
Abstract
Cynomolgus macaques (Macaca fascicularis) have become an important animal model for biomedical research. In particular, it is the animal model of choice for the development of vaccine candidates associated with emerging dangerous pathogens. Despite their increasing importance as animal models, the cynomolgus macaque genome is not fully characterized, hindering molecular studies for this model. More importantly, the lack of knowledge about the immunoglobulin (IG) locus organization directly impacts the analysis of the humoral response in cynomolgus macaques. Recent advances in next generation sequencing (NGS) technologies to analyze IG repertoires open the opportunity to deeply characterize the humoral immune response. However, the IG locus organization for the animal is required to completely dissect IG repertoires. Here, we describe the localization and organization of the rearranging IG heavy (IGH) genes on chromosome 7 of the cynomolgus macaque draft genome. Our annotation comprises 108 functional genes which include 63 variable (IGHV), 38 diversity (IGHD), and 7 joining (IGHJ) genes. For validation, we provide RNA transcript data for most of the IGHV genes and all of the annotated IGHJ genes, as well as proteomic data to validate IGH constant genes. The description and annotation of the rearranging IGH genes for the cynomolgus macaques will significantly facilitate scientific research. This is particularly relevant to dissect the immune response during vaccination or infection with dangerous pathogens such as Ebola, Marburg and other emerging pathogens where non-human primate models play a significant role for countermeasure development.
Similar content being viewed by others


Introduction
Non-human primates (NHP) are one of the most important animal models for human biomedical research. This is a result of the strong similarities across physiological, developmental, immunological responses, and genetics between NHP and humans (Carlsson et al. 2004; Palermo et al. 2013). The rhesus macaque (Macaca mulatta) has been used extensively as an animal model for several infectious diseases, especially for hepatitis C virus (HCV) and human immunodeficiency virus (HIV) infections (Bukh 2012; Desrosiers 1995). This species has been well characterized at the genomic and transcriptomic level (Gibbs et al. 2007; Zimin et al. 2014). Key immunological molecules of the rhesus macaques were compared with humans, including IGH locus (Olivieri and Gambon-Deza 2015; Thullier et al. 2010), TRB locus (Li et al. 2015) and MHC/KIR coevolution (de Groot et al. 2015). However, the limitations in breeding these animals in captivity and restrictions in capture on animals from the wild for research usage have created the necessity of utilizing other NHP species as possible animal models. Within those, the use of cynomolgus macaques (Macaca fascicularis) and African green monkeys (Chlorocebus sabaeus) in biomedical research is rising rapidly. Cynomolgus macaques have become the primordial animal model for several infectious diseases (Higgs and Ziegler 2010; Nakayama and Saijo 2013; Pripuzova et al. 2013; Reynaud and Horvat 2013). Although, two draft genomes (Ebeling et al. 2011; Higashino et al. 2012; Yan et al. 2011) and the transcriptome (Lee et al. 2014) were published for cynomolgus macaques recently, its annotation, gene organization and characterization are not at the level of other animal models, including rhesus macaques.
Immunoglobulins (IG), or antibodies, are a key component of the humoral immune response either as membrane B cell receptors (BcR) or as soluble antibodies. IG are generated by the combination of two identical heavy (H) chains with two identical light (L) kappa or lambda chains (Lefranc 2014a; Lefranc 2014b; Lefranc and Lefranc 2001; Lucas 2003; Schroeder and Cavacini 2010). The IG heavy (IGH), the IG kappa (IGK), and the IG lambda (IGL) loci comprise multiple variable (V), diversity (D) (only for IGH), joining (J), and constant (C) genes. The V-(D)-J junction resulting from the rearrangement of the V, D, and J genes forms the complementarity determining region CDR3 which in combination with the CDR1 and CDR2 encoded by the germline V-region, define the paratope of the IG, and consequently the specificity and affinity for the antigen it recognizes. The C gene defines the isotype and biological function of the immunoglobulin. Recent advances in deep sequencing technologies of IG repertoires encoded by B cells are improving our knowledge of humoral immune responses (Georgiou et al. 2014). These technologies utilize high-throughput DNA sequencing (HTS) to analyze the V-(D)-J rearrangement on the B cell transcripts and define the B cell repertoire. However, the analysis of the IG repertoire by HTS requires a well-characterized gene annotation and IG locus organization. The extensive annotation of human, mouse, rat and rhesus macaques IG loci permits direct application of HTS and IG analysis using the IGH, IGK, and IGL germline V, D, and J annotation (Alamyar et al. 2012; Giudicelli et al. 2005, 2015). However, while draft genomes have been announced for cynomolgus macaques, there is insufficient information about the IG locus organization to characterize B cell repertoires and the humoral responses against specific epitopes of pathogens in infections or in vaccinations.
