated renal damage depended on the formation of the anaphylotoxin C5a but not on the formation of C5b-9. The role of C5 was supported by the reduction of proteinuria and glomerular neutrophil accumulation after treatment with an antibody against C5.
These observations suggest that anti-C5 treatment could serve as a treatment option in MPGN type II.
Complement and the atypical haemolytic uremic syndrome
The haemolytic uremic syndrome is characterised by microangiopathic haemolytic anemia, consumptive thrombocytopaenia and the formation of microvascular thrombi. The vascular damage is particularly severe in the kidney and can lead to acute renal failure. Most cases of HUS are associated with diarrhoea and are caused by the verotoxin-producing Escherichia coli strain O157:H7. The less common form that is not associated with diarrhoea is referred to as atypical HUS (aHUS). Especially in children, the outcome and prognosis of diarrhoea-associated HUS is good, whereas atypical HUS is associated with substantial chronic renal failure and mortality.
The familial occurrence of aHUS [56] and the occasional finding of complement consumption [57] and deposition [58] in familial aHUS suggested a hereditary defect in alternative pathway activation or control. Importantly low levels of C3 persisted in patients with familial HUS after remission of the disease and low C3 levels were also detected in unaffected relatives. The discovery of fH mutations in families with aHUS confirmed this hypothesis [59, 60]. Until now, more than more than 100 fH mutations have been described [61]. These can be searched in an interactive HUS database (http://www.FH-HUS.org). The fH mutation frequency is 38% in familial forms of aHUS and 20% in sporadic forms [62]. In contrast to the fH mutations in patients with MPGN type II, patients with aHUS are usually heterozygous for the fH mutation. The penetrance is around 60%. Patients usually have normal levels of circulating fH protein, but reduced C3 levels are found in about 50% of the aHUS cases with a fH mutation [62]. Also in contrast with the mutations associated with MPGN, the fH mutations in patients with aHUS are located in the C-terminal region which is important for binding to cellular surfaces [63–65] via an interaction with C3b deposited in the surface of these cells [66]. Because mutated fH cannot bind to surface-bound C3b, circulating factor B can associate with the C3b and C3 convertase is formed, leading to unopposed complement activation on the endothelium.
The creation of a transgenic mouse that lacks the exons encoding for the C-terminal region of fH that is responsible for the binding to cellular surfaces has resulted in an aHUS model that is highly similar to the human disease [67]. These mice have a preserved capacity to regulate fluid phase complement activation and did not develop glomerulonephritis. The absence of systemic complement depletion in the presence of defective endothelial protection against complement attack led to a typical picture of HUS including the formation of glomerular microthrombi, fragmentocytes in peripheral blood and thrombocytopenia.
Next to fH mutations, other mutations in complement regulatory proteins have been discovered in patients with aHUS. MCP mutations are found in approximately 14% of the patients with aHUS [62] and until now 43 mutations have been reported [61]. The course of the disease is milder in patients with MCP mutations, and plasma therapy does not seem to contribute to the outcome.
Factor I mutations are quite rare and are found in about 4.5% of the patients with aHUS. More recently, two factor B mutations have been discovered [68]. One of these mutations increases the affinity of factor B for C3b, while the other increases the half-life of C3bBb. Both mutations result in an increased activity of the alternative pathway.
Taken together, the clinical and experimental findings clearly point towards an important role of complement regulation in the pathogenesis of aHUS. However, until now, mutations of complement-regulatory proteins are only found in about 50% of the affected patients and family members of affected patients can share the mutations without manifesting aHUS. It seems that both additional predisposing factors and triggering circumstances, e.g. infections, are necessary to initiate the full-blown microangiopathy of aHUS.
Nevertheless, it is clinically useful to screen patients with aHUS for the known mutations because the findings may influence the prognosis and therapeutic decisions. Although evidence is lacking, patients with fH deficiency are usually treated with plasma therapy [69]. On the other hand, MCP is a membrane-bound protein and there is no rationale for plasma substitution in patients with MCP mutations and aHUS. As mentioned above, retrospective data do not indicate that plasma therapy results in improved outcomes in these patients [62]. However, patients with MCP mutations do well after kidney transplantation [70] as functional MCP is present on the endothelium of the transplanted kidney whereas patients with fH mutations have a very high rate of disease recurrence and graft loss after kidney transplantation [71]. Preemptive plasma therapy may be an option in these patients, but data are lacking to support this approach. Others have attempted combined liver and kidney transplantation in children with fH-associated HUS, but liver transplantation is associated with greatly increased risks when compared with kidney transplantation alone, and this procedure has not been uniformly successful until now [72–74].
