
Abstract
Single-domain antibody (sdAb) or nanobody possesses specific features non-accessible for conventional antibodies that make them suitable for research and biotechnological applications. Porcine epidemic diarrhea virus (PEDV) causes lethal diarrhea in piglets, resulting in great economic losses all over the world. To detect and isolate PEDV rapidly and accurately is important for the control and further research of the clinical PEDV strains. In this study, four sdAb fragments (sdAb-Mc19/29/30/37) targeting the membrane (M) protein of PEDV were selected from sdAb library that was constructed through M protein-immunized Camelus bactrianus. The selected sdAb-Mcs were solubly expressed in Escherichia coli. The functional characteristics analysis revealed that the recombinant sdAb-Mcs have excellent binding activity and specificity to M protein but have no neutralizing activity to PEDV. For further application, sdAb-Mc37 was conjugated with quantum dots to synthesize a nanoprobe for imaging PEDV in vero cells. The observed fluorescence in vero cells clearly reflects that PEDV virions can be reliably recognized and labeled by the nanoprobe. Furthermore, the sdAb-Mc29 was conjugated with superparamagnetic nanobeads to construct immunomagnetic nanobeads (IMNBs) used to isolate PEDV. One PEDV strain was successfully isolated from clinical fecal sample, suggesting IMNBs as a novel and efficient tool suitable for PEDV isolation from clinical samples. This study provided a novel application and substantiated the suitability of sdAb as a specific binder for the isolation of viruses.
Similar content being viewed by others


Introduction
Single-domain antibody (sdAb), also called nanobodies, is variable domain of the heavy-chain antibody (HcAb) found in the serum of camels, llamas, and sharks (Hamers-Casterman et al. 1993). HcAb devoids of light chains naturally, but keeps the function of conventional antibodies depend on three complementarity determining regions in variable domain. The sdAb displays very significant characteristics including a small molecular weight of 15 kDa, ease of expression in Escherichia coli (E. coli) and purification, high expression yields, high solubility and thermostability, and stability at extreme pH conditions that are superior to conventional antibodies (Anderson et al. 2017; Dumoulin et al. 2002). In addition, sdAb is easily selected and cloned by selection systems such as phage, yeast, or ribosomal display (Harmsen and De Haard 2007; Muyldermans et al. 2009). These attributes make sdAbs suitable for many medical, biotechnological, diagnostic, and therapeutic applications (Arbabi-Ghahroudi 2017; Bannas et al. 2017; Beghein and Gettemans 2017; Gonzalez-Sapienza et al. 2017).
For the applications of antibodies in biotechnology or diagnostics, indicative markers such as horseradish peroxidase (HRP), quantum dots (QDs), and nanomaterial, were conjugated commonly for different purposes. QDs have emerged as interesting fluorescent particles due to their high fluorescence quantum yield, size-tunable emission band, and photostability (Shirasaki et al. 2013). The conjugate of QDs and antibodies as a powerful bioprobe has been used widely in immunoassays t(Pathak et al. 2007; Zhang et al. 2016).
Nanosized superparamagnetic beads functionalized with specific surface are versatile materials used for various applications in chemical assay, biosensors, bioseparation systems, and biomedical studies (Rampini et al. 2015; Rampini et al. 2016; Ran et al. 2014). To provide specificity, magnet beads can be decorated with ligands, such as antibodies, aptamers, and peptides, qualifying them as useful tools for the purification, detection, and separation of biological analytes from complex samples (Muzard et al. 2012).
To combine the properties of sdAb, QDs, and magnet beads to detect and separate of virus, we took porcine epidemic diarrhea virus (PEDV) membrane protein (M) as example. PEDV, a member of the genus Coronavirus, family Coronaviridae, is the causative agent of porcine epidemic diarrhea (International Committee on Taxonomy of Viruses. and King 2012), the most economically important disease affecting the global swine industry today (Van Diep et al. 2018). PEDV encodes four major structural proteins, membrane (M) (27–32 kDa), envelope (E) (7 kDa), spike (S) (180–220 kDa), and nucleocapsid (N) (55–58 kDa) (Utiger et al. 1995; Yeo et al. 2003). The M protein is the most abundant of structural proteins and is involved in the assembly of viral nucleocapsid and membrane. In addition, the nucleotide sequence of the M gene is highly conserved among different PEDV strains but shows little homology with other coronaviruses (Kim and Chae 2000). Thus, the M protein has been used as a target antigen for PEDV detection (Ren et al. 2011; Shenyang et al. 2007).
In this study, sdAbs specific to PEDV M protein were identified and selected from the immunized Camelus bactrianus (C. bactrianus) sdAb library by phage display technique. The selected sdAbs were characterized and conjugated with CdSe/ZnS core-shell QDs, a highly specific, newly developed nanoprobe, to achieve targeting PEDV in vero cells. In addition, an sdAb was used for functionalization of magnetic nanobeads to provide an efficient tool for PEDV isolation from complex clinical samples.
Materials and methods
C. bactrianus immunization and sdAb library construction
PEDV strain CV777, a commercial attenuated vaccine strain, preserved in Lanzhou veterinary research institute CAAS, was used as a standard strain for the research. Truncated M protein (GenBank: AF353511) of CV777 was expressed in E. coli with histidine tag (His-tag, M-His) and glutathione S-transferase tag (GST-tag, M-GST). A 1-year-old male C. bactrianus was immunized with 800 μg M-His emulsified in 206 oil adjuvant (SEPPIC, France). On days 21, 35, and 49 after the first immunization, the camel was injected with 500 μg emulsified M antigen to boost its immunity. Serum samples were collected from the camel after each immunization and used for monitoring humoral immune response by an indirect enzyme-linked immunosorbent assay (ELISA) based on M-GST. Pre-immune serum was collected as negative control. Peripheral blood of 50 mL was collected in EDTA-coated tubes from the jugular vein when the camel exhibited strong humoral immune response. The lymphocytes were isolated with lymphoprep (Solarbio®, Beijing, China) from the blood and stored at − 80 °C until further use.
Total RNA was extracted by the TRIzol (Invitrogen, USA) from approximately 107 peripheral blood lymphocytes (PBLCs). First-strand cDNA was synthesized using the superscript II reverse transcriptase (Invitrogen, USA) with oligodT (18) primer. Camel antibody encoding cDNA was specifically amplified and sdAb library was constructed as described previously (Yang et al. 2014; Yin et al. 2013).