Several public databases provide sequence information and feature annotation describing cynomolgus macaques IGH locus. NCBI contains a draft genome of a female from “Tinjil” (RefSeq Assembly GCF_000364345.1) that is annotated by the NCBI Eukaryotic Genome Annotation Pipeline, which describes nine “C region-like” and 46 “V region-like” IGH features. In addition, GenBank contains 72 primary submissions describing IGH features, most of which are mRNA transcripts without genomic placement. IMGT/GENE-DB, the gene database of IMGT®, the international ImMunoGeneTics information system® (IMGT) includes five genes and 11 alleles for the immunoglobulin alpha and gamma heavy chain constant regions but it does not include a single reference sequence or annotation for the variable region (Giudicelli et al. 2005; Lefranc et al. 2015). In contrast, the IMGT/LIGM-DB includes 141 associated IGH sequences (Giudicelli et al. 2006). Overall, the annotation on the IGH locus of M. fascicularis is minimal compared to rhesus. In comparison, there are 1914 GenBank records, 98 genes and 136 alleles in IMGT/GENE-DB, and 1028 sequences in the IMGT/LIGM-DB database, describing the IGH locus for M. mulatta. This deficit in sequence data and annotation of the IGH locus compromises immunological studies focused on immune response modeling and vaccine development.
Using the draft genome GCA_000364345.1, NCBI M. fascicularis assembly 5.0, we described the IGH locus on chromosome 7. Within the IGH locus, 150 IGHV genes were found. 63 of these IGHV genes are functional, 80 are pseudogenes, and 7 are open reading frames (ORF). On chromosome 7 we also found 40 IGHD genes, 38 of those are functional and two are pseudogenes. Downstream from the IGHD genes, 8 IGHJ genes were located. Seven of these IGHJ genes are functional and one is a pseudogene. Similarly to rhesus macaques, cynomolgus macaques underwent a duplication of IGHJ5 (Link et al. 2002). At the 3′ end of the locus, we also identified 7 IGHC genes. Five of those are located at the chromosome 7 (IGHM, IGHD, IGHG putative subclasses 1, 2, and 3) and the other two (IGHA and IGHE) at an unlocalized scaffold within chromosome 7.
We envision that the annotation and announcement of the IGH locus from cynomolgus macaques will permit researches to analyze the B cell repertoire utilizing next generation sequencing (NGS) protocols, to better characterize the humoral immune response of these animals against a vaccine candidate or exposure to a pathogen.
Results
IGH locus
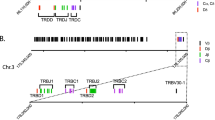
The genome assembly of the cynomolgus macaque (crab-eating macaque—M. fascicularis 5.0, GCA_000364345.1) was produced in June 2013 by Washington University. It consists of 22 chromosomes (20 autosomal, X, and mitochondrial), 7107 unplaced scaffolds and 518 unlocalized scaffolds. Bioinformatic analysis identified variable and constant IGH genes on chromosome 7q near the telomere on the minus strand, as well as several genes at an unlocalized scaffold on chromosome 7 and an unplaced scaffold. Specifically, 55 IGHV, 40 IGHD, 8 IGHJ, and 5 IGHC genes were found within a 600-kb region of the M. fascicularis chromosome 7 genomic scaffold KE145476.1. Two additional IGHC genes were located on an unlocalized genomic scaffold KE145669.1 (49 kb) placed on chromosome 7, and 95 additional IGHV genes were found within a 700-kb region of an unplaced genomic scaffold KE145882.1 (808 kb).
The location of IGHV, IGHD, and IGHJ genes is shown in Fig. 1 and in Online Resource 1. Nucleotide sequence data reported are available in the Third Party Annotation Section of the DDBJ/EMBL/GenBank databases under the accession numbers TPA: BK008911-BK009108, and will be made available in IMGT/LIGM-DB.