Complement and progressive renal damage
Independent of the underlying renal disease, proteinuria is associated with tubulointerstitial fibrosis and a progressive loss of renal function [75–77]. A number of mechanisms by which proteinuria may cause renal function loss have been suggested. These include oxidative damage induced by transferrin, lysosomal rupture caused by overload of the resorptive capacity for urinary proteins and pro-inflammatory effects of albumin-bound free fatty acids [78–81].
For many years, complement deposition along the brush border has been noted in proteinuric renal disease [82]. C5b-9 is found in urine from patients with various proteinuric renal diseases including diabetic nephropathy [83]. The C5b-9 in the urine is very probably generated within the tubulular lumen due to an intrinsic complement-activating property of the tubular cells [84, 85]. The exact mechanism of tubular complement activation is not understood, but tubular ammonia production [86] and a low expression of complement regulatory proteins on the apical cell surface [87] are thought to contribute.
The insertion of sublytic amounts of C5b-9 in the cell membrane of tubular cells leads to the production of pro-inflammatory cytokines [88, 89] and collagen. In vivo evidence for the role of complement in proteinuria-mediated renal damage has been derived from studies in C6-deficient rats. Rats with an inherited C6 deficiency were protected against tubulointerstitial damage in both the remnant kidney model [90] and the puromycin model of proteinuric renal disease [91]. A therapeutic intervention with either the murine complement inhibitor Crry or CD59 targeted to the renal tubulus resulted in improved renal function and less interstitial damage when compared with untreated animals [92].
Conclusions
Increasing knowledge about the complement system has taught us about both the protective and harmful roles of complement in renal disease. In the course of this review, it has repeatedly become clear that complement inhibition early on in both the classical and alternative pathways is associated with the risk of increased deposition of immune complexes and the resulting damage may outweigh the benefit. On the other hand, it seems that complement inhibition distal of the formation of the C3 convertases is safe and offers more promising therapeutic options for renal diseases for which no satisfying treatment has been established until now.
Independently of these promising therapeutic prospects, complement has become an invaluable tool in the diagnosis and monitoring of renal disease and results of complement studies have a strong impact on day-to-day decision making in the care of our patients with renal disease.
References
Walport MJ (2001) Complement. First of two parts. N Engl J Med 344:1058–1066
Walport MJ (2001) Complement. Second of two parts. N Engl J Med 344:1140–1144
Brown KM, Sacks SH, Sheerin NS (2007) Mechanisms of disease: the complement system in renal injury—new ways of looking at an old foe. Nat Clin Pract Nephrol 3:277–286
Berger SP, Roos A, Daha MR (2005) Complement and the kidney: what the nephrologist needs to know in 2006? Nephrol Dial Transplant 20:2613–2619
Pillemer L, Blum L, Lepow IH, Ross OA, Todd EW, Wardlaw AC (1954) The properdin system and immunity. I. Demonstration and isolation of a new serum protein, properdin, and its role in immune phenomena. Science 120:279–285
Fearon DT, Austen KF (1975) Properdin: binding to C3b and stabilization of the C3b-dependent C3 convertase. J Exp Med 142:856–863
Hourcade DE (2006) The role of properdin in the assembly of the alternative pathway C3 convertases of complement. J Biol Chem 281:2128–2132