Library panning and selection of M protein specific sdAb fragments
The sdAb library was subjected to four rounds of panning on microtiter wells (Sigma, USA) coated with M-GST. M-GST was coated as follows: 50, 30, 15, and 5 μg/mL for rounds 1, 2, 3, and 4, respectively. Blank wells were maintained during panning by coating wells with phosphate-bufferered saline (PBS). Wells were blocked with block buffer (100 μL, 1% w/v casein in PBS). After phage incubation for 2 h at 37 °C, the antigen and control wells were washed 10 times with PBST (0.1% Tween-20 in PBS). Washing stringency was increased according to the concentration of Tween-20 in PBST with 0.2, 0.3, and 0.4% for rounds 2, 3, and 4 of panning, respectively. Bound phages were eluted by adding 100 μL of freshly prepared 100 mM HCl-Glycin (pH 2.2). The eluent was neutralized after incubation for 30 min by adding 100 μL of 1.0 M Tris-HCl (pH 9.1).
Enrichment factors were calculated on the basis of the input and output phages after each panning round. Individual clones were randomly selected from the fourth rounds of panning and were subjected to single-phage ELISA to obtain clone target to M-GST (Yang et al. 2014). Briefly, phage clones were added to M-GST-coated microtiter plate wells for 2-h incubation at 37 °C. After six times washing with PBST, rabbit anti-M13 IgG (1:10,000) was added and for another 1-h incubation at 37 °C. The phages bound to M protein were detected by the addition of HRP-conjugated goat anti-rabbit IgG. The color reaction was conducted by the addition of o-phenylenediamine dihydrochloride substrate solution and stopped by 2 M H2SO4. The absorbance was measured in the microplate reader at 490 nm. GST coated and blank wells were used as control. Antigen-reactive clones were sequenced and analyzed for the selection of unique clones.
Expression of sdAb fragments
The DNA sequences of sdAb fragments (named sdAb-Mc19/29/30/37) were subcloned into the expression vector PET-32a+ with 6×His tag. The expression vector was then transformed into the E. coli BL21-Codon-Plus (DE3)-RIL strain. The E. coli cells containing recombinant 32a-sdAb plasmids were cultured in luria bertani medium (containing 34 μg/mL chloramphenicol and 100 μg/mL ampicillin) and incubated at 37 °C until OD600 = 0.6–0.8. Then, the culture was induced using isopropy β-D-thiogalactopyranoside at a final concentration of 0.5 mM and further incubated for 20 h at 18 °C with shaking at 220 rpm/min.
The cells were harvested by 7000×g centrifugation for 10 min. The cell pellet was resuspended in 50 mM Tris-HCl buffer (pH 7.4) and disrupted through ultrasonication on ice-bath. The supernatant was collected after centrifuging at 12,000×g for 30 min at 4 °C and transferred to a His-tag protein purification column (containing Ni2+ affinity resins, Qiagen, Germany) according to the instructions. The target protein was identified by 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and Western blot by using a HRP conjugate anti-His mouse monoclonal antibody (Kangwei, China). The concentration of purified sdAb-Mc19/29/30/37 was determined by using Nanodrop2000 (NAX10DROP2000) and stored at 4 °C for further analysis.
Binding activity
The binding activity of recombinant sdAb-Mcs with M-GST was determined by an indirect ELISA. The microplate wells were coated with M-GST at concentration of 10 μg/mL and blocked with blocking buffer (2% bull serum albumin in PBS, pH 7.4). Then, serial dilutions of sdAb-Mcs (10, 5, 2.5, 1.25, 0.63, 0.32, 0.16, and 0 μg/mL) were added to the wells and incubated for 1 h at 37 °C. A recombinant sdAb (psdAb-c1) specific for porcine circovirus type 2 was used as negative control (Yang et al. 2014). After five times washing, the binding sdAb-Mcs were detected by the HRP conjugate anti-His mouse monoclonal antibody and tetramethyl benzidine (TMB). The absorbance was measured in a microplate reader at 450 nm (Bio-Rad, USA). Meanwhile, the binding activity of recombinant sdAb-Mcs with M-GST was detected by Western blot. M-GST was transferred to a PVDF membrane and incubated with sdAb-Mcs. Polyclonal serum against PEDV obtained from PEDV-free and -infected pig was used as positive and negative control.
The binding activity of sdAb-Mcs with PEDV was analyzed using transmission electron microscope (TEM). sdAb-Mcs were combined with Ni2+ affinity resins (Qiagen, Germany) and incubated with supernatant of PEDV strain CV777-infected Vero cells at 37 °C for 1 h. Then, resin-sdAb-virus composites were centrifuged at 2000g for 5 min and washing with Tris-HCl buffer (pH 7.4) for five times. The sdAb-virus composites were eluted with 50 mM Tris-HCl buffer containing 100 mM imidazole (pH 7.4). After centrifuging at 2000g for 5 min, the supernatant was observed with TEM.
Specificity
The specificity of recombinant sdAb-Mcs combined with PEDV M protein was examined and evaluated through an indirect ELISA. The recombinant structural proteins of several viruses, which was common pathogens of some pig diseases and available in our lab, such as classical swine fever (CSF) E2 protein, porcine circovirus type 2 (PCV2) Cap protein, porcine reproductive and respiratory syndrome virus (PRRSV) GP5 protein (Yang et al. 2015), and foot-and-mouth disease virus (FMDV) VP1 protein (Guo et al. 2013) were used for the evaluation. M protein was used as positive control. Recombinant psdAb-c1 was used as negative control. The recombinant sdAb-Mcs were added into the wells of the microplate coated with the structural proteins of several viruses (10 μg/mL) and incubated for 1 h at 37 °C. After four times washing with PBST, the binding sdAb-Mcs was detected by the HRP conjugate anti-His mouse monoclonal antibody and TMB substrate.
Neutralizing activity
The purified sdAb-Mcs were heat-inactivated (30 min, 56 °C) and used in a microtiter neutralization assay on vero cells. sdAb-Mcs, at final concentrations of 10, 20, and 100 μg/mL, were incubated with 100× tissue culture infectious dose 50 (TCID50) of CV777 at 37 °C for 1 h, followed by infection of monolayers of vero cells in 96-well plates for 72 h. Thereafter, the cells were examined for PEDV-specific cytopathic effect and the neutralization activity of sdAb-Mcs was calculated by comparing the changes of TCID50 value.