IGH locus annotation for M. fascicularis genome assembly GCA_000364345.1. a IGHV genes found on the unplaced scaffold KE 145882.1. In this scaffold, 95 IGHV genes were found, of which 39 genes are functional (green), 52 pseudogenes (orange) and 4 open reading frames (ORF) (yellow). b 55 IGHV genes, 40 IGHD genes, and 8 IGHJ genes were found on scaffold KE 145476.1, which was placed on chromosome 7q. Within the IGHV genes, 24 are functional, 28 pseudogenes, and 3 ORF. 38 of the IGHD genes are functional and 2 pseudogenes. 7 IGHJ genes are functional and one is a pseudogene
Constant genes
Seven constant IGHC genes encoding the constant region of the H chain isotypes (H-mu, H-delta, H-gamma1, H-gamma2, H-gamma3, H-epsilon, and H-alpha) and associated exons were mapped onto scaffolds KE145476.1 and KE145669.1 of assembly GCA_000364345.1 (Online Resource 1). The IGHM, IGHD, and IGHG genes were found downstream the V, D, and J genes on chromosome 7 KE145476.1 scaffolds. The IGHE and IGHA genes were found on the KE145669.1 unlocalized scaffold. We were not able to identify the complete CDS for IGHG genes due to large assembly gaps in KE145476.1. However, using proteomic analysis on purified immunoglobulins from cynomolgus macaque serum, we were able to identify 46 peptides for IGHG1, IGHG2, IGHG3, IGHA, and IGHM, confirming that these genes are functional and likely encode for the corresponding isotypes H-gamma1, H-gamma2, and H-gamma3 (Online Resource 2).
Variable genes
A total of 150 IGHV genes were found at the genome assembly (Fig. 1a, b). Fifty-five of those genes were found within a 500-kb region on the chromosome 7 genomic scaffold KE145476.1. The additional 95 IGHV genes were localized within a 700-kb region on the unplaced genomic scaffold KE145882.1. Among those 150 IGHV genes, only 63 are functional. Interspersed within the functional genes, 7 ORF and 80 pseudogenes were also found (Table 1A and B).
In total, 63 IGHV functional genes were identified and annotated in the cynomolgus macaque genome assembly. Distribution of the genes within the IGHV subgroups was not even, similar to that observed in other species (Table 1C). Most of the functional IGHV genes belong to the IGHV3 subgroup (35 in total, 16 at the chromosome 7 and 19 at the unplaced scaffold). This subgroup is also the most prevalent in rhesus macaque and human IGH annotation. Fifteen functional IGHV genes belong to the IGHV4 subgroup (1 at Chromosome 7 and 14 at the unplaced scaffold), compared to the 11 genes found in the human IGH locus. There is only one IGHV4 annotated at IMGT/GENE-DB in the rhesus IGH locus, which might indicate a need to improve the rhesus annotation. The other IGHV subgroups were represented by a low number of functional genes, five for IGHV1, three for IGHV2, two for IGHV5 and IGHV7, and only one for IGHV6. Phylogenetic analysis of the functional IGHV genes found in the genome assembly is shown in Fig. 2.

Phylogenetic analysis of the functional IGHV genes found within the M. fascicularis genome. IGHV genes sequence alignments and phylogenetic trees were constructed and analyzed with Geneious® 7.1.9, using a global alignment with free end gaps, and a Tamura-Nei distance model, with neighbor-joining method. IGHV genes cluster according to their subgroups. Only functional genes were incorporated in this analysis
Diversity genes
IGHD genes were found on chromosome 7q of the Macaca fascicularis-5.0 assembly, located between the IGHV and the IGHJ genes (Fig. 1b), as expected. Thirty-eight functional IGHD genes and two pseudogenes were found on chromosome 7 genomic scaffold KE145476.1. These genes belong to six subgroups (IGHD1 to IGHD6), structured in gene clusters similar to the human locus organization (Table 2). IGHD genes missing in the clusters are most likely due to gaps in the genome assembly since there is an assembly gap at the location of every missing IGHD gene (Fig. 1). Phylogenetic analysis of the IGHD genes is shown in Fig. 3. Alignment of the functional IGHD genes, with the 5′D-RS and 3′D-RS is provided in Online Resource 3.

Phylogenetic analysis of the IGHD genes found within the M. fascicularis genome. IGHD genes sequence alignments and phylogenetic trees were constructed and analyzed with Geneious® 7.1.9, using a global alignment with free end gaps, and a Tamura-Nei distance model, with neighbor-joining method. IGHDV genes cluster according to their subgroups. Only functional genes were incorporated in this analysis
Joining genes
Eight IGHJ genes were located downstream of the IGHD genes. Seven are functional and one is a pseudogene. Similar to rhesus macaques (M. mulatta) the IGHJ5 gene is duplicated (IGHJ5–1 and IGHJ5–2) (Link et al. 2002). Functional IGHJ genes are IGHJ1, IGHJ2, IGHJ3, IGHJ4, IGHJ5–1, IGHJ5–2, and IGHJ6. The pseudogene IGHJ3P (without a correct ORF) was located between IGHJ5–2 and IGHJ6. Phylogenetic analysis of all the IGHJ genes is shown in Fig. 4a. Nucleotide and amino acid alignment of the functional IGHJ genes is shown in Fig. 4B. The reading frame of the functional IGHJ genes encodes the expected motif WGXG (tryptophan, glycine, any amino acid, and glycine). The WGXG motif is highlighted in red in all the IGHJ sequences described (Fig. 4b).