Nielsen EW, Waage C, Fure H, Brekke OL, Sfyroera G, Lambris JD, Mollnes TE (2007) Effect of supraphysiologic levels of C1-inhibitor on the classical, lectin and alternative pathways of complement. Mol Immunol 44:1819–1826
Yoon SH, Fearon DT (1985) Characterization of a soluble form of the C3b/C4b receptor (CR1) in human plasma. J Immunol 134:3332–3338
Helmy KY, Katschke KJ Jr, Gorgani NN, Kljavin NM, Elliott JM, Diehl L, Scales SJ, Ghilardi N, van Lookeren CM (2006) CRIg: a macrophage complement receptor required for phagocytosis of circulating pathogens. Cell 124:915–927
Katschke KJ Jr, Helmy KY, Steffek M, Xi H, Yin J, Lee WP, Gribling P, Barck KH, Carano RA, Taylor RE, Rangell L, Diehl L, Hass PE, Wiesmann C, van Lookerenb CM (2007) A novel inhibitor of the alternative pathway of complement reverses inflammation and bone destruction in experimental arthritis. J Exp Med 204:1319–1325
Cochrane CG, Unanue ER, Dixon FJ (1965) A role of polymorphonuclear leukocytes and complement in nephrotoxic nephritis. J Exp Med 122:99–116
Salant DJ, Belok S, Madaio MP, Couser WG (1980) A new role for complement in experimental membranous nephropathy in rats. J Clin Invest 66:1339–1350
Baker PJ, Ochi RF, Schulze M, Johnson RJ, Campbell C, Couser WG (1989) Depletion of C6 prevents development of proteinuria in experimental membranous nephropathy in rats. Am J Pathol 135:185–194
Brandt J, Pippin J, Schulze M, Hansch GM, Alpers CE, Johnson RJ, Gordon K, Couser WG (1996) Role of the complement membrane attack complex (C5b-9) in mediating experimental mesangioproliferative glomerulonephritis. Kidney Int 49:335–343
Couser WG, Johnson RJ, Young BA, Yeh CG, Toth CA, Rudolph AR (1995) The effects of soluble recombinant complement receptor 1 on complement-mediated experimental glomerulonephritis. J Am Soc Nephrol 5:1888–1894
Trendelenburg M, Fossati-Jimack L, Cortes-Hernandez J, Turnberg D, Lewis M, Izui S, Cook HT, Botto M (2005) The role of complement in cryoglobulin-induced immune complex glomerulonephritis. J Immunol 175:6909–6914
Sheerin NS, Springall T, Carroll M, Sacks SH (1999) Altered distribution of intraglomerular immune complexes in C3-deficient mice. Immunology 97:393–399
Clynes R, Dumitru C, Ravetch JV (1998) Uncoupling of immune complex formation and kidney damage in autoimmune glomerulonephritis. Science 279:1052–1054
Sylvestre D, Clynes R, Ma M, Warren H, Carroll MC, Ravetch JV (1996) Immunoglobulin G-mediated inflammatory responses develop normally in complement-deficient mice. J Exp Med 184:2385–2392
Abrera-Abeleda MA, Xu Y, Pickering MC, Smith RJ, Sethi S (2007) Mesangial immune complex glomerulonephritis due to complement factor D deficiency. Kidney Int 71:1142–1147
Bao L, Haas M, Boackle SA, Kraus DM, Cunningham PN, Park P, Alexander JJ, Anderson RK, Culhane K, Holers VM, Quigg RJ (2002) Transgenic expression of a soluble complement inhibitor protects against renal disease and promotes survival in MRL/lpr mice. J Immunol 168:3601–3607
Bao L, Haas M, Kraus DM, Hack BK, Rakstang JK, Holers VM, Quigg RJ (2003) Administration of a soluble recombinant complement C3 inhibitor protects against renal disease in MRL/lpr mice. J Am Soc Nephrol 14:670–679
Pickering MC, Botto M, Taylor PR, Lachmann PJ, Walport MJ (2000) Systemic lupus erythematosus, complement deficiency, and apoptosis. Adv Immunol 76:227–324
Mitchell DA, Pickering MC, Warren J, Fossati-Jimack L, Cortes-Hernandez J, Cook HT, Botto M, Walport MJ (2002) C1q deficiency and autoimmunity: the effects of genetic background on disease expression. J Immunol 168:2538–2543
Paul E, Pozdnyakova OO, Mitchell E, Carroll MC (2002) Anti-DNA autoreactivity in C4-deficient mice. Eur J Immunol 32:2672–2679