QDs-605 labeled sdAb-Mc37 as probes imaging PEDV in vero cells
Quantum dots (QD605nm, ZnS-capped CdSe), modified with thioglycolic acid, were activated and conjugated with sdAb-Mc37 to obtain the QDs-sdAb-Mc37 probe according to a previously described protocol (Wang et al. 2015; Yin et al. 2013). The probes were used at a direct immunofluorescence assay for the imaging of PEDV in vero cells. Vero cells (CCL-81) were seeded on 6-well plates and infected when cells reached 80% confluency with CV777 at 10 times of 50% tissue culture infectious dose (TCID50). After 1 h of incubation, the supernatant was removed, and the monolayer was washed thrice with fresh Dulbecco’s modified Eagle medium (DMEM). Then, the cells were incubated in DMEM containing 1% fetal bovine serum at 37 °C with 5% CO2. At 24 h post-infection (h.p.i.), the cells were fixed with 4% paraformaldehyde (Solarbio®, Beijing, China) for 10 min and permeabilized with 0.1% TritonX-100 in PBS (Sigma, USA) for 20 min at 37 °C. QDs-sdAb-Mc37 probes were diluted in PBS (1:1000) and incubated with the fixed cells overnight at 4 °C at gentle shaking. After five times washing with PBST, the cell nuclei were stained with 4, 6-diamidino-2-phenylindole (DAPI) solution for 5 min at room temperature and then washed five times with PBST, at 5 min each time. Stained cells were visualized using an inverted fluorescence microscope at wavelength of 605 nm.
In the parallel assay, a quantitative real-time polymerase chain reaction (qRT-PCR) and an indirect immunofluorescence assay based on PEDV antibody positive porcine serum and fluorescein isothiocyanate (FITC) conjugate anti-pig antibody were performed on the PEDV-free and -infected vero cells. For indirect immunofluorescence assay, the fixed cells were stained using PEDV antibody positive porcine serum and incubated at 37 °C for 1 h. After five times washing with Tris-Buffered Saline and Tween 20 (TBST), FITC conjugated goat anti-pig IgG (BSZH, USA) was diluted in Tris-Buffered Saline (1:1000) and added. After another 1-h incubation, cell nucleus were stained with DAPI (1 μg/mL) for 5 min at room temperature. After five times washing with TBST, cells were visualized using an inverted fluorescence microscope at wavelength of 488 nm.
For qRT-PCR, cells were collected at 6, 12, 24, 36, and 48 hpi with PEDV. Total RNA was extracted using RNA extraction kit (Qiagen, Germany), and cDNA was synthesized by using cDNA reverse transcription kit (ThermoFisher, USA) with oligo dt primer. The cDNA was then subjected to real-time PCR with primer pairs specific for PEDV and β-actin (Cao et al. 2015). The β-actin gene served as the endogenous control. The relative quantities of RNA were evaluated based on 2−ΔΔCt method compared with 6 hpi results. Each sample was run in triplicate.
Binding activity of immunomagnetic nanobeads
Immunomagnetic nanobeads (IMNBs) were produced by conjugation of sdAb-Mc29 with superparamagnetic nanobeads (200 nm in radius, 3 mg/mL, purchased from Ademtech SA, Pessac, France) that was activated by 1-ethyl-3-[3-dimethyl aminopropyl] carbodiimide hydrochloride and N-hydroxy succinimide. The IMNBs were blocked with 0.1% (m/v) bovine serum albumin and resuspended in 1 mL of PBS for further use.
The binding activity of IMNBs with M protein was detected by SDS-PAGE and Western blot. IMNBs (10 μL, 3 mg/mL) were incubated with recombinant M protein (50 μL, 2 mg/mL) at 37 °C for 1 h in a tube. Then, IMNBs-M composites were separated with a magnetic scaffold to remove the supernatant and washed 5 times with PBST (0.5% Tween-20 in PBS, pH 7.2). The separated IMNBs-M composites were detected by SDS-PAGE and Western blot based on PEDV positive porcine serum.
The binding activity of IMNBs with PEDV particles was directly detected using a TEM. IMNBs (10 μL, 3 mg/mL) were incubated with supernatant of PEDV infected vero cells (1 mL) at 37 °C for 1 h in a tube. IMNBs-virus composites were separated with magnetic scaffold. After 5 times washing with PBST, the separated IMNBs-virus composites were stained by sodium phosphotungstate (3%, pH 7.4) and observed by TEM.
Isolation of PEDV from clinical fecal sample using IMNBs
Fecal matter collected (Collection No.PEDJX2018-01) from a PEDV-infected piglet (confirmed by PCR assay) in a pig farm of China was suspended in PBS buffer (pH 7.4) and incubated with IMNBs at 37 °C for 1 h. Magnetic beads not functionalized with sdAb were used as control. IMNBs-virus composites were separated with magnetic scaffold and washed five times with DMEM (containing 100 IU penicillin and 100 μg/mL streptomycin). Then, IMNBs-virus composites were added to the confluent vero cells grown in 6-well plates for another 1-h incubation at 37 °C under 5% CO2. Subsequently, 2 mL of DMEM was supplemented with 100 IU penicilin and 100 μg/mL streptomycin, 0.3% tryptose phosphate broth, 0.02% yeast extract (Sigma, USA), 10 mM HEPES (Invitrogen), and 5 μg/mL of trypsin (Sigma, USA) (Lee et al. 2015). The inoculated cells were maintained at 37 °C under 5% CO2 and monitored daily for cytopathic effects. When 80% cytopathic effects occurred, the inoculated cells were subjected to three rounds of freezing and thawing. The culture supernatants were then collected and centrifuged at 500g for 10 min. The clarified supernatants were used for the virus propagation and spike protein gene clone. Spike protein gene was aligned with subtype sequences G1 and G2 published in GenBank, and phylogenetic tree was constructed using the mega software (version 6.0, https://www.megasoftware.net). The growth kinetics of isolates in vero cells were analyzed by calculating the TCID50 value at different time points post-infection and different passages.
The virus-infected cells were detected by immunofluorescence assay based on QDs-sdAb probe and qPCR assay mentioned above. Virion in virus-infected vero cells was observed by TEM directly. Cells were collected and fixed in 2.5% glutaraldehyde overnight at 4 °C. The cell pellet was used to make thin sections for observation under TEM, as described previously (Sun et al. 2017).
Statistical methods
Student’s t test was performed to assess the statistical significance of the TCID50 values for sdAb-Mcs treatment and the control groups.
Results
sdAb-Ms library construction, screening, and selection of sdAb fragment
M protein was recombinantly expressed in E. coli with His (M-His) and GST (M-GST) tag (Fig. S1). Camel was immunized using M-His, and the immune reaction against the M protein in camel was monitored and evaluated by an ELISA based on M-GST. When the results of ELISA showed high OD450 nm value (Fig. S2), the PBLCs were collected 5 days after the last immunization and lymphocytes were isolated and used for RNA extraction. sdAb-encoding cDNA was amplified and cloned into phage vector pHEN2 and transformed into E. coli TG1 cells. The antibody library of sdAb against M protein was generated by scraping the clones from the plated cells with 2×YT-ampicillin. Colony screening by PCR showed that 27 out of 30 of the clones (90%) contained a plasmid with an insert of the expected size for camel sdAb gene. The capacity of the library was 107 of that calculated by the positive clone numbers. After infection with M13K07 helper phage, the phage display library was obtained. Approximately 1011 phage particles from the phage library were screened against M-GST by four rounds of panning. Under stringent panning conditions, the enriching factor of output and input phages increased steadily, with more than 103-fold increment of phage recovery after the fourth round of panning compared with that from the first one. However, the enriching factor did not dramatically increase in the control well. This result indicated an efficient enrichment of specific sdAbs (Table 1).