Analysis of the IGHJ genes found within the M. fascicularis genome. a Phylogenetic analysis of the IGHJ genes. IGHJ genes sequence alignments and phylogenetic trees were constructed and analyzed with Geneious® 7.1.9, using a global alignment with free end gaps, and a Tamura-Nei distance model, with neighbor-joining method. IGHJ genes cluster according to their subgroups. The seven functional genes and the pseudogene (IGHJ3P) were included in the analysis. b IGHJ alignment at the nucleotide and amino acid level. The WGXG motif is highlighted in red in all the IGHJ sequences described
IGHV and IGHJ genes validation
To validate the IG locus annotation, RNA was extracted from five cynomolgus macaque animals and B cell transcripts were obtained using a 5′RACE protocol. IG transcripts were used to validate functionality of the IGHV and IGHJ genes annotated in the M. fascicularis genome. Using this methodology we were able to validate all the functional IGHJ genes and 76 % of the functional IGHV genes (Online Resource 4). All the genes from the IGHV1 (5/5), IGHV2 (3/3), IGHV6 (1/1), and IGHV7 (2/2) subgroups were validated as functional by the IG transcripts. 72 % of the IGHV3 genes (25/35) and 80 % of the IGHV4 genes (12/15) were also validated. No IG transcript was found containing IGHV5 gene (0/2) in their rearrangement. IGHV gene usage showed preferential usage of some genes. The most abundant IGHV subgroup found in the IG transcripts from these five M. fascicularis was the IGHV4 subgroup, which represents 63.5 % of the IGHV usage. Within the IGV4 subgroup, IGHV4S7 (15.5 % usage) and IGHV4S11 (18.4 % usage) were the most prevalent. IGHV1 subgroup usage was 16.1 % and IGHV3 subgroup was 12.7 %. For all other IGHV subgroups the usage was below 6 %. There is also preferential usage of IGHJ genes in non-infected animals. IGHJ4 genes represented almost half of the IGHJ usage, found in 46.8 % of the IG transcripts rearranged. IGHJ5–1 was found in 20.3 % of the IGH transcripts. All the other IGHJ genes were found in less than 10 % of the IGH transcripts (7.9, 2.45, 9.6, 5.1, and 7.8 % for IGHJ1, IGHJ2, IGHJ3, IGHJ5–2, and IGHJ6, respectively).
Discussion
Nonhuman primates are one of the closest evolutionary related animal models for human biomedical research. The strong physiological, developmental, immunological and genetic similarities with humans make these animals an important experimental model for human diseases and therapeutics development. This model is especially important for infectious diseases involving life threatening pathogens as well as biological warfare agents (BWA) like filoviruses, flaviviruses, arenaviruses, and alphaviruses. For several of these pathogens, where human efficacy trials are not feasible and ethical, development and testing of therapeutics should be done by the FDA Animal Efficacy Rule. The rhesus macaque has been used extensively as an animal model for several infectious diseases, especially for hepatitis C virus (HCV) and human immunodeficiency virus (HIV) (Carlsson et al. 2004; Palermo et al. 2013). Recently, both rhesus and cynomolgus macaques have also been utilized for BWA countermeasure development (Bente et al. 2009; Herrero et al. 2011; Moraz and Kunz, 2011; Pena and Ho 2015; Reed et al. 2005; Vallender and Miller 2013). Rhesus gene loci involved in adaptive immune responses have been described, including the major histocompatibility complex (MHC) locus organization (Wiseman et al. 2013), the IG loci (Gibbs et al. 2007; Link et al. 2002; Sundling et al. 2012), T cell receptor (TR) loci (Greenaway et al. 2009) and the MHC/KIR coevolution (de Groot et al. 2015). Recently, two draft genomes and an annotated transcriptome were published for cynomolgus macaques (Ebeling et al. 2011; Higashino et al. 2012; Lee et al. 2014; Yan et al. 2011) but the knowledge of gene organization and characterization for this animal is not at the level of the rhesus macaque or human genome.