Botto M, Dell’Agnola C, Bygrave AE, Thompson EM, Cook HT, Petry F, Loos M, Pandolfi PP, Walport MJ (1998) Homozygous C1q deficiency causes glomerulonephritis associated with multiple apoptotic bodies. Nat Genet 19:56–59
Nauta AJ, Trouw LA, Daha MR, Tijsma O, Nieuwland R, Schwaeble WJ, Gingras AR, Mantovani A, Hack EC, Roos A (2002) Direct binding of C1q to apoptotic cells and cell blebs induces complement activation. Eur J Immunol 32:1726–1736
Taylor PR, Carugati A, Fadok VA, Cook HT, Andrews M, Carroll MC, Savill JS, Henson PM, Botto M, Walport MJ (2000) A hierarchical role for classical pathway complement proteins in the clearance of apoptotic cells in vivo. J Exp Med 192:359–366
Seelen MA, Trouw LA, Daha MR (2003) Diagnostic and prognostic significance of anti-C1q antibodies in systemic lupus erythematosus. Curr Opin Nephrol Hypertens 12:619–624
Moroni G, Trendelenburg M, Del Papa N, Quaglini S, Raschi E, Panzeri P, Testoni C, Tincani A, Banfi G, Balestrieri G, Schifferli JA, Meroni PL, Ponticelli C (2001) Anti-C1q antibodies may help in diagnosing a renal flare in lupus nephritis. Am J Kidney Dis 37:490–498
Trendelenburg M, Lopez-Trascasa M, Potlukova E, Moll S, Regenass S, Fremeaux-Bacchi V, Martinez-Ara J, Jancova E, Picazo ML, Honsova E, Tesar V, Sadallah S, Schifferli J (2006) High prevalence of anti-C1q antibodies in biopsy-proven active lupus nephritis. Nephrol Dial Transplant 21:3115–3121
Seelen MA, Trouw LA, van der Hoorn JW, Fallaux-van den Houten FC, Huizinga TW, Daha MR, Roos A (2003) Autoantibodies against mannose-binding lectin in systemic lupus erythematosus. Clin Exp Immunol 134:335–343
Trouw LA, Seelen MA, Duijs JM, Benediktsson H, van Kooten C, Daha MR (2003) Glomerular deposition of C1q and anti-C1q antibodies in mice following injection of antimouse C1q antibodies. Clin Exp Immunol 132:32–39
McCoy RC, Abramowsky CR, Tisher CC (1974) IgA nephropathy. Am J Pathol 76:123–144
Rauterberg EW, Lieberknecht HM, Wingen AM, Ritz E (1987) Complement membrane attack (MAC) in idiopathic IgA-glomerulonephritis. Kidney Int 31:820–829
Lhotta K, Wurzner R, Konig P (1999) Glomerular deposition of mannose-binding lectin in human glomerulonephritis. Nephrol Dial Transplant 14:881–886
Endo M, Ohi H, Ohsawa I, Fujita T, Matsushita M, Fujita T (1998) Glomerular deposition of mannose-binding lectin (MBL) indicates a novel mechanism of complement activation in IgA nephropathy. Nephrol Dial Transplant 13:1984–1990
Matsuda M, Shikata K, Wada J, Sugimoto H, Shikata Y, Kawasaki T, Makino H (1998) Deposition of mannan binding protein and mannan binding protein-mediated complement activation in the glomeruli of patients with IgA nephropathy. Nephron 80:408–413
Endo M, Ohi H, Ohsawa I, Fujita T, Matsushita M (2000) Complement activation through the lectin pathway in patients with Henoch–Schonlein purpura nephritis. Am J Kidney Dis 35:401–407
Hisano S, Matsushita M, Fujita T, Iwasaki H (2005) Activation of the lectin complement pathway in Henoch–Schonlein purpura nephritis. Am J Kidney Dis 45:295–302
Roos A, Bouwman LH, Gijlswijk-Janssen DJ, Faber-Krol MC, Stahl GL, Daha MR (2001) Human IgA activates the complement system via the mannan-binding lectin pathway. J Immunol 167:2861–2868
Roos A, Rastaldi MP, Calvaresi N, Oortwijn BD, Schlagwein N, Gijlswijk-Janssen DJ, Stahl GL, Matsushita M, Fujita T, van Kooten C, Daha MR (2006) Glomerular activation of the lectin pathway of complement in IgA nephropathy is associated with more severe renal disease. J Am Soc Nephrol 17:1724–1734
Appel GB, Cook HT, Hageman G, Jennette JC, Kashgarian M, Kirschfink M, Lambris JD, Lanning L, Lutz HU, Meri S, Rose NR, Salant DJ, Sethi S, Smith RJ, Smoyer W, Tully HF, Tully SP, Walker P, Welsh M, Wurzner R, Zipfel PF (2005) Membranoproliferative glomerulonephritis type II (dense deposit disease): an update. J Am Soc Nephrol 16:1392–1403