The binding activity of 56 clones from the fourth elution was detected by the single-phage ELISA based on M-GST and 12 of them that showed high OD490 nm value were subjected to sequencing (Fig. S3). Four unique sdAb gene fragments, named as sdAb-Mc19, sdAb-Mc29, sdAb-Mc30, and sdAb-Mc37 (GenBank: sdAb-Mc19/ 29/ 30/ 37, MH550664/ MH550665/MH550666/MH550667), were identified through sequence alignment. Their deduced amino acid sequences are illustrated in Fig. 1a. The sequence of the framework region was defined according to a published scheme (Saerens et al. 2005). All selected sdAb-Mc19/29/ 30/ 37 have hallmark amino acid of sdAb in FR2 region, including 37Phe, 44Glu, 45Arg, and 47Gly (Kang et al. 1991). All selected sdAb-Mc19/29/30/37 shared same 15 amino acids in CDR3 but showed some different amino acids in CDR1 and CDR2.

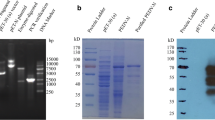
Alignments, expression, and purification of sdAb-Mcs. (A)a The differences of amino acids at CDR region were underlined. The specific amino acids of sdAb were showed in the line boxes. The framework region was defined according to a published scheme (Saerens et al. 2005). b Expression, purification, and verification by Western blot. Lanes 1–4, the expression of sdAb-c19, sdAb-c29, sdAb-c30, and sdAb-c37 in E. coli. Lanes 5–8, the purification of sdAb-c19, sdAb-c29, sdAb-c30, and sdAb-c37. Lanes 9–12, the verification by Western blot. The expressed sdAb was detected by using HRP conjugate anti-His mouse monoclonal antibody. Lane M, protein MW markers
Expression and purification of recombinant sdAb-Mcs
The gene of sdAb-Mcs was cloned into PET-32a+ vector and expressed in E. coli. SDS-PAGE analysis revealed that the expressed and purified recombinant sdAb-Mcs had the expected molecular weights of 35 kDa (Fig. 1b). Moreover, the recombinant sdAb-Mcs were identified via Western blot analysis, and specific bands were observed in recombinant sdAb-Mcs protein transfer membrane (Fig. 1b).
Binding activity
The binding activity of sdAb-Mcs with M protein of PEDV was analyzed through an indirect ELISA and Western blot. Indirect ELISA showed that OD450 nm values increased with increasing sdAb-Mcs concentration (Fig. 2a). The result suggested that sdAb-Mcs can recognize the M antigen. Western blot revealed that sdAb-Mcs can bind to M similarly as the polyclonal serum against PEDV (Fig. 2b). The PEDV can be observed directly by TEM in the supernatant of sdAb-virus composites (Fig. 2c). This result suggested that sdAb-Mcs have binding activity to PEDV.

Binding activity of recombinant sdAb-Mcs. a Binding activity of sdAb-Mcs with M protein was analyzed by indirect ELISA. The series dilution of each sdAb-Mcs was used for detection M-GST protein by indirect ELISA with HRP conjugated anti-His-antibody as the secondary antibody. psdAb-c1 against PCV2 was used as negative control. Results are expressed as the means of three wells and error bars represent ±SD. b Binding activity of sdAb-Mcs with M protein was analyzed by Western blot. The M protein in PVDF membrane was detected by sdAb-Mcs and HRP conjugated anti-His-antibody. PEDV-specific antibody positive and negative serum were used as positive (lane C+) and negative control (lane C−). c Binding activity of sdAb-Mcs with virions of CV777. The virion was incubated with sdAb-Mcs coupling in His-tag binding resin. After washing, the sdAb-Mcs-virions composites was eluted with Tris-Hcl buffer containing imidazole and detected by TEM. The blank resin was used as a control. Some virus particles are indicated by arrows
Specificity
ELISA experiments were performed to determine the specificity of sdAb-Mc19, sdAb-Mc29, sdAb-Mc30, and sdAb-Mc37. The cross-reaction assay showed that all sdAb-Mcs can specifically bind with the M protein of PEDV and do not cross-react with PCV2 Cap, PRRSV GP5, FMDV VP1, and CSF E2 proteins. As the negative control, PCV2-specific psdAb-c1 do not cross-react with M protein (Fig. 3).

Specificity. The cross-reaction of sdAb-Mcs was examined by ELISA. sdAb-Mcs were added into the microplates that were coated with recombinant structural proteins of CSFV, PCV2, PRRSV, FMDV, and PEDV, respectively. The bound sdAb-Mcs were detected by HRP conjugated anti-His-antibody. psdAb-c1against PCV2 was used as negative control. Results are expressed as the means of three wells and error bars represent ±SD
Neutralizing activity
The changes of infectious titer of CV777 after incubating with sdAb-Mcs were analyzed by TCID50 assay. The CV777 incubated with 10, 20, 100 μg/mL of sdAb-Mcs showed TCID50/0.1 mL range from 106.5 to 106.75. This value was not significantly different that resulting from control incubated with PBS alone showed a TCID50/0.1 mL value range from 106.75 to 107.6 (Fig. 4).

Neutralizing activity assay. The neutralizing activity of sdAb-Mcs to CV777 was analyzed in vero cells. The different concentration of sdAb-Mcs was incubated with CV777 and the TCID50 value was calculated by Reed-muench methods. PBS buffer was incubated with CV777 as control. Results are expressed as the means of triplicate experiments and error bars represent ±SD
PEDV imaging in vero cells with sdAb-Mc37 probe
The DIFA result indicated that PEDV can be targeted by sdAb-Mc37 probe in vero cells (Fig. 5a, b). To confirm the result of imaging based on sdAb-Mc37 probe, an indirect immunofluorescence assay based on PEDV antibody positive serum and a qRT-PCR were performed. In the indirect immunofluorescence assay, PEDV can be detected in vero cells that were consistent with the direct immunofluorescence assay (Fig. 5c, d). The results of qRT-PCR assay indicated an increase of PEDV gene copies with the increase of infection time. This result demonstrates that PEDV was replicating in the vero cells (Fig. 5e).