Advancing NGS technologies are being used to characterize the repertoire of the antigen receptors of B and T cells, following vaccination or infection. This novel technology allows for a longitudinal analysis of the B and T cell clonotypes to identify and characterize the cells that specifically respond and expand in response to the antigen or pathogen. In the case of the B cell receptor sequencing, this analysis can characterize the B cells participating in the specific humoral response, IGHV and IGHJ usages, clonotype frequency, somatic hypermutation rates and paratope composition and structure (Alamyar et al. 2012; Georgiou et al. 2014; Giudicelli et al. 2005; Giudicelli et al. 2015). Several publications have reported this successful analysis in mice, humans and rhesus macaques (Jackson et al. 2014; Lavinder et al. 2014; Li et al. 2013; Parameswaran et al. 2013). The IGH, IGK and IGL loci for these species are well-characterized and the germline genes and alleles have been described (Giudicelli et al. 2005; Link et al. 2002). In contrast, there is little to no information about the cynomolgus macaque IG locus organization. Without information on germline genes, the analysis on the V-(D)-J rearrangement is limited. Here we present the identification and annotation of 108 functional IGH rearranging genes (63 IGHV, 38 IGHD, and 7 IGHJ) with the goal of upgrading the level of characterization of the gene organization of the cynomolgus macaque IGH locus and permit deep analysis of IGH VDJ rearrangement in this animal model.
Given the many gene duplication and deletion events which occur during the evolution of multigene loci with highly homologous genes, it is not surprising that genome assembly of the IGH locus is challenging and that several IGHV genes were found in an unplaced scaffold. The assembly method used to generate the draft genome for the cynomolgus macaque was a hybrid method. The rhesus genome was used as a reference, which will preferentially favor the assembly of IGH regions of homology between both macaques. Thus, the location of several IGHV genes (95 genes) in an unplaced scaffold can be explained by two possible mechanisms: the failure of the genome assembly in areas where rhesus and cynomolgus macaques have a high degree of heterogeneity, or a result of the diploid nature of the genome, given that the draft genome for cynomolgus macaque resulted in the assembled of only one chromosome 7. If this female individual is the offspring of relatively genetic distant parents, then the two sets of IGHV alleles could be very different at each gene and the unplaced scaffold might be a reflection of the second unassembled chromosome. This is in line with the current genomic approach in which assemblies are built, if possible, from a single chromosome from a single individual. The cynomolgus macaque IGH locus assembly, by being built from a single chromosome, parallels the human IGH locus assembly in NCBI GR38Ch.p2 which results from the tiling of clones from the haploid hydatiform mole CHORI-17 BAC library. The M. fascicularis reference assembly will allow the description of copy number variations (CNV) by insertion/deletion polymorphisms, from different individuals and/or different haplotypes, based on IMGT-ONTOLOGY (Lefranc 2014a) as this has been done recently for the human IGH locus (Watson et al. 2013). IGH alleles of the M. fascicularis reference assembly will be entered in IMGT/GENE-DB, IMGT/V-QUEST and IMGT/HighV-QUEST, as this has been done for human alleles (Lefranc et al. 2015). If new alleles are discovered they will be similarly added if they fulfill the IMGT criteria. Future efforts to improve the resolution of the IGH locus could include: (1) BAC library shot-gun sequencing to get deeper coverage in the region; (2) more locus-specific assembly strategies, algorithms, and parameters could be used in all the steps of assembly process; and (3) longer reads or/and DNA pull-down experiments, to better resolve this region of the genome. Also, additional studies in more animals will be needed to describe the alleles at the population level. Cynomolgus macaques most likely express several IGHG isotypes encoded by different IGHG genes, similar to other primates (Attanasio et al. 2002; Lefranc and Lefranc 2001; Scinicariello et al. 2004). The fact that only partial IGHG subclasses were found so far suggests that even at the level of the constant genes, the genomic assembly needs to be improved.
In summary, we describe here the IGH locus annotation for cynomolgus macaque, containing 63 functional V genes, 38 functional D genes and 7 functional J genes and we provide preliminary data on seven constant genes (IGHM, IGHD, IGHG1, IGHG2, IGHG3, IGHE, and IGHA).
Methods
Macaca fascicularis sequence
Macaca fascicularis (cynomolgus macaque) genome data were obtained from the NCBI draft genome GCA_000364345.1. For the genome assembly, the DNA is from a 5.8 years old female from “Tinjil”, an island off the south coast of Java that was seeded with monkeys by the Washington National Primate Center. Sequences were generated on the Illumina HiSeq, with genome coverage for each paired end read type is as follows: 50 × 300–500 bp inserts, 10 × 3 kb insert, and 2 × 8 kb insert. The genome assembly was built using a hybrid approach of assisted and de novo assembly. For assisted assembly, the published assembly (MMUL_1) of rhesus macaque was used as a reference. In Macaca fascicularis_5.0, there were 102,878 contigs with an N50 contig length of 85 kb. There were 7627 scaffolds with the N50 scaffold length of 144 Mb. The IGH locus was found on chromosome 7, on scaffolds KE145476.1 localized on the 7q region near the telomere, an unlocalized scaffold KE145669, and an unplaced scaffold KE145882.1 using an in-house pipeline based on the Basic Local Alignment Search Tool (BLAST) and Fuzznuc (Altschul et al. 1990; Rice et al. 2000).