Schwertz R, Rother U, Anders D, Gretz N, Scharer K, Kirschfink M (2001) Complement analysis in children with idiopathic membranoproliferative glomerulonephritis: a long-term follow-up. Pediatr Allergy Immunol 12:166–172
Daha MR, Fearon DT, Austen KF (1976) C3 nephritic factor (C3NeF): stabilization of fluid phase and cell-bound alternative pathway convertase. J Immunol 116:1–7
Weiler JM, Daha MR, Austen KF, Fearon DT (1976) Control of the amplification convertase of complement by the plasma protein beta1H. Proc Natl Acad Sci U S A 73:3268–3272
Mathieson PW, Wurzner R, Oliveria DB, Lachmann PJ, Peters DK (1993) Complement-mediated adipocyte lysis by nephritic factor sera. J Exp Med 177:1827–1831
Levy M, Halbwachs-Mecarelli L, Gubler MC, Kohout G, Bensenouci A, Niaudet P, Hauptmann G, Lesavre P (1986) H deficiency in two brothers with atypical dense intramembranous deposit disease. Kidney Int 30:949–956
Pickering MC, Cook HT, Warren J, Bygrave AE, Moss J, Walport MJ, Botto M (2002) Uncontrolled C3 activation causes membranoproliferative glomerulonephritis in mice deficient in complement factor H. Nat Genet 31:424–428
Hageman GS, Anderson DH, Johnson LV, Hancox LS, Taiber AJ, Hardisty LI, Hageman JL, Stockman HA, Borchardt JD, Gehrs KM, Smith RJ, Silvestri G, Russell SR, Klaver CC, Barbazetto I, Chang S, Yannuzzi LA, Barile GR, Merriam JC, Smith RT, Olsh AK, Bergeron J, Zernant J, Merriam JE, Gold B, Dean M, Allikmets R (2005) A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc Natl Acad Sci U S A 102:7227–7232
Edwards AO, Ritter R III, Abel KJ, Manning A, Panhuysen C, Farrer LA (2005) Complement factor H polymorphism and age-related macular degeneration. Science 308:421–424
Klein RJ, Zeiss C, Chew EY, Tsai JY, Sackler RS, Haynes C, Henning AK, SanGiovanni JP, Mane SM, Mayne ST, Bracken MB, Ferris FL, Ott J, Barnstable C, Hoh J (2005) Complement factor H polymorphism in age-related macular degeneration. Science 308:385–389
Haines JL, Hauser MA, Schmidt S, Scott WK, Olson LM, Gallins P, Spencer KL, Kwan SY, Noureddine M, Gilbert JR, Schnetz-Boutaud N, Agarwal A, Postel EA, Pericak-Vance MA (2005) Complement factor H variant increases the risk of age-related macular degeneration. Science 308:419–421
Pickering MC, Warren J, Rose KL, Carlucci F, Wang Y, Walport MJ, Cook HT, Botto M (2006) Prevention of C5 activation ameliorates spontaneous and experimental glomerulonephritis in factor H-deficient mice. Proc Natl Acad Sci U S A 103:9649–9654
Kaplan BS, Chesney RW, Drummond KN (1975) Hemolytic uremic syndrome in families. N Engl J Med 292:1090–1093
Carreras L, Romero R, Requesens C, Oliver AJ, Carrera M, Clavo M, Alsina J (1981) Familial hypocomplementemic hemolytic uremic syndrome with HLA-A3,B7 haplotype. JAMA 245:602–604
Gonzalo A, Mampaso F, Gallego N, Bellas C, Segui J, Ortuno J (1981) Hemolytic uremic syndrome with hypocomplementemia and deposits of IgM and C3 in the involved renal tissue. Clin Nephrol 16:193–199
Warwicker P, Goodship TH, Goodship JA (1997) Three new polymorphisms in the human complement factor H gene and promoter region. Immunogenetics 46:437–438
Richards A, Buddles MR, Donne RL, Kaplan BS, Kirk E, Venning MC, Tielemans CL, Goodship JA, Goodship TH (2001) Factor H mutations in hemolytic uremic syndrome cluster in exons 18–20, a domain important for host cell recognition. Am J Hum Genet 68:485–490
Saunders RE, Abarrategui-Garrido C, Fremeaux-Bacchi V, Goicoechea dJ, Goodship TH, Lopez TM, Noris M, Ponce CI, Remuzzi G, Rodriguez dC, Sanchez-Corral P, Skerka C, Zipfel PF, Perkins SJ (2007) The interactive Factor H-atypical hemolytic uremic syndrome mutation database and website: update and integration of membrane cofactor protein and Factor I mutations with structural models. Hum Mutat 28:222–234
Caprioli J, Noris M, Brioschi S, Pianetti G, Castelletti F, Bettinaglio P, Mele C, Bresin E, Cassis L, Gamba S, Porrati F, Bucchioni S, Monteferrante G, Fang CJ, Liszewski MK, Kavanagh D, Atkinson JP, Remuzzi G (2006) Genetics of HUS: the impact of MCP, CFH, and IF mutations on clinical presentation, response to treatment, and outcome. Blood 108:1267–1279
Manuelian T, Hellwage J, Meri S, Caprioli J, Noris M, Heinen S, Jozsi M, Neumann HP, Remuzzi G, Zipfel PF (2003) Mutations in factor H reduce binding affinity to C3b and heparin and surface attachment to endothelial cells in hemolytic uremic syndrome. J Clin Invest 111:1181–1190
Jokiranta TS, Cheng ZZ, Seeberger H, Jozsi M, Heinen S, Noris M, Remuzzi G, Ormsby R, Gordon DL, Meri S, Hellwage J, Zipfel PF (2005) Binding of complement factor H to endothelial cells is mediated by the carboxy-terminal glycosaminoglycan binding site. Am J Pathol 167:1173–1181
Jozsi M, Heinen S, Hartmann A, Ostrowicz CW, Halbich S, Richter H, Kunert A, Licht C, Saunders RE, Perkins SJ, Zipfel PF, Skerka C (2006) Factor H and atypical hemolytic uremic syndrome: mutations in the C-terminus cause structural changes and defective recognition functions. J Am Soc Nephrol 17:170–177
Jokiranta TS, Jaakola VP, Lehtinen MJ, Parepalo M, Meri S, Goldman A (2006) Structure of complement factor H carboxyl-terminus reveals molecular basis of atypical haemolytic uremic syndrome. EMBO J 25:1784–1794
Pickering MC, de Jorge EG, Martinez-Barricarte R, Recalde S, Garcia-Layana A, Rose KL, Moss J, Walport MJ, Cook HT, de Cordoba SR, Botto M (2007) Spontaneous hemolytic uremic syndrome triggered by complement factor H lacking surface recognition domains. J Exp Med 204:1249–1256
Goicoechea dJ, Harris CL, Esparza-Gordillo J, Carreras L, Arranz EA, Garrido CA, Lopez-Trascasa M, Sanchez-Corral P, Morgan BP, Rodriguez dC (2007) Gain-of-function mutations in complement factor B are associated with atypical hemolytic uremic syndrome. Proc Natl Acad Sci U S A 104:240–245
Noris M, Remuzzi G (2005) Genetic abnormalities of complement regulators in hemolytic uremic syndrome: how do they affect patient management? Nat Clin Pract Nephrol 1:2–3
Richards A, Kathryn LM, Kavanagh D, Fang CJ, Moulton E, Fremeaux-Bacchi V, Remuzzi G, Noris M, Goodship TH, Atkinson JP (2007) Implications of the initial mutations in membrane cofactor protein (MCP; CD46) leading to atypical hemolytic uremic syndrome. Mol Immunol 44:111–122
Zimmerhackl LB, Scheiring J, Prufer F, Taylor CM, Loirat C (2007) Renal transplantation in HUS patients with disorders of complement regulation. Pediatr Nephrol 22:10–16
Remuzzi G, Ruggenenti P, Codazzi D, Noris M, Caprioli J, Locatelli G, Gridelli B (2002) Combined kidney and liver transplantation for familial haemolytic uraemic syndrome. Lancet 359:1671–1672
Remuzzi G, Ruggenenti P, Colledan M, Gridelli B, Bertani A, Bettinaglio P, Bucchioni S, Sonzogni A, Bonanomi E, Sonzogni V, Platt JL, Perico N, Noris M (2005) Hemolytic uremic syndrome: a fatal outcome after kidney and liver transplantation performed to correct factor h gene mutation. Am J Transplant 5:1146–1150
Saland JM, Emre SH, Shneider BL, Benchimol C, Ames S, Bromberg JS, Remuzzi G, Strain L, Goodship TH (2006) Favorable long-term outcome after liver–kidney transplant for recurrent hemolytic uremic syndrome associated with a factor H mutation. Am J Transplant 6:1948–1952
Breyer JA, Bain RP, Evans JK, Nahman NS Jr, Lewis EJ, Cooper M, McGill J, Berl T (1996) Predictors of the progression of renal insufficiency in patients with insulin-dependent diabetes and overt diabetic nephropathy. The Collaborative Study Group. Kidney Int 50:1651–1658
Peterson JC, Adler S, Burkart JM, Greene T, Hebert LA, Hunsicker LG, King AJ, Klahr S, Massry SG, Seifter JL (1995) Blood pressure control, proteinuria, and the progression of renal disease. The Modification of Diet in Renal Disease Study. Ann Intern Med 123:754–762
The GISEN Group (Gruppo Italiano di Studi Epidemiologici in Nefrologia) (1997) Randomised placebo-controlled trial of effect of ramipril on decline in glomerular filtration rate and risk of terminal renal failure in proteinuric, non-diabetic nephropathy. Lancet 349:1857–1863
Tang S, Leung JC, Abe K, Chan KW, Chan LY, Chan TM, Lai KN (2003) Albumin stimulates interleukin-8 expression in proximal tubular epithelial cells in vitro and in vivo. J Clin Invest 111:515–527
Wang Y, Chen J, Chen L, Tay YC, Rangan GK, Harris DC (1997) Induction of monocyte chemoattractant protein-1 in proximal tubule cells by urinary protein. J Am Soc Nephrol 8:1537–1545
Dixon R, Brunskill NJ (1999) Activation of mitogenic pathways by albumin in kidney proximal tubule epithelial cells: implications for the pathophysiology of proteinuric states. J Am Soc Nephrol 10:1487–1497
Arici M, Brown J, Williams M, Harris KP, Walls J, Brunskill NJ (2002) Fatty acids carried on albumin modulate proximal tubular cell fibronectin production: a role for protein kinase C. Nephrol Dial Transplant 17:1751–1757
Camussi G, Stratta P, Mazzucco G, Gaido M, Tetta C, Castello R, Rotunno M, Vercellone A (1985) In vivo localization of C3 on the brush border of proximal tubules of kidneys from nephrotic patients. Clin Nephrol 23:134–141
Morita Y, Ikeguchi H, Nakamura J, Hotta N, Yuzawa Y, Matsuo S (2000) Complement activation products in the urine from proteinuric patients. J Am Soc Nephrol 11:700–707
Camussi G, Rotunno M, Segoloni G, Brentjens JR, Andres GA (1982) In vitro alternative pathway activation of complement by the brush border of proximal tubules of normal rat kidney. J Immunol 128:1659–1663
Peake PW, Pussell BA, Mackinnon B, Charlesworth JA (2002) The effect of pH and nucleophiles on complement activation by human proximal tubular epithelial cells. Nephrol Dial Transplant 17:745–752
Nath KA, Hostetter MK, Hostetter TH (1985) Pathophysiology of chronic tubulo-interstitial disease in rats. Interactions of dietary acid load, ammonia, and complement component C3. J Clin Invest 76:667–675
Ichida S, Yuzawa Y, Okada H, Yoshioka K, Matsuo S (1994) Localization of the complement regulatory proteins in the normal human kidney. Kidney Int 46:89–96
Abe K, Li K, Sacks SH, Sheerin NS (2004) The membrane attack complex, C5b-9, upregulates collagen gene expression in renal tubular epithelial cells. Clin Exp Immunol 136:60–66
David S, Biancone L, Caserta C, Bussolati B, Cambi V, Camussi G (1997) Alternative pathway complement activation induces proinflammatory activity in human proximal tubular epithelial cells. Nephrol Dial Transplant 12:51–56
Nangaku M, Pippin J, Couser WG (2002) C6 mediates chronic progression of tubulointerstitial damage in rats with remnant kidneys. J Am Soc Nephrol 13:928–936
Nangaku M, Pippin J, Couser WG (1999) Complement membrane attack complex (C5b-9) mediates interstitial disease in experimental nephrotic syndrome. J Am Soc Nephrol 10:2323–2331
He C, Imai M, Song H, Quigg RJ, Tomlinson S (2005) Complement inhibitors targeted to the proximal tubule prevent injury in experimental nephrotic syndrome and demonstrate a key role for C5b-9. J Immunol 174:5750–5757
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Berger, S.P., Daha, M.R. Complement in glomerular injury. Semin Immunopathol 29, 375–384 (2007). https://doi.org/10.1007/s00281-007-0090-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00281-007-0090-3