QDs605 labeled sdAb-Mc37 as probes imaging PEDV in vero cells. The vero cells were immobilized at 24 h post PEDV infection. PEDV in vero cell was detected by immunofluorescence based on QDs-sdAb-Mc37 (red a) and PEDV antibody positive serum and FITC labeled second antibody (green c). Nucleus were stained with DAPI. PEDV free vero cells were used as negative control (b, d). The merged images show the localization of PEDV. e Comparative analysis of PEDV genome by qRT-PCR. Cells were collected at 6, 12, 24, 36, and 48 h post PEDV infection. Total RNA was extracted and detected by a quantitative SYBR green RT-PCR. PEDV genome level was compared with 6 h post PEDV infection, β-actin gene served as an endogenous control. Error bars represent ±SD
Capture activity of IMNBs
Functional IMNBs were incubated with M protein and CV777. IMNBs–M composites were identified through SDS-PAGE and Western blot. Specific bands were observed (Fig. 6a), revealing that IMNBs can bind with the recombinant M protein. IMNBs-virus particles were observed by TEM directly. The result showed that the size approximately 100 nm of virions was binding on the IMNB surface (Fig. 6b).

The binding capability analysis of IMNBs. a The binding capability of IMNBs to M protein was analyzed by SDS-PAGE and Western blot. IMNBs were incubated with M protein. The IMNBs-M composites were collected by a magnetic scaffold. M protein binding with IMNBs (lane 1), input (lane 2), and in supernatant (lane 3) was showed by SDS-PAGE and evaluated by Western blot (lanes 4–6) based on PEDV positive porcine serum and HRP conjugated anti-pig antibody. b The binding capability of IMNBs to PEDV virions was examined by TEM. IMNBs were incubated with supernatant of PEDV-infected vero cells. The IMNBs-virions composites were collected by a magnetic scaffold and observed by TEM. IMNBs and virus particles are indicated by arrows
Isolation of a PEDV clinical strain by IMNBs
IMNBs were used to isolate PEDV clinical strain from fecal sample of the piglet. The cytopathic effects were observed in the IMNB-virus composite-inoculated Vero cells at 12 and 24 hpi (Fig. 7a), but cannot be observed in magnetic bead control. Meanwhile, the immunofluorescence assay showed PEDV positive in these vero cells (Fig. 7b). In addition, IMNBs-virus composite-inoculated vero cells were detected by qRT-PCR and TEM. The virus gene level increased gradually in vero cells at 6, 12, 24, 36, and 48 hpi (Fig. 7c), and the virus particles can be observed in ultrathin sections of IMNBs-virus composite-inoculated vero cells (Fig. 7d). The alignment and phylogenetic analysis showed the isolated PEDV strain JXDV1801 located within the G1 cluster (Fig. S4). The growth kinetics study showed that JXDV1801 can replicate efficiently and propagate stably in vero cells, reaching a maximum titer near 107 TCID50/0.1 mL (Fig. S5). The isolated strains were deposited and professional management in Lanzhou veterinary research institute CAAS.

Isolation of a PEDV from clinical feces sample by IMNBs. IMNBs were incubated with feces sample, then collected by a magnetic scaffold and inoculated to vero cells. a Cytopathic effects in vero cells after isolated PEDV infection. b Detection by direct immunofluorescence assay based on QDs-sdAb-Mc37 probe. c Comparative analysis of PEDV genome by qRT-PCR. Virus genome level was compared with 6 hpi, β-actin gene served as an endogenous control. Error bars represent ±SD. d Ultrathin sections of isolates infected vero cells at 24 hpi showing accumulated of virus particles in the cytoplasm (magnification 30,000× and 120,000×, bar = 500 and 100 nm). Some virus particles are indicated by arrows.
Discussion
sdAb has been widely used in an increasing variety of fields with the discovery of its potentials. For biotechnological application, sdAb has been used as reagent for the specific detection of bacteria, parasite, and virus, and for the human therapy of virus infection due to its neutralizing activity (de Marco 2011). In addition, sdAb can be used as a reagent for purification and bioseparation for targeting protein to simplify complex multistep procedures to a single step protocol, reducing production costs and time (Bossi et al. 2010). Considering this meaningful application as example, sdAb specific for PEDV was isolated from immunized C. bactrianus sdAb library by phage display technique in this study.
PEDV is the causative agent of porcine epidemic diarrhea disease which is a highly contagious enteric disease of swine characterized by severe enteritis, watery diarrhea, and high mortality in piglets. Porcine epidemic diarrhea disease has been the most economically important disease affecting the global swine industry today. PEDV M proteins are relatively conserved that show high levels of homology at the nucleotide and amino acid level at various PEDV strains (Chen et al. 2014; Wang et al. 2013). Meanwhile, M protein is the most abundant of structural proteins and contains neutralizing epitopes. Thus, M protein is the optimal antigen for the making of antibodies that have potential using in specific detection and research of PEDV. In view of these, M protein was expressed in E. coli and used as an antigen for immunization of camel and screening of sdAbs in this study.
After four rounds of panning, four clones showing different sequences were selected and expressed in E. coli. The amino acid sequences of all four binders showed characteristic substitutions at four FR2 positions (Val37Phe, Gly44Glu, Leu45Arg, and Trp47Gly from conventional heavy chain variable domains to sdAb), which are conserved in conventional heavy chain variable domains (Muyldermans et al. 1994; Vu et al. 1997). In addition, the selected sdAb-Mcs were expressed solubly in E. coli at a high yield of 5 mg per liter of bacterial culture (data not shown) that is in accordance with the unique property of sdAb. In practice, the high yield and solubility of sdAb are well within the range needed for most applications.
To assess the characteristics of sdAb-Mcs, the binding activity, specificity, affinity, and the neutralizing activity to M protein and PEDV strain CV777 were analyzed. ELISA and Western blot assay indicated that sdAb-Mc19, sdAb-Mc29, sdAb-Mc30, and sdAb-Mc37 display excellent binding activity to M protein. Further evaluation, the affinity by ITC 200 system (Supplementary material) showed that sdAb-Mcs unfold equilibrium binding constant value from 8.13 × 10−9 to 357 × 10−9 M to M protein (Table S1) that was similar with the published sdAbs (Harmsen et al. 2006; Li et al. 2012). Meanwhile, the binding activity to PEDV was also demonstrated by the TEM analysis. Furthermore, sdAb-Mcs could specifically bind with the M protein but did not cross-react with PCV2 Cap, PRRSV GP5, FMDV VP1, and CSF E2 proteins, indicating that sdAb-Mcs have good specificity. This analysis suggested that the selected sdAb-Mcs have potential application on detection and research of PEDV, but cannot be used as therapeutic antibody because all selected sdAb-Mcs do not show neutralizing activity to CV777.