Identification of M. fascicularis IGHV, IGHD, and IGHJ genes
Annotated sequences of IGHV, IGHD, and IGHJ genes and recombination signals (RS) of all available organisms were downloaded in FASTA from the IMGT web site (http://www.imgt.org) (Lefranc et al. 2015). Using an in-house pipeline based on BLAST and Fuznuc, sequences for each chromosome, unlocalized and unplaced scaffold were searched on both strands for V and J gene sequences using Mega-BLAST (e value 0.001, word size 14). Only two scaffolds were found to have V and J hits. Further analysis was focused on these two scaffolds. For V, D, and J gene search, BLAST was used to find the V, D, and J genes. All the hits on the references are ranked by the e value and percentage of base identity and only the top hit at any given site was recorded. For V gene identification, the leader, intron, and V-region sequences, and V-RS were identified separately and assembled as a whole V-GENE-UNIT. For RS identification using Fuzznuc, all the RS annotated for all available species and RS for Human and rhesus at NCBI were used as reference sequences. The nonamers and heptamers were identified separately, then a whole RS was assembled if the spacer sequence has the correct length. All RS and other gene sequence features were annotated on the sequences submitted to NCBI TPA database.
Functional analysis
The IGHV genes were analyzed for translation start sites, open reading frames (ORF), consensus splice sites, V-RS, and conserved framework amino acids 1st-CYS (C23), conserved-TRP (W41), hydrophobic 89 and 2nd-CYS (C104), in the IMGT unique numbering (Lefranc et al. 2003), based on IMGT-ONTOLOGY concepts (Giudicelli and Lefranc 2012) and IMGT immunoinformatic standards (Lefranc 2014a). For phylogenetic analyses, the FR1-FR3 regions (corresponding to amino acids 1–104 in the IMGT unique numbering; (Elemento and Lefranc 2003)) were aligned. Trees were constructed in Geneious® 7.1.9 (HKY genetic distance model, neighbor-joining, bootstrap resampling with 100,000 replicates). Chicken IGHV was used as the outgroup. Genes sharing at least 75 % sequence identity and clustering together in the phylogenetic tree were assigned to the same IGHV subgroup (Giudicelli and Lefranc 1999, 2012; Lefranc 2014a). All the gene features and functional analysis results were annotated on the sequences submitted to NCBI TPA database.
Identification and annotation of M. fascicularis IGHC genes
The IGHC genes (IGHM, IGHD, IGHG1, IGHE, and IGHA) and associated exons were mapped onto scaffolds KE145476.1 and KE145669.1 of assembly GCA_000364345.1 based on BLAST (Altschul et al. 1990) bl2Seq pairwise alignments with reference sequences obtained from IMGT or NCBI for cynomolgus and rhesus macaques, default parameters. All reference sequences are listed in Online Resource 1; note that the majority of annotations for IGHC reference sequences were not genomic level but were from clones and transcripts. Specific genomic CH exon annotation was manually adjusted to reflect the reference sequence annotation provided by IMGT and shared motifs conserved across species as described in Scinicariello et al. (2004). 5′RACE experiments described below produced five transcripts containing constant domain sequences that were used as input into NCBI SPlign (Kapustin et al. 2008), EBI Clustal Omega (Sievers et al. 2011), and SIB Bioinformatics Resource Portal ExPASy Translate tools (http://web.expasy.org/translate/) to guide manual annotation of coding sequences alongside reference sequence alignments.
Sequence and phylogenetic analysis
Sequence alignments and phylogenetic trees were constructed and analyzed with Geneious® 7.1.9, using a global alignment with free end gaps, and a Tamura-Nei distance model, with neighbor-joining method.
Generation of RNA transcripts from Macaca fascicularis
Peripheral blood was obtained from five healthy cynomolgus macaques. Peripheral blood mononuclear cells (PBMCs) were purified by Ficoll® gradient. RNA was extracted from the cells and cDNA synthesis performed by 5′RACE, using IGG specific primers (Druar et al. 2005). Further PCR amplification was done using the RACE and the IGG specific primers. IG amplicons were prepared for sequencing according the Illumina® protocol and sequenced on an Illumina® MiSeq using a 600 cycle kit (2 × 300 cycles). Paired reads were merged and used to validate IGHV and IGHJ genes.
All animals were cared for according to the regulations and guidelines of the Institutional Care and Use Committees at their respective institutions.
Immunoglobulin sample preparation
Immunoglobulins were affinity purified from plasma of a single cynomolgus macaque using the Montage Antibody Purification Kit with Prosep-G Media (Milipore, MA, USA) as per the manufacturer’s instructions. 10 μg of purified IgG was reduced and fractionated by SDS-PAGE onto a 4–12 % Nupage Novex Bis-tris gel (Thermo Fisher Scientific, MA) in MOPS buffer and visualized by BioSafe Coommassie G-250 (BioRad, CA). The heavy chain was excised from the gel and washed with 50 % acetonitrile followed by dehydration by 100 % acetonitrile. The proteins were then reduced by 10 mM dithiothreitol (DTT) (Shelton Scientific, IA) in 100 mM NH4HCO3 pH 8.2 then alkylated by 100 mM iodoacetamide (IAA) (Sigma-Aldrich, MO) in 100 mM NH4HCO3. The gel piece was sequentially washed several times in 100 % acetonitrile followed by 100 mM NH4HCO3 before being evaporated to dryness in a SpeedVac. The IG heavy chain was incubated overnight at 37 °C with 30 μL of a 12.5 ng/μL mass spectrometry grade Trypsin Gold solution (Promega, Madison, WI) in 50 mM NH4HCO3. Peptides were then extracted by incubating in 50 % acetonitrile, 0.1 % formic acid and the combined digest dried to completion by SpeedVac.
Liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis and protein search
Individual gel digests were suspended in 25 μL of 0.1 % formic acid (mobile phase A). Five microliters of each gel digest was injected onto a Dionex Ultimate 3000 RSLCnano system housing a 15 cm × 75 um ID Easy Spray C18 (3 μm particle, 100 Å porosity) column (Thermo Fisher Scientific). The tryptic peptides were eluted with a gradient elution of 5–65 % mobile phase B (80 % acetonitrile in 0.1 % formic acid) at a flow rate of 300 nL/min. The chromatographic eluent was connected to an Easy Spray nanospray source (Thermo Fisher Scientific) and subjected to an electrospray ionization potential of 2.2 kV. An Orbitrap Elite mass spectrometer (Thermo Scientific) was used with an ion-transfer tube temperature of 2750 °C and an S-lens voltage of 50 %. A 400–2000 amu survey scan with a resolution of 30,000 (FWHM at m/z 445) was followed by 15 Rapid ms/ms scans of the most abundant ions. The data dependent scan settings used a default precursor ion charge state of 2 with an isolation width of 2.0 amu. A collision dissociation (CID) energy of 32 % with an isolation q of 0.250 was used to generate ms/ms fragment ions. Searches were performed with Proteome Discoverer 2.0 using a custom cross-species database composed of 165 GenBank records describing constant immunoglobulin domains appended to a 27,849 sequence Aedes albopictus subset of the NCBI protein non-redundant database. Static modifications include Carbamidomethyl (C) to account for alkylation of cysteines. Variable modifications used were Acetyl (K) and Oxidation (M). The delta Cn was set at 0.05 maximum and the false discovery rate (FDR) was set at 0.01 % for strict and 0.05 % for relaxed parameters. Mass tolerances were 10 ppm for the MS1 scan and 200 ppm for all ms/ms scans.
References
Alamyar E, Giudicelli V, Li S, Duroux P, Lefranc MP (2012) IMGT/HighV-quest: the IMGT® web portal for immunoglobulin (Ig) or sntibody snd T cell ceceptor (TR) analysis from NGS high throughput and deep sequencing. Immunome Research 8:15
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410
Attanasio R, Jayashankar L, Engleman CN, Scinicariello F (2002) Baboon immunoglobulin constant region heavy chains: identification of four IGHG genes. Immunogenetics 54:556–561
Bente D, Gren J, Strong JE, Feldmann H (2009) Disease modeling for Ebola and Marburg viruses. Dis Model Mech 2:12–17
Bukh J (2012) Animal models for the study of hepatitis C virus infection and related liver disease. Gastroenterology 142(1279–1287):e3
Carlsson HE, Schapiro SJ, Farah I, Hau J (2004) Use of primates in research: a global overview. Am J Primatol 63:225–237
de Groot NG, Blokhuis JH, Otting N, Doxiadis GG, Bontrop RE (2015) Co-evolution of the MHC class I and KIR gene families in rhesus macaques: ancestry and plasticity. Immunol Rev 267:228–245
Desrosiers RC (1995) Non-human primate models for AIDS vaccines. AIDS 9(Suppl A):S137–S141
Druar C, Saini SS, Cossitt MA, Yu F, Qiu X, Geisbert TW, Jones S, Jahrling PB, Stewart DI, Wiersma EJ (2005) Analysis of the expressed heavy chain variable-region genes of Macaca fascicularis and isolation of monoclonal antibodies specific for the Ebola virus’ soluble glycoprotein. Immunogenetics 57:730–738
Ebeling M, Kung E, See A, Broger C, Steiner G, Berrera M, Heckel T, Iniguez L, Albert T, Schmucki R, Biller H, Singer T, Certa U (2011) Genome-based analysis of the nonhuman primate Macaca fascicularis as a model for drug safety assessment. Genome Res 21:1746–1756
Elemento O, Lefranc MP (2003) IMGT/PhyloGene: an on-line tool for comparative analysis of immunoglobulin and T cell receptor genes. Dev Comp Immunol 27:763–779
Georgiou G, Ippolito GC, Beausang J, Busse CE, Wardemann H, Quake SR (2014) The promise and challenge of high-throughput sequencing of the antibody repertoire. Nat Biotechnol 32:158–168
Gibbs RA, Rogers J, Katze MG, Bumgarner R, Weinstock GM, Mardis ER, Remington KA, Strausberg RL, Venter JC, Wilson RK, Batzer MA, Bustamante CD, Eichler EE, Hahn MW, Hardison RC, Makova KD, Miller W, Milosavljevic A, Palermo RE, Siepel A, Sikela JM, Attaway T, Bell S, Bernard KE, Buhay CJ, Chandrabose MN, Dao M, Davis C, Delehaunty KD, Ding Y, Dinh HH, Dugan-Rocha S, Fulton LA, Gabisi RA, Garner TT, Godfrey J, Hawes AC, Hernandez J, Hines S, Holder M, Hume J, Jhangiani SN, Joshi V, Khan ZM, Kirkness EF, Cree A, Fowler RG, Lee S, Lewis LR, Li Z, Liu YS, Moore SM, Muzny D, Nazareth LV, Ngo DN, Okwuonu GO, Pai G, Parker D, Paul HA, Pfannkoch C, Pohl CS, Rogers YH, Ruiz SJ, Sabo A, Santibanez J, Schneider BW, Smith SM, Sodergren E, Svatek AF, Utterback TR, Vattathil S, Warren W, White CS, Chinwalla AT, Feng Y, Halpern AL, Hillier LW, Huang X, Minx P, Nelson JO, Pepin KH, Qin X, Sutton GG, Venter E, Walenz BP, Wallis JW, Worley KC, Yang SP, Jones SM, Marra MA, Rocchi M, Schein JE, Baertsch R, Clarke L, Csuros M, Glasscock J, Harris RA, Havlak P, Jackson AR, Jiang H, et al. (2007) Evolutionary and biomedical insights from the rhesus macaque genome. Science 316:222–234
Giudicelli V, Chaume D, Lefranc MP (2005) IMGT/GENE-DB: a comprehensive database from human and mouse immunoglobulin and T cell receptor genes. Nucl. Acids Res. 33:D256–D261
Giudicelli V, Duroux P, Ginestoux C, Folch D, Jabado-Michaloud J, Chaume D, Lefranc MP (2006) IMGT/LIGM-DB, the IMGT(R) comprehensive database of immunoglobulin and T cell receptor nucleotide sequences. Nucl Acids Res 34:D781–D784
Giudicelli V, Duroux P, Lavoie A, Aouinti S, Lefranc MP, Kossida S (2015) From IMGT-ONTOLOGY to IMGT/HighVQUEST for NGS immunoglobulin (IG) and T cell receptor (TR) repertoires in autoimmune and infectious diseases. Autoimmun Infect Dis 1
Giudicelli V, Lefranc MP (1999) Ontology for immunogenetics: the IMGT-ONTOLOGY. Bioinformatics 15:1047–1054
Giudicelli V, Lefranc MP (2012) IMGT-ONTOLOGY Front Genet 3:79
Greenaway HY, Kurniawan M, Price DA, Douek DC, Davenport MP, Venturi V (2009) Extraction and characterization of the rhesus macaque T-cell receptor beta-chain genes. Immunol Cell Biol 87:546–553
Herrero L, Nelