
Abstract
Swine acute diarrhea syndrome coronavirus (SADS-CoV) is an emerging swine enteric alphacoronavirus that can cause acute diarrhea, vomiting, dehydration, and death of newborn piglets. In this study, we developed a double-antibody sandwich quantitative enzyme-linked immunosorbent assay (DAS-qELISA) for detection of SADS-CoV by using an anti-SADS-CoV N protein rabbit polyclonal antibody (PAb) and a specific monoclonal antibody (MAb) 6E8 against the SADS-CoV N protein. The PAb was used as the capture antibodies and HRP-labeled 6E8 as the detector antibody. The detection limit of the developed DAS-qELISA assay was 1 ng/mL of purified antigen and 101.08TCID50/mL of SADS-CoV, respectively. Specificity assays showed that the developed DAS-qELISA has no cross-reactivity with other swine enteric coronaviruses, such as porcine epidemic diarrhea virus (PEDV), transmissible gastroenteritis virus (TGEV), and porcine deltacoronavirus (PDCoV). Three-day-old piglets were challenged with SADS-CoV and collected anal swab samples which were screened for the presence of SADS-CoV by using DAS-qELISA and reverse transcriptase PCR (RT-PCR). The coincidence rate of the DAS-qELISA and RT-PCR was 93.93%, and the kappa value was 0.85, indicating that DAS-qELISA is a reliable method for applying antigen detection of clinical samples.
Key points
• The first double-antibody sandwich quantitative enzyme-linked immunosorbent assay for detection SADS-CoV infection.
• The custom ELISA is useful for controlling the SADS-CoV spread.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Swine acute diarrhea syndrome coronavirus (SADS-CoV) belongs to the family Coronaviridae and is closely related with bat-HUK2 coronavirus (Gong et al. 2017; Pan et al. 2017; Wang et al. 2018b). So far, besides porcine epidemic diarrhea virus (PEDV), transmissible gastroenteritis virus (TGEV), and porcine deltacoronavirus (PDCoV), SADS-CoV is a newly discovered swine enteric coronavirus (Cui et al. 2019). The clinical signs and pathogenesis of these four viruses are similar, including acute diarrhea, vomiting, dehydration, and death of newborn piglets. SADS-CoV’s first outbreak was in Guangdong province, China, in 2017 and re-emerged in southern China in 2019 with high mortality up to 90% in five days or younger piglets (Gong et al. 2017; Pan et al. 2017; Zhou et al. 2019). In 2020, epidemiology investigation showed that SADS-CoV infection had existed in other provinces, such as Shanxi, Yunnan, Guangdong, Jiangxi, Henan, Hubei, Hebei, Hunan, Qinghai, Anhui, and Shanxi in China (Peng et al. 2021). In May 2021, a large-scale fatal swine diarrhea disease outbreak of SADS-CoV in an intensive scale pig farm in Guangxi province was reported (Sun et al. 2022). The latest report showed that SADS-CoV had a wide range of cellular fitness in vitro, including various rodent and human cell lines, suggesting that the virus has a potential threat of cross-species transmission to humans (Yang et al. 2019b). Therefore, besides strict biosecurity measures, the development of a rapid and sensitive method to monitor the epidemic of SADS-CoV in pig herds is vitally important to prevent the spread of SADS.
Currently, multiple detection methods of SADS-CoV have been developed to monitor the epidemic, which can be classified molecular and serological methods. Molecular methods including TaqMan-based real-time RT-PCR assay (Zhou et al. 2018a), real-time reverse transcription loop-mediated isothermal amplification method (RT-LAMP) (Wang et al. 2018a), SYBR green-based real-time RT-PCR assay (Ma et al. 2019), TaqMan-probe-based multiplex real-time PCR (Huang et al. 2019; Pan et al. 2020), microfluidic-RT-LAMP chip (Zhou et al. 2020), a novel reverse transcription droplet digital PCR assay (Zhang et al. 2022), and CRISPR-Cas12a combined with multiplex RT-LAMP (Liu et al. 2022) have been developed for detecting SADS-CoV. All of these assays detect viral nucleic acid, and the sensitivity heavily depends on the quality of the samples, the specificity of the primer, the instrument, and professional knowledge, but also to prevent nucleic acid contamination, otherwise prone to false-positive accuracy. The most common serological assays for SADS-CoV detection are the indirect fluorescent assay (IFA) and enzyme-linked immunosorbent assay (ELISA). IFAs were mainly used to detect SADS-CoV replication and infection in the laboratory research. ELISA was used to detect the level of antibodies against SADS-CoV (Peng et al. 2021; Yang et al. 2019a; Zhou et al. 2018b).
ELISA has been widely used in the detection of human and animal diseases because of its simple operation, strong specificity, and high sensitivity. In this study, we target the highly conserved N gene of SADS-CoV and expressed using the prokaryotic expression system. The purified recombinant N (rN) protein was used as an immunogen to immunize mouse and rabbit to obtain the monoclonal and polyclonal antibodies. A double-antibody sandwich quantitative ELISA (DAS-qELISA) was then established using a high-affinity rabbit polyclonal antibody and horseradish peroxidase (HRP)-labeled monoclonal antibody (MAb) as capture and detection antibodies, respectively. The established DAS-qELISA has high sensitivity, specificity, and reproducibility, which is a reliable method for applying SADS-CoV antigen detection of clinical samples.
Materials and methods
Cells and viruses
The SP2/0 and Huh7 cells were kept in our laboratory. The SP2/0 hybridoma cell strain 6E8 and Huh7 cells were cultured in Dulbecco minimum essential medium (DMEM; Thermo Fisher Scientific, Waltham, MA, USA) containing 10% fetal bovine serum (FBS; Thermo Fisher Scientific, Waltham, MA, USA). SADS-CoV strain GDS04 (GenBank Accession No. MF167434.1) was kindly provided by the professor of Sun Yat-Sen University of Yongchang Cao (Gong et al. 2017; Xu et al. 2019). Huh7 cells were used to propagate SADS-CoV, and virus titers were determined by the Reed–Muench method. PEDV, TGEV, and PDCoV were kept in our laboratory.
Production and purification of MAb and PAb against N protein
The complete N (1128 bp) gene was inserted into the pET32a vector, the recombinant plasmids expressed in Escherichia coli (E. coli) BL21 (DE3). The recombinant pET32a-N protein (rN) was purified by Ni-chelating affinity chromatography. To produce the N protein antibodies, the purity immunized the 6 to 6-week-old female BALB/c mice and 2 to 12-week-old female rabbit (Lanzhou, China). In the first immunization, the purified rN protein (30 μg/mouse and 300 μg/rabbit) was emulsified with the equal volume of Freund’s complete adjuvant (Sigma-Aldrich, St. Louis, MO, USA) and injected subcutaneously in sites on the belly of mouse or the back of rabbit. Freund’s incomplete adjuvant (Sigma-Aldrich, St. Louis, MO, USA) was used in the booster immunization at 2-week intervals. The method and dose of booster immunization were the same as those of the first immunization. After 4 times of immunization, mouse spleen cells were isolated and fused with mouse myeloma SP2/0 cells as previously described (Köhler and Milstein 1975). Hybridoma culture supernatants were tested by indirect ELISA and indirect immunofluorescence assay (IFA). Positive clones were subcloned 3 times by limiting dilution and injected into Freund’s incomplete adjuvant-treated BALB/c mice to obtain ascetic fluid. For rabbit, the blood samples were obtained according to the animal ethical and moral standards and stored at 4 °C overnight. The blood was centrifuged at 12,000 rpm/min for 30 min for the next day, and the supernatant was the rabbit PAb. Immunoglobulin G (IgG) from MAb and PAb was purified using protein G sepharose (GE Healthcare, Chicago, IL, USA). The purity of the purified MAb and PAb was identified by SDS-PAGE, and the titers were determined by indirect ELISA.
Indirect ELISA
The ELISA plates were coated with 50 μL of the culture supernatants of SADS-CoV-infected Huh7 cells (1:1) diluted in carbonated coating buffer (pH 9.6) at 4 °C overnight. The plates were washed 4 times with PBST (phosphate-buffered saline (PBS) containing 0.05% Tween 20) and blocked with 2% trehalose in PBST at 4 °C for 10 h. For positive hybridoma screen, 50 μL of the supernatants of hybridoma was added to each well. For the titer of the purified anti-N MAb and PAb detection, the antibodies were serially diluted (1:100–1:102,400) in PBST and added into ELISA plates (50 μL/well). The plates were incubated for 30 min at 37 °C. After washing, the HRP-conjugated goat anti-mouse IgG or HRP-conjugated goat anti-rabbit IgG (Abcam, Cambridge, MA, USA) diluted at 1:20,000 in PBST was added (50 μL/well) and incubated at 37 °C for 30 min. After washing, 50 μL/well of freshly prepared TMB substrate solution was added and reacted for 10 min at 37 °C in the dark. The reaction was stopped by adding 2 M H2SO4 (50 μL/well). The absorbance was measured at 450 nm by a multifunctional plate reader (BioTek Synergy HTX, Winooski, VT, USA). The supernatant of SP2/0 cells, unrelated MAb, and unimmunized rabbit serum was used as negative control (NC). Sample is considered to be positive if OD450 of sample divided by OD450 of negative control > 2.
Immunofluorescence assay
Huh7 cells were seeded in 96-well plates and infected with SADS-CoV at a multiplicity of infection (MOI) of 0.001 for 36 h after grown in monolayers. At room temperature, the cells were fixed with 4% paraformaldehyde for 30 min and permeabilized with 0.1% Triton X-100 for 10 min. The cells were washed 3 times with PBS and incubated with the supernatants of hybridoma or SP2/0 cells (50 μL/well) at 37 °C for 1 h. After washing, the cells were incubated with a FITC-conjugated anti-mouse IgG secondary antibody (Sigma-Aldrich, St. Louis, MO, USA; 1:100, 50 μL/well) at 37 °C for 30 min. Cells were washed with PBS again, and the nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI, Sigma-Aldrich, St. Louis, MO, USA) for 10 min at room temperature in a dark. The fluorescence was observed by the fluorescent microscope (AMG EVOS F1; Advanced Microscopy Group, Mill Creek, WA, USA).
Establishment and optimization of DAS-qELISA
The purified MAb as detection antibody was labeled with horseradish peroxidase (HRP) using HRP conjugation kit according to manufacturer’s instruction (Sigma-Aldrich, St. Louis, MO, USA). To determine the optimal concentrations of capture and detection antibodies, the checkerboard titrations were used. The purified rabbit PAb as capture antibody was serially diluted (10, 5, 2.5, 1.25, 0.625, 0.3125, and 0.15625 μg/mL) in carbonated coating buffer (pH 9.6), and 50 μL was horizontally added into ELISA plates and incubated at 37 °C for 1 h. The plates were washed 4 times with PBST and then blocked with 2% (w/v) trehalose in PBST at 37 °C for 1 h. After washing with PBST, 50 μL of SADS-CoV-infected or mock-infected culture supernatants was added into each well and incubated at 37 °C for 30 min. After washing, HRP-labeled MAb was diluted at 1:1000, 1:2000, 1:4000, 1:8000, and 1:16,000 in PBST and longitudinally added to the wells (50 μL/well) and then incubated at 37 °C for 30 min. The plates were washed again and developed with TMB (50 μL/well) at 37 °C for 10 min, stopped by using 2 M H2SO4 (50 μL/well). The absorbance was measured at 450 nm. The optimal capture and detection antibody concentrations were determined by the highest positive-to-negative (P/N) ratio.
After the conditions mentioned above were determined, the optimal concentration of rabbit PAb was coated at 37 °C for 1, 2, 3, and 4 h or 4 °C for 12 h. Then, the blocking conditions were optimized as follows. The rabbit PAb-coated plates were blocked with 5% (w/v) skimmed milk, 5% (w/v) BSA, or 2% (w/v) trehalose in PBST at 37 °C for 1 h. To determine the optimal blocking time, the rabbit PAb-coated plates were blocked in the optimal blocking solution at 37 °C for 1, 2, 3, and 4 h or 4 °C for 12 h. Next, the detection antigens (SADS-CoV-infected or mock-infected culture supernatants) were incubated at 37 °C for 0.5, 1, 2, or 3 h. The incubation time of HRP-MAb was optimized at 37 °C for 0.5, 1, 1.5, or 2 h. The reactions were stopped and optimized by assessing 5, 10, 15, and 20 min, respectively. All experiments were assessed by the P/N ratio.
Determination of the cutoff value of DAS-qELISA
A total of 68 SADS-CoV-negative anal swabs were obtained from healthy piglets. The anal swabs were diluted with 1 mL PBS (0.01 M, pH 7.2), vortexed oscillation 30 s, and inactivated at 56 °C for 30 min, and then, the supernatant was collected after centrifugation at 12,000 × g for 10 min. The samples were detected by the established DAS-qELISA with the determined optimal conditions. The OD450 value of sample-to-negative (\({\varvec{S}}/{\varvec{N}}\)) ratio was calculated. Next, the cutoff value was calculated as follows: \(\overline{{\varvec{X}} }\)+3SD. \(\overline{{\varvec{X}} }\) represents the mean value of \({\varvec{S}}/{\varvec{N}}\) value of 68 negative samples, and 3SD represents three standard deviations.
Specificity, sensitivity, and reproducibility
The specificity, sensitivity, and reproducibility test was carried out as described previously (Li et al. 2021). PEDV, TGEV, and PDCoV were utilized to evaluate the specificity of DAS-qELISA. The culture supernatants of SADS-CoV-infected or mock cells were used for positive and negative control.
To evaluate the sensitivity of DAS-qELISA, the purified rN protein was diluted with PBS to 16, 8, 4, 2, 1, 0.5, 0.25, and 0 ng/mL, and the culture supernatants of SADS-CoV-infected (106.5/mL TCID50) or mock-infected were double diluted to 1:2–1:16,777,216, respectively, and the \({\varvec{S}}/{\varvec{N}}\) value was calculated.
To test intra-assay reproducibility, 6 SADS-CoV-positive anal swabs were assayed 3 times. Three batches of DAS-qELISA were used to confirm interassay reproducibility. The average value of \({\varvec{S}}/{\varvec{N}}\) (\(\overline{{\varvec{X}} }\)) and the SD value were calculated. The coefficient of variation (CV) was calculated as \((\mathbf{S}\mathbf{D}/\overline{{\varvec{X}} })\times 100\mathbf{\%}\).
Comparison of DAS-qELISA and RT-PCR
To effectively apply the DAS-qELISA prepared in this study to clinical practice tests, three 3-day-old piglets (numbered as 3, 9, and 10) were challenged with SADS-CoV (3.4 × 107 copies/piglet) by oral gavage, and two (numbered as 2 and 6) were given DMEM orally as negative control. The anal swab samples were taken for each at 0 ~ 10 d and diluted in 1 mL PBS. SADS-CoV in the anal swab samples were detected by RT-PCR and DAS-qELISA. 200 μL suspensions of the anal swab samples were extracted RNA using the RNeasy mini kit according to manufacturer’s instruction (Qiagen Sciences, Hilden, Germany). RNA was reverse transcribed into cDNA using a PrimeScriptII 1st-strand cDNA synthesis kit (TakaRa, Dalian, China). The RT-PCR specific primers were designed based on the sequence of SADS-COV N gene, forward primer: 5′-CAGTCTGTTGACATTGTTGC-3′, reverse primer: 5′-TGATTGCGAGAACGAGACTG-3′. The PCR program was as follows: 5 min at 95 °C, followed by 30 cycles of 10 s at 95 °C, 30 s at 56 °C, and 15 s at 72 °C; elongation for 5 min at 72 °C. The consistency of DAS-qELISA and RT-PCR was evaluated by coincidence rate and kappa value (\(\boldsymbol{\rm K}\)) (Landis and Koch 1977).
Results
Development of antibodies
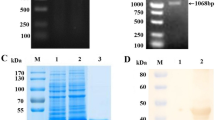
To obtain the antibodies of N protein, the purified rN was immunized with mouse and rabbit, respectively. One specific monoclonal antibody against SADS-CoV N protein (designated as 6E8) was obtained by hybridoma cell fusion technique. The reactivity of MAb 6E8 and rabbit PAb against N protein with SADS-CoV was identified by IFA. As shown in Fig. 1A, the MAb 6E8 and rabbit PAb against SADS-CoV N protein could specifically react with SADS-CoV, but not the SP/20 cell supernatant and unimmunized rabbit serum. The antibodies were purified by protein G sepharose, and the purity was determined by SDS-PAGE, which showed two clear bands with the expected molecular weight masses of heavy (about 55 kDa) and light (about 25 kDa) chains (Fig. 1B, C). By indirect ELISA, the binding titers of the purified MAb 6E8 and rabbit PAb P/N ≥ 2were determined to be 1:51,200 and 1:25,600, respectively (Fig. 1D).

Preparation and purification of the MAb and PAb against SADS-CoV N protein. A The reactivity of the MAb and PAb with SADS-CoV was identified by IFA. Cells were infected with SADS-CoV for 36 h. Cells were fixed and strained with the MAb 6E8 and rabbit PAb against SADS-CoV N protein, the SP/20 cell supernatant, and unimmunized rabbit serum as negative control. The nuclei were stained with DAPI. Purification of the MAb 6E8 (B) and rabbit PAb (C) against SADS-CoV N protein analyzed by SDS-PAGE. Lane M, protein molecular weight marker; lane 1, purified the MAb 6E8 and rabbit PAb. D Determination for the titration of the MAb 6E8 and rabbit PAb against SADS-CoV N protein by indirect ELISA. The purity MAb 6E8, rabbit PAb against SADS-CoV N protein, unrelated MAb, and unimmunized rabbit serum were diluted (1:100, 1:200, 1:400, 1:800, 1:1600, 1:3200, 1:6400, 1:12,800, 1:51,200, and 1:102,400) in PBST and then detected by indirect ELISA. The OD450 value of positive-to-negative (\(P/N\)) ratio was calculated
Establishment of the DAS-qELISA
To establish the DAS-qELISA, firstly, optimal reaction conditions of the rabbit PAb and HRP-labeled 6E8 MAb were determined by checkerboard. As shown in Fig. 2A, the capture antibody of 2.5 μg/mL and detection antibody diluted in 1:8000 were the best pair for detection the SADS-CoV.

Optimal conditions of DAS-qELISA. A Optimum concentration of the capture and detecting antibodies. B Optimum coating time and temperature. Optimum blocking buffer (C), incubation time, and temperature (D). Optimum reaction time of antigen (E) and detection antibody (F). G Optimum developing time. SADS-CoV-infected or mock-infected culture supernatants as the positive and negative antigens were detected. The OD450 value of positive-to-negative (\(P/N\)) ratio was calculated
Secondly, the time and temperature for the coating of the rabbit anti-N PAb onto the ELISA plate were optimized. The data showed that the \({\varvec{P}}/{\varvec{N}}\) ratio was higher at 4 °C for 12 h than at 37 °C for 1, 2, 3, and 4 h (Fig. 2B).
Thirdly, the blocking buffer and the optimal blocking time were evaluated. As compared with 5% BSA and 5% skimmed milk, the \({\varvec{P}}/{\varvec{N}}\) ratios had obviously increased in 2% trehalose (Fig. 2C). The effect of blocking time at 37 °C for 1, 2, and 3 h or 4 °C for 12 h is shown in Fig. 2D. It was found that the \({\varvec{P}}/{\varvec{N}}\) ratio was 10.25 at 4 °C for 12 h and higher than 8.78, 9.07, and 9.70 at 37 °C for 1, 2, and 3 h. Thus, the optimal blocking buffer was 2% trehalose, and the optimal blocking time was 4 °C for 12 h.
Fourthly, the optimal binding time of SADS-CoV antigen and detection antibody was investigated. As shown in Fig. 2E, the \({\varvec{P}}/{\varvec{N}}\) ratios (9.28, 11.60, 11.76, and 11.79) had gradually increased with antigen incubation time increased (0.5–3 h). However, there was no significant difference when the incubation time was 1, 2, and 3 h. Thus, 1 h was selected as the optimal binding time of SADS-CoV antigen. As can be seen from Fig. 2F, the \({\varvec{P}}/{\varvec{N}}\) ratios had gradually decreased with detection antibody incubation time increased (0.5–3 h), so the optimal reaction time of detection antibody was 0.5 h.
Finally, effects of the developing time were observed, and the experimental results suggested that the \({\varvec{P}}/{\varvec{N}}\) ratio was the highest at 10 min (Fig. 2G).
Determination of the cutoff value of DAS-qELISA
A total of 68 SADS-CoV-negative anal swabs were used to determine the cutoff value (Fig. 3). The mean (\(\overline{{\varvec{X}} }\)) was 1.068 and the standard deviation (SD) was 0.302; thus, the critical value (\(\overline{{\varvec{X}} }\) + 3SD) was 1.973. Therefore, the sample was judged positive when \({\varvec{S}}/{\varvec{N}}>1.973\), while \({\varvec{S}}/{\varvec{N}}\le 1.973\) was judged to be negative.

Determine the cutoff value. The optimized DAS-qELISA was employed to assay 68 SADS-CoV-negative anal swabs. The OD450 value of sample-to-negative (\(S/N\)) ratio was calculated. The cutoff value was calculated as follows: \(\overline{{\varvec{X}} }\) + 3SD
Specificity of DAS-qELISA
The clinical and pathological symptoms of SADS-CoV are similar to those of PEDV, TGEV, and PDCoV, making it difficult to differentiate among them. To determine the specificity of the DAS-qELISA, the cell culture supernatants of SADS-CoV, PEDV, TGEV, and PDCoV were tested. The test results showed that the \({\varvec{S}}/{\varvec{N}}\) value was 9.796 for SADS-CoV-infected cell culture, whereas it was lower than the cutoff value (1.973) for other porcine coronaviruses (Fig. 4). These results suggested that the DAS-qELISA for the detection of SADS-CoV had good specificity and no cross-reactivity with PEDV, TGEV, and PDCoV.

Specificity of the DAS-qELISA for SADS-CoV. The cell culture supernatants of SADS-CoV, PEDV, TGEV, and PDCoV were detected by DAS-qELISA. The \(S/N\) ratio was calculated
Sensitivity of DAS-qELISA
The purified rN (16 ng/mL) and the cell culture supernatants of SADS-CoV (106.5TCID50/mL) were twofold diluted serially in an ELISA plate to assay the sensitivity of the established DAS-qELISA. The line standard curve was obtained between the \({\varvec{S}}/{\varvec{N}}\) and the concentration of the purified rN protein, and the linear equation was \({\varvec{Y}}=0.4927{\varvec{X}}+1.5433\), R2 = 0.9948 (Fig. 5A). A good linear relationship of this method was ranged from 0.25 to 16 ng/mL, and the sensitivity was up to 1 ng/mL. For SADS-CoV, it was diluted with 1:262,144 (101.08 TCID50/mL); the \({\varvec{S}}/{\varvec{N}}\) ratio was 2.02 which was above the cutoff value, suggesting that the detection limit was as 101.08 TCID50/mL (Fig. 5B).

Sensitivity of the DAS-qELISA. A The standard curve of the DAS-qELISA. The purified rN protein and unrelated protein were twofold serially diluted (16, 8, 4, 2, 1, 0.5, 0.25, and 0 ng/mL) in PBS and assayed by DAS-qELISA. The OD450 value of rN-to-unrelated protein (\(S/N\)) ratio was calculated. The standard curve was calculated using a linear relationship between the \(S/N\) values and concentrations. B Sensitivity of the DAS-qELISA for SADS-CoV. SADS-CoV-infected or mock-infected culture supernatants were twofold diluted in PBS and detected by DAS-qELISA. The \(S/N\) ratio was calculated
Reproducibility of DAS-qELISA
The intra-assay reproducibility was calculated by analyzing 6 positive anal swabs for SADS-CoV for a total of 3 times in a single batch. Moreover, three batches of the DAS-qELISA were analyzed to confirm interassay reproducibility. As shown in Table 1, the intra-assay CV was < 10% and the interassay CV was < 5%, suggesting good reproducibility of the DAS-qELISA.
Sample analysis
A total of 55 anal swab samples were collected from five 3-day-old piglets after challenging with DMEM or SADS-CoV (0 ~ 10 d). 23 of the 55 anal swab samples were determined to be positive by using DAS-qELISA (Table 2), whereas 24 samples were positive by RT-PCR (the data are not shown). As shown in Table 3, in the total 33 anal swab samples in SADS-CoV challenging groups, 23 were positive and 9 were negative, as determined using these two methods. The accuracy of these two detection methods was 93.93%. In addition, the kappa value was 0.85, suggesting a high consistency between the DAS-qELISA and RT-PCR methods.
Discussion
SADS-CoV as an emerging virus, jumping from bat to pigs, caused a huge number of piglet infections and deaths in China (Sun et al. 2022; Wang et al. 2018b; Zhou et al. 2019, 2018b). So far, there is no vaccine or clinically approved drugs to prevent and cure SADS-CoV, nor commercial diagnostic kits. Therefore, to prevent SADS-CoV infection, it is very important to develop a rapid and reliable detection for SADS-CoV. DAS-qELISA is a traditional detection technology with high specificity and sensitivity and simple operation that can precisely quantify antigens and has been broadly applied for pathogen diagnosis. Previous studies have reported the use of similar assays for the detection of canine distemper virus (Zhang et al. 2020), hepatitis C virus (Qin et al. 2020), PEDV (Fan et al. 2020), PDCoV (Wang et al. 2021), koi herpesvirus (Li et al. 2021), Crimean-Congo hemorrhagic fever virus (Shrivastava et al. 2021), and so on. To date, there is no report of a DAS-qELISA for detection of SADS-CoV antigens.
SADS-CoV contains four structural proteins: the spike surface glycoprotein (S), membrane glycoprotein (M), envelope protein (E), and nucleocapsid protein (N) (Scarpa et al. 2021). The N protein has multiple functions in pathogenesis, viral replication or transcription, and viral evasion of immune system (Cong et al. 2020; Liu et al. 2021; McBride et al. 2014; Zhou et al. 2021). Based on the characters of the high immunogenicity, conservation, and abundantly expressed during viral infection, the N protein is an ideal antigen for development of diagnostic methods. In this study, SADS-CoV N protein is highly expressed by E. coli and purified by Ni-chelating affinity chromatography. The purified rN as an antigen immunized mouse and rabbit to obtain MAbs and PAbs. The sensitivity and specificity of DAS-qELISA depend on the characters of antibodies. The MAb 6E8 and rabbit PAbs have a good reaction with SADS-CoV. As the capability of PAbs to recognize the multiple epitopes, the rabbit PAbs against SADS-CoV N protein were selected as capture antibodies which can increase the sensitivity of the assay. As for the detection antibodies, the HPR-labeled MAb 6E8 against SADS-CoV N protein were used, which both ensured the specificity and raised the sensitivity of detection assay. The date showed that the detection limit of the developed DAS-qELISA assay was 1 ng/mL with the purified rN protein and 101.08 TCID50/mL with SADS-CoV culture supernatants, respectively.
Diseases brought by porcine enteric coronaviruses (TGEV, PEDV, SADS-CoV, and PDCoV) have similar clinical symptoms and pathogenesis and cannot be differentiated without laboratory diagnosis. Most commonly, porcine enteric coronaviruses exist coinfection and often aggravate the disease process (Chen et al. 2018; Zhang et al. 2013). The developed DAS-qELISA in this study could differentiate SADS-CoV from PEDV, TGEV, and PDCoV, no cross-reactivity among them, suggesting that the DAS-qELISA for SADS-CoV was highly specific. The outbreak of SADS-CoV was sporadic, and the collection of clinical samples was limited. Therefore, the piglets were challenged with SADS-CoV by oral gavage to monitor the detoxification. The data showed that the virions could be detected 1 ~ 8 d after challenging with SADS-CoV. Furthermore, the results of anal swab sample detection reveal a high consistency between DAS-qELISA and RT-PCR. To the best of our knowledge, this is the first report of the DAS-qELISA for detection of SADS-CoV antigens; our data showed that this method has a high specificity, sensitivity, and reproducibility. Thus, the established DAS-qELISA for detection of SADS-CoV antigens has a potential application value in evaluating the viral infection in clinical samples. The surveillance of SADS-CoV epidemic situation in pig herds has important public health significance, so we will timely collect clinical diarrhea samples to monitor the infection of SADS-CoV in the future.
Data Availability
Data is not applicable.
References
Chen Q, Wang L, Zheng Y, Zhang J, Guo B, Yoon KJ, Gauger PC, Harmon KM, Main RG, Li G (2018) Metagenomic analysis of the RNA fraction of the fecal virome indicates high diversity in pigs infected by porcine endemic diarrhea virus in the United States. Virology Journal 15(1):95. https://doi.org/10.1186/s12985-018-1001-z
Cong Y, Ulasli M, Schepers H, Mauthe M, V’kovski P, Kriegenburg F, Thiel V, de Haan CAM, Reggiori F, (2020) Nucleocapsid protein recruitment to peplication-transcription complexes plays a crucial role in coronaviral life cycle. J Virol 94(4):e01925-19. https://doi.org/10.1128/JVI.01925-19
Cui J, Li F, Shi ZL (2019) Origin and evolution of pathogenic coronaviruses. Nat Rev Microbiol 17(3):181–192. https://doi.org/10.1038/s41579-018-0118-9
Fan B, Sun J, Zhu L, Zhou J, Zhao Y, Yu Z, Sun B, Guo R, He K, Li B (2020) Development of a novel double antibody sandwich quantitative enzyme-linked immunosorbent assay for detection of porcine epidemic diarrhea virus antigen. Frontiers Vet Sci 7:540248. https://doi.org/10.3389/fvets.2020.540248
Gong L, Li J, Zhou Q, Xu Z, Chen L, Zhang Y, Xue C, Wen Z (2017) Cao Y (2017) A new bat-HKU2-like coronavirus in swine, China. Emerg Infect Dis 23(9):1607–1609. https://doi.org/10.3201/eid2309.170915
Huang X, Chen J, Yao G, Guo Q, Wang J, Liu G (2019) A TaqMan-probe-based multiplex real-time RT-qPCR for simultaneous detection of porcine enteric coronaviruses. Appl Microbiol Biotechnol 103(12):4943–4952. https://doi.org/10.1007/s00253-019-09835-7
Köhler G, Milstein C (1975) Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 256(5517):495–497. https://doi.org/10.1038/256495a0
Landis JR, Koch GG (1977) The measurement of observer agreement for categorical data. Biometrics 33(1):159–174
Li Y, Wang Q, Hu F, Wang Y, Bergmann SM, Zeng W, Yin J, Shi C (2021) Development of a double-antibody sandwich enzyme-linked immunosorbent assay (DAS-ELISA) for the detection of KHV. J Fish Dis 44(7):913–921. https://doi.org/10.1111/jfd.13351
Liu Y, Liang QZ, Lu W, Yang YL, Chen R, Huang YW, Wang B (2021) A comparative analysis of coronavirus nucleocapsid (N) proteins reveals the SADS-CoV N protein antagonizes IFN-β production by inducing ubiquitination of RIG-I. Frontiers in immunology 12:688758. https://doi.org/10.3389/fimmu.2021.688758
Liu J, Tao D, Chen X, Shen L, Zhu L, Xu B, Liu H, Zhao S, Li X, Liu X, Xie S, Niu L (2022) Detection of Four Porcine Enteric Coronaviruses Using CRISPR-Cas12a Combined with Multiplex Reverse Transcriptase Loop-Mediated Isothermal Amplification Assay. Viruses 14(4):833. https://doi.org/10.3390/v14040833
Ma L, Zeng F, Cong F, Huang B, Huang R, Ma J, Guo P (2019) Development of a SYBR green-based real-time RT-PCR assay for rapid detection of the emerging swine acute diarrhea syndrome coronavirus. J Virol Methods 265:66–70. https://doi.org/10.1016/j.jviromet.2018.12.010
McBride R, van Zyl M, Fielding BC (2014) The coronavirus nucleocapsid is a multifunctional protein. Viruses 6(8):2991–3018. https://doi.org/10.3390/v6082991
Pan Y, Tian X, Qin P, Wang B, Zhao P, Yang YL, Wang L, Wang D, Song Y, Zhang X, Huang YW (2017) Discovery of a novel swine enteric alphacoronavirus (SeACoV) in southern China. Vet Microbiol 211:15–21. https://doi.org/10.1016/j.vetmic.2017.09.020
Pan Z, Lu J, Wang N, He WT, Zhang L, Zhao W, Su S (2020) Development of a TaqMan-probe-based multiplex real-time PCR for the simultaneous detection of emerging and reemerging swine coronaviruses. Virulence 11(1):707–718. https://doi.org/10.1080/21505594.2020.1771980
Peng P, Gao Y, Zhou Q, Jiang T, Zheng S, Huang M, Xue C, Cao Y, Xu Z (2021) Development of an indirect ELISA for detecting swine acute diarrhoea syndrome coronavirus IgG antibodies based on a recombinant spike protein. Transbound Emerg Dis. https://doi.org/10.1111/tbed.14196
Qin YJ, Sha RC, Feng YC, Huang YC (2020) Comparison of double antigen sandwich and indirect enzyme-linked immunosorbent assay for the diagnosis of hepatitis C virus antibodies. J Clin Lab Anal 34(11):23481. https://doi.org/10.1002/jcla.23481
Scarpa F, Sanna D, Azzena I, Cossu P, Giovanetti M, Benvenuto D, Coradduzza E, Alexiev I, Casu M, Fiori PL, Ciccozzi M (2021) Update on the phylodynamics of SADS-CoV. Life (Basel). 11(8):820. https://doi.org/10.3390/life11080820
Shrivastava N, Kumar JS, Yadav P, Shete AM, Jain R, Shrivastava A, Dash PK (2021) Development of double antibody sandwich ELISA as potential diagnostic tool for rapid detection of Crimean-Congo hemorrhagic fever virus. Sci Rep 11(1):14699. https://doi.org/10.1038/s41598-021-93319-0
Sun Y, Xing J, Xu ZY, Gao H, Xu SJ, Liu J, Zhu DH, Guo YF, Yang BS, Chen XN, Zheng ZZ, Wang H, Lang G, Holmes EC (2021) Zhang GH (2022) Re-emergence of Severe Acute Diarrhea Syndrome Coronavirus (SADS-CoV) in Guangxi, China. J Infect 85(5):e130–e133. https://doi.org/10.1016/j.jinf.2022.08.020
Wang H, Cong F, Zeng F, Lian Y, Liu X, Luo M, Guo P, Ma J (2018) Development of a real time reverse transcription loop-mediated isothermal amplification method (RT-LAMP) for detection of a novel swine acute diarrhea syndrome coronavirus (SADS-CoV). J Virol Methods 260:45–48. https://doi.org/10.1016/j.jviromet.2018.06.010
Wang L, Su S, Bi Y, Wong G, Gao GF (2018) Bat-origin coronaviruses expand their host range to pigs. Trends Microbiol 26(6):466–470. https://doi.org/10.1016/j.tim.2018.03.001
Wang W, Li J, Fan B, Zhang X, Guo R, Zhao Y, Zhou J, Zhou J, Sun D, Li B (2021) Development of a novel double antibody sandwich ELISA for quantitative detection of porcine deltacoronavirus antigen. Viruses 13(12):2403. https://doi.org/10.3390/v13122403
Xu Z, Zhang Y, Gong L, Huang L, Lin Y, Qin J, Du Y, Zhou Q, Xue C, Cao Y (2019) Isolation and characterization of a highly pathogenic strain of porcine enteric alphacoronavirus causing watery diarrhoea and high mortality in newborn piglets. Transbound Emerg Dis 66(1):119–130. https://doi.org/10.1111/tbed.12992
Yang YL, Liang QZ, Xu SY, Mazing E, Xu GH, Peng L, Qin P, Wang B, Huang YW (2019a) Characterization of a novel bat-HKU2-like swine enteric alphacoronavirus (SeACoV) infection in cultured cells and development of a SeACoV infectious clone. Virology 536:110–118. https://doi.org/10.1016/j.virol.2019.08.006
Yang YL, Qin P, Wang B, Liu Y, Xu GH, Peng L, Zhou J, Zhu SJ, Huang YW (2019b) Broad cross-species infection of cultured cells by bat HKU2-related swine acute diarrhea syndrome coronavirus and identification of its replication in murine dendritic cells in vivo highlight its potential for diverse interspecies transmission. J Virol 93(24):e01448-19–19. https://doi.org/10.1128/JVI.01448-19
Zhang Q, Hu R, Tang X, Wu C, He Q, Zhao Z, Chen H, Wu B (2013) Occurrence and investigation of enteric viral infections in pigs with diarrhea in China. Adv Virol 158(8):1631–1636. https://doi.org/10.1007/s00705-013-1659-x
Zhang Y, Xu G, Zhang L, Zhao J, Ji P, Li Y, Liu B, Zhang J, Zhao Q, Sun Y, Zhou EM (2020) Development of a double monoclonal antibody-based sandwich enzyme-linked immunosorbent assay for detecting canine distemper virus. Appl Microbiol Biotechnol 104(24):10725–10735. https://doi.org/10.1007/s00253-020-10997-y
Zhang Z, Wang N, Liu X, Lv J, Jing H, Yuan X, Chen D, Lin X, Wu S (2022) A novel reverse transcription droplet digital PCR assay for the sensitive detection of combing severe acute respiratory syndrome coronavirus 2 with swine acute diarrhea syndrome coronavirus. J AOAC Int. https://doi.org/10.1093/jaoacint/qsac039
Zhou L, Sun Y, Wu JL, Mai KJ, Chen GH, Wu ZX, Bai Y, Li D, Zhou ZH, Cheng J, Wu RT, Zhang XB, Ma JY (2018) Development of a TaqMan-based real-time RT-PCR assay for the detection of SADS-CoV associated with severe diarrhea disease in pigs. J Virol Methods 255:66–70. https://doi.org/10.1016/j.jviromet.2018.02.002
Zhou P, Fan H, Lan T, Yang XL, Shi WF, Zhang W, Zhu Y, Zhang YW, Xie QM, Mani S, Zheng XS, Li B, Li JM, Guo H, Pei GQ, An XP, Chen JW, Zhou L, Mai KJ, Wu ZX, Li D, Anderson DE, Zhang LB, Li SY, Mi ZQ, He TT, Cong F, Guo PJ, Huang R, Luo Y, Liu XL, Chen J, Huang Y, Sun Q, Zhang XL, Wang YY, Xing SZ, Chen YS, Sun Y, Li J, Daszak P, Wang LF, Shi ZL, Tong YG, Ma JY (2018) Fatal swine acute diarrhoea syndrome caused by an HKU2-related coronavirus of bat origin. Nature 556(7700):255–258. https://doi.org/10.1038/s41586-018-0010-9
Zhou L, Li QN, Su JN, Chen GH, Wu ZX, Luo Y, Wu RT, Sun Y, Lan T, Ma JY (2019) The re-emerging of SADS-CoV infection in pig herds in Southern China. Transbound Emerg Dis 66(5):2180–2183. https://doi.org/10.1111/tbed.13270
Zhou L, Chen Y, Fang X, Liu Y, Du M, Lu X, Li Q, Sun Y, Ma J, Lan T (2020) Microfluidic-RT-LAMP chip for the point-of-care detection of emerging and re-emerging enteric coronaviruses in swine. Anal Chim Acta 1125:57–65. https://doi.org/10.1016/j.aca.2020.05.034
Zhou Z, Sun Y, Xu J, Tang X, Zhou L, Li Q, Lan T, Ma J (2021) Swine acute diarrhea syndrome coronavirus nucleocapsid protein antagonizes interferon-β production via blocking the interaction between TRAF3 and TBK1. Frontiers Immunol 12:573078. https://doi.org/10.3389/fimmu.2021.573078
Funding
This work was supported by the Key Research and Development Program of Sichuan Province (2022YFN0008), the Sichuan Province Fund for Distinguished Young Scholars (21JCQN0175 to Q.W.), the Elite Youth Program of the Chinese Academy of Agricultural Sciences (Q.W., http://www.caas.cn/en/scientists/talent_program), and the Chinese Academy of Agricultural Science and Technology Innovation Project (ASTIP2021-34-IUA-07, ASTIP2022-34-IUA-07, CAAS-ASTIP-2021-LVRI).
Author information
Authors and Affiliations
Contributions
Liyan Cao: conceptualization, methodology, software, investigation, formal analysis, writing—original draft; Xiangyu Kong: data curation, methodology, software; Yu Zhang: methodology, software; Xuepeng Suo: data curation; Xiangtong Li: visualization, data curation, methodology, software; Yueyue Duan: data curation; Cong Yuan: data curation; Haixue Zheng: funding acquisition, resources; Qi Wang: conceptualization, funding acquisition, resources, supervision, writing—review and editing.
Corresponding authors
Ethics declarations
Ethics approval
All animal experimental procedures have been reviewed and approved by the Animal Care and Use Committee of Lanzhou Veterinary Research Institute of the Chinese Academy of Agricultural Sciences (approval ID: SYXK(Gan) 2015–0003).
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Cao, L., Kong, X., Zhang, Y. et al. Development of a novel double-antibody sandwich quantitative ELISA for detecting SADS-CoV infection. Appl Microbiol Biotechnol 107, 2413–2422 (2023). https://doi.org/10.1007/s00253-023-12432-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-023-12432-4