The conjugate of QDs and antibodies has been recognized as a powerful bioprobe and applied widely in ELISA and immunofluorescence (Goldman et al. 2002; Umakoshi et al. 2018). Given the excellent property of sdAb in terms of structure, stability, and production costs, sdAbs have been used successfully as an attractive alternative to conjugate with QDs for the imaging and diagnostic platforms (Sukhanova et al. 2012; Wang et al. 2015; Yin et al. 2013). In the current study, sdAb-Mc37 showed high Kd value, conjugated with a ZnS-capped CdSe QDs. Moreover, the ability of QD-sdAb to act as targeting probes for imaging PEDV in vero cells was evaluated by direct immunofluorescence assay. The observed fluorescence in vero cells stained with QD-sdAb probe nicely reflected that PEDV virions can be reliably recognized and labeled by QD-sdAb probe that was further confirmed by an indirect immunofluorescence assay and qRT-PCR. As a diagnostic reagent, QD-sdAb probe used in direct immunofluorescence assay showed high efficiency with lesser steps and shorter times compared with indirect immunofluorescence assay. Additionally, as a molecular tracer, sdAb has been used to tracing antigens in different subcellular compartments in living cells (Rothbauer et al. 2006), targeting brain epitopes by transmigrating through blood-brain barrier (Abulrob et al. 2005) and detecting the activity of cell proteins in vivo (Iqbal et al. 2010). Thus, QD-sdAbs possess potential application for the dynamic tracing of PEDV in virus-host interaction.
In recent years, functionalized magnetic nanoparticles are successfully applied in bioseparation for cells such as cancer and bacterial cells, biological molecules such as proteins, peptides, and nucleic acids (Jain 2003; Lee et al. 2018; Mostafaei et al. 2018), and viruses (Li et al. 2011; Zhao et al. 2012). For the viruses, however, most applications focus on efficient and rapid detection. Whether the viruses separated by nanoparticles can be propagated in cells and used for further research is still not clear. Thus, magnetic nanobeads were functionalized with sdAb-Mc29 and were tried to be used as a tool for the isolation of virus in this study. The characteristics of binders were responsible for the final application of its functionalized nanobeads. Fortunately, sdAb-Mc29 showed excellent specificity and affinity but did not have neutralizing activity, which perfectly fit the destination of this research. For the analysis of the capture activity, IMNBs were incubated with recombinant M protein and CV777. The captured M protein was clearly reflected by SDS-PAGE and Western blot. Meanwhile, virions around the IMNBs were observed directly by TEM, suggesting that PEDV can be separated from mixture of cells by the IMNBs.
To evaluate the practicability, IMNBs were used for PEDV isolation from clinical fecal sample from PEDV-infected piglet. Interestingly, the cytopathic effects were observed at 12 h post-incubation of IMNBs-virus composite with vero cells that was not observed on the control cells. The further assay of direct immunofluorescence assay, qPCR, TEM, and spike protein gene sequencing confirmed that one epidemic strain of PEDV named JXDV1801 was successfully isolated by using IMNBs. The growth kinetics analysis indicated that JXDV1801 can replicate efficiently and propagate stably in vero cells. Thus, a novel, practicable, and efficient tool for PEDV isolation from clinical complex samples was developed in this research. Our research expanded the spectrum of application of sdAb and provided a novel method for isolation of virus from clinical samples.
References
Abulrob A, Sprong H, Van Bergen EN, Henegouwen P, Stanimirovic D (2005) The blood-brain barrier transmigrating single domain antibody: mechanisms of transport and antigenic epitopes in human brain endothelial cells. J Neurochem 95(4):1201–1214. https://doi.org/10.1111/j.1471-4159.2005.03463.x
Anderson GP, Liu JH, Zabetakis D, Liu JL, Goldman ER (2017) Thermal stabilization of anti-alpha-cobratoxin single domain antibodies. Toxicon 129:68–73. https://doi.org/10.1016/j.toxicon.2017.02.008
Arbabi-Ghahroudi M (2017) Camelid single-domain antibodies: historical perspective and future outlook. Front Immunol 8:1589. https://doi.org/10.3389/fimmu.2017.01589
Bannas P, Hambach J, Koch-Nolte F (2017) Nanobodies and Nanobody-based human heavy chain antibodies as antitumor therapeutics. Front Immunol 8:1603. https://doi.org/10.3389/fimmu.2017.01603
Beghein E, Gettemans J (2017) Nanobody technology: a versatile toolkit for microscopic imaging, protein-protein interaction analysis, and protein function exploration. Front Immunol 8:771. https://doi.org/10.3389/fimmu.2017.00771
Bossi S, Ferranti B, Martinelli C, Capasso P, de Marco A (2010) Antibody-mediated purification of co-expressed antigen-antibody complexes. Protein Expr Purif 72(1):55–58. https://doi.org/10.1016/j.pep.2010.01.003
Cao L, Ge X, Gao Y, Zarlenga DS, Wang K, Li X, Qin Z, Yin X, Liu J, Ren X, Li G (2015) Putative phage-display epitopes of the porcine epidemic diarrhea virus S1 protein and their anti-viral activity. Virus Genes 51(2):217–224. https://doi.org/10.1007/s11262-015-1234-5
Chen Q, Li GW, Stasko J, Thomas JT, Stensland WR, Pillatzki AE, Gauger PC, Schwartz KJ, Madson D, Yoon KJ, Stevenson GW, Burrough ER, Harmon KM, Main RG, Zhang JQ (2014) Isolation and characterization of porcine epidemic diarrhea viruses associated with the 2013 disease outbreak among swine in the United States. J Clin Microbiol 52(1):234–243. https://doi.org/10.1128/Jcm.02820-13
de Marco A (2011) Biotechnological applications of recombinant single-domain antibody fragments. Microb Cell Factories 10(1):44. https://doi.org/10.1186/1475-2859-10-44
Dumoulin M, Conrath K, Van Meirhaeghe A, Meersman F, Heremans K, Frenken LG, Muyldermans S, Wyns L, Matagne A (2002) Single-domain antibody fragments with high conformational stability. Protein Sci 11(3):500–515. https://doi.org/10.1110/ps.34602
Goldman ER, Balighian ED, Kuno MK, Labrenz S, Tran PT, Anderson GP, Mauro JM, Mattoussi H (2002) Luminescent quantum dot-adaptor protein-antibody conjugates for use in fluoroimmunoassays. Phys Status Solidi B 229(1):407–414. https://doi.org/10.1002/1521-3951(200201)229:1<407::Aid-Pssb407>3.0.Co;2-S
Gonzalez-Sapienza G, Rossotti MA, Tabares-da Rosa S (2017) Single-domain antibodies as versatile affinity reagents for analytical and diagnostic applications. Front Immunol 8:977. https://doi.org/10.3389/fimmu.2017.00977
Guo HC, Sun SQ, Jin Y, Yang SL, Wei YQ, Sun DH, Yin SH, Ma JW, Liu ZX, Guo JH, Luo JX, Yin H, Liu XT, Liu DX (2013) Foot-and-mouth disease virus-like particles produced by a SUMO fusion protein system in Escherichia coli induce potent protective immune responses in guinea pigs, swine and cattle. Vet Res 44:48. https://doi.org/10.1186/1297-9716-44-48
Hamers-Casterman C, Atarhouch T, Muyldermans S, Robinson G, Hamers C, Songa EB, Bendahman N, Hamers R (1993) Naturally occurring antibodies devoid of light chains. Nature 363(6428):446–448. https://doi.org/10.1038/363446a0
Harmsen MM, De Haard HJ (2007) Properties, production, and applications of camelid single-domain antibody fragments. Appl Microbiol Biotechnol 77(1):13–22. https://doi.org/10.1007/s00253-007-1142-2
Harmsen MM, van Solt CB, van Zijderveld Bemmel AM, Niewold TA, van Zijderveld FG (2006) Selection and optimization of proteolytically stable llama single-domain antibody fragments for oral immunotherapy. Appl Microbiol Biotechnol 72(3):544–551. https://doi.org/10.1007/s00253-005-0300-7
International Committee on Taxonomy of Viruses., King AMQ (2012) Virus taxonomy: Classification and nomenclature of viruses: ninth report of the International Committee on Taxonomy of Viruses. Academic Press, London; Waltham, MA
Iqbal U, Trojahn U, Albaghdadi H, Zhang J, O'Connor-McCourt M, Stanimirovic D, Tomanek B, Sutherland G, Abulrob A (2010) Kinetic analysis of novel mono- and multivalent VHH-fragments and their application for molecular imaging of brain tumours. Br J Pharmacol 160(4):1016–1028. https://doi.org/10.1111/j.1476-5381.2010.00742.x
Jain KK (2003) Nanodiagnostics: application of nanotechnology in molecular diagnostics. Expert Rev Mol Diagn 3(2):153–161. https://doi.org/10.1586/14737159.3.2.153
Kang AS, Barbas CF, Janda KD, Benkovic SJ, Lerner RA (1991) Linkage of recognition and replication functions by assembling combinatorial antibody Fab libraries along phage surfaces. Proc Natl Acad Sci U S A 88(10):4363–4366. https://doi.org/10.1073/pnas.88.10.4363
Kim O, Chae C (2000) In situ hybridization for the detection and localization of porcine epidemic diarrhea virus in the intestinal tissues from naturally infected piglets. Vet Pathol 37(1):62–67. https://doi.org/10.1354/vp.37-1-62
Lee S, Kim Y, Lee C (2015) Isolation and characterization of a Korean porcine epidemic diarrhea virus strain KNU-141112. Virus Res 208:215–224. https://doi.org/10.1016/j.virusres.2015.07.010
Lee DW, Fatima H, Kim KS (2018) Preparation of silica coated magnetic nanoparticles for bioseparation. J Nanosci Nanotechnol 18(2):1414–1418. https://doi.org/10.1166/jnn.2018.14888
Li D, Wang J, Wang R, Li Y, Abi-Ghanem D, Berghman L, Hargis B, Lu H (2011) A nanobeads amplified QCM immunosensor for the detection of avian influenza virus H5N1. Biosens Bioelectron 26(10):4146–4154. https://doi.org/10.1016/j.bios.2011.04.010
Li T, Bourgeois JP, Celli S, Glacial F, Le Sourd AM, Mecheri S, Weksler B, Romero I, Couraud PO, Rougeon F, Lafaye P (2012) Cell-penetrating anti-GFAP VHH and corresponding fluorescent fusion protein VHH-GFP spontaneously cross the blood-brain barrier and specifically recognize astrocytes: application to brain imaging. FASEB J 26(10):3969–3979. https://doi.org/10.1096/fj.11-201384
Mostafaei M, Hosseini SN, Khatami M, Javidanbardan A, Sepahy AA, Asadi E (2018) Isolation of recombinant hepatitis B surface antigen with antibody-conjugated superparamagnetic Fe3O4/SiO2 core-shell nanoparticles. Protein Expr Purif 145:1–6. https://doi.org/10.1016/j.pep.2017.12.004
Muyldermans S, Atarhouch T, Saldanha J, Barbosa JA, Hamers R (1994) Sequence and structure of VH domain from naturally occurring camel heavy chain immunoglobulins lacking light chains. Protein Eng 7(9):1129–1135
Muyldermans S, Baral TN, Retamozzo VC, De Baetselier P, De Genst E, Kinne J, Leonhardt H, Magez S, Nguyen VK, Revets H, Rothbauer U, Stijlemans B, Tillib S, Wernery U, Wyns L, Hassanzadeh-Ghassabeh G, Saerens D (2009) Camelid immunoglobulins and nanobody technology. Vet Immunol Immunopathol 128(1–3):178–183. https://doi.org/10.1016/j.vetimm.2008.10.299
Muzard J, Platt M, Lee GU (2012) M13 bacteriophage -activated superparamagnetic beads for affinity separation. Small 8(15):2403–2411. https://doi.org/10.1002/smll.201200099
Pathak S, Davidson MC, Silva GA (2007) Characterization of the functional binding properties of antibody conjugated quantum dots. Nano Lett 7(7):1839–1845. https://doi.org/10.1021/nl062706i
Rampini S, Kilinc D, Li P, Monteil C, Gandhi D, Lee GU (2015) Micromagnet arrays for on-chip focusing, switching, and separation of superparamagnetic beads and single cells. Lab Chip 15(16):3370–3379. https://doi.org/10.1039/c5lc00581g
Rampini S, Li P, Lee GU (2016) Micromagnet arrays enable precise manipulation of individual biological analyte-superparamagnetic bead complexes for separation and sensing. Lab Chip 16(19):3645–3663. https://doi.org/10.1039/c6lc00707d
Ran YF, Fields C, Muzard J, Liauchuk V, Carr M, Hall W, Lee GU (2014) Rapid, highly sensitive detection of herpes simplex virus-1 using multiple antigenic peptide-coated superparamagnetic beads. Analyst 139(23):6126–6134. https://doi.org/10.1039/c4an00774c
Ren X, Suo S, Jang YS (2011) Development of a porcine epidemic diarrhea virus M protein-based ELISA for virus detection. Biotechnol Lett 33(2):215–220. https://doi.org/10.1007/s10529-010-0420-8
Rothbauer U, Zolghadr K, Tillib S, Nowak D, Schermelleh L, Gahl A, Backmann N, Conrath K, Muyldermans S, Cardoso MC, Leonhardt H (2006) Targeting and tracing antigens in live cells with fluorescent nanobodies. Nat Methods 3(11):887–889. https://doi.org/10.1038/nmeth953
Saerens D, Pellis M, Loris R, Pardon E, Dumoulin M, Matagne A, Wyns L, Muyldermans S, Conrath K (2005) Identification of a universal VHH framework to graft non-canonical antigen-binding loops of camel single-domain antibodies. J Mol Biol 352(3):597–607. https://doi.org/10.1016/j.jmb.2005.07.038
Shenyang G, Enhui Z, Baoxian L, Xinyuan Q, Lijie T, Junwei G, Yijing L (2007) High-level prokaryotic expression of envelope exterior of membrane protein of porcine epidemic diarrhea virus. Vet Microbiol 123(1–3):187–193. https://doi.org/10.1016/j.vetmic.2007.03.027
Shirasaki Y, Supran GJ, Bawendi MG, Bulović V (2013) Emergence of colloidal quantum-dot light-emitting technologies. Nat Photonics 7(1):13–23. https://doi.org/10.1038/nphoton.2012.328
Sukhanova A, Even-Desrumeaux K, Kisserli A, Tabary T, Reveil B, Millot JM, Chames P, Baty D, Artemyev M, Oleinikov V, Pluot M, Cohen JH, Nabiev I (2012) Oriented conjugates of single-domain antibodies and quantum dots: toward a new generation of ultrasmall diagnostic nanoprobes. Nanomedicine 8(4):516–525. https://doi.org/10.1016/j.nano.2011.07.007
Sun M, Ma J, Yu Z, Pan Z, Lu C, Yao H (2017) Identification of two mutation sites in spike and envelope proteins mediating optimal cellular infection of porcine epidemic diarrhea virus from different pathways. Vet Res 48(1):44. https://doi.org/10.1186/s13567-017-0449-y
Umakoshi T, Udaka H, Uchihashi T, Ando T, Suzuki M, Fukuda T (2018) Quantum-dot antibody conjugation visualized at the single-molecule scale with high-speed atomic force microscopy. Colloids Surf B Biointerfaces 167:267–274. https://doi.org/10.1016/j.colsurfb.2018.04.015
Utiger A, Tobler K, Bridgen A, Ackermann M (1995) Identification of the membrane protein of porcine epidemic diarrhea virus. Virus Genes 10(2):137–48 doi: 10.1007/BF01702594
Van Diep N, Sueyoshi M, Norimine J, Hirai T, Myint O, Teh APP, Izzati UZ, Fuke N, Yamaguchi R (2018) Molecular characterization of US-like and Asian non-S INDEL strains of porcine epidemic diarrhea virus (PEDV) that circulated in Japan during 2013-2016 and PEDVs collected from recurrent outbreaks. BMC Vet Res 14(1):96. https://doi.org/10.1186/s12917-018-1409-0
Vu KB, Ghahroudi MA, Wyns L, Muyldermans S (1997) Comparison of llama VH sequences from conventional and heavy chain antibodies. Mol Immunol 34(16–17):1121–1131. https://doi.org/10.1016/S0161-5890(97)00146-6
Wang XM, Niu BB, Yan H, Gao DS, Yang X, Chen L, Chang HT, Zhao J, Wang CQ (2013) Genetic properties of endemic Chinese porcine epidemic diarrhea virus strains isolated since 2010. Arch Virol 158(12):2487–2494. https://doi.org/10.1007/s00705-013-1767-7
Wang D, Yang S, Yin S, Shang Y, Du P, Guo J, He J, Cai J, Liu X (2015) Characterization of single-domain antibodies against foot and mouth disease virus (FMDV) serotype O from a camelid and imaging of FMDV in baby hamster kidney-21 cells with single-domain antibody-quantum dots probes. BMC Vet Res 11:120. https://doi.org/10.1186/s12917-015-0437-2
Yang S, Shang Y, Yin S, Tian H, Chen Y, Sun S, Jin Y, Liu X (2014) Selection and identification of single-domain antibody fragment against capsid protein of porcine circovirus type 2 (PCV2) from C. bactrianus. Vet Immunol Immunopathol 160(1–2):12–19. https://doi.org/10.1016/j.vetimm.2014.03.004
Yang S, Shang Y, Yin S, Wang D, Cai J, Gong Z, Serge M, Liu X (2015) A phage-displayed single domain antibody fused to alkaline phosphatase for detection of porcine circovirus type 2. J Virol Methods 213:84–92. https://doi.org/10.1016/j.jviromet.2014.11.023
Yeo SG, Hernandez M, Krell PJ, Nagy EE (2003) Cloning and sequence analysis of the spike gene of porcine epidemic diarrhea virus Chinju99. Virus Genes 26(3):239–246. https://doi.org/10.1023/A:1023447732567
Yin S, Yang S, Shang Y, Sun S, Zhou G, Jin Y, Tian H, Wu J, Liu X (2013) Characterization of Asia 1 sdAb from camels bactrianus (C. bactrianus) and conjugation with quantum dots for imaging FMDV in BHK-21 cells. PLoS One 8(5):e63500. https://doi.org/10.1371/journal.pone.0063500
Zhang B, Yu J, Liu C, Wang J, Han H, Zhang P, Shi D (2016) Improving detection sensitivity by oriented bioconjugation of antibodies to quantum dots with a flexible spacer arm for immunoassay. RSC Adv 6(55):50119–50127. https://doi.org/10.1039/c6ra09279a
Zhao W, Zhang WP, Zhang ZL, He RL, Lin Y, Xie M, Wang HZ, Pang DW (2012) Robust and highly sensitive fluorescence approach for point-of-care virus detection based on immunomagnetic separation. Anal Chem 84(5):2358–2365. https://doi.org/10.1021/ac203102u
Acknowledgments
We are grateful for the help from all staff in the public instrument center of Lanzhou Veterinary Research Institute of Chinese Academy of Agricultural Sciences.
Funding
This work was supported by the Agricultural Science and Technology Innovation Program (CAAS-ASTIP-2014-LVRI-09).
Author information
Authors and Affiliations
Contributions
Yang SL and Li L participated in the planning of the study, conducted most of the experiments, and drafted the manuscript. Yin SH participated in the immunization of the camel. He XY, Yuan L, and Gao X helped for the ELISA assay. Shang YJ, Liu XT, Umar, and Cai JP contributed to the interpretation of the findings and revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics statement
Blood samples from a 1-year-old C. bactrianus, grown in a farm of Gobi desert, northwest of China, were collected according to the Animal Ethics Procedures and Guidelines of the People’s Republic of China. The current study was approved by the Animal Ethics Committee of Lanzhou Veterinary Research Institute, Chinese Academy of Agricultural Sciences (No. LVRIAEC2012-006).
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
ESM 1
(PDF 779 kb)
Rights and permissions
About this article

Cite this article
Yang, S., Li, L., Yin, S. et al. Single-domain antibodies as promising experimental tools in imaging and isolation of porcine epidemic diarrhea virus. Appl Microbiol Biotechnol 102, 8931–8942 (2018). https://doi.org/10.1007/s00253-018-9324-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-018-9324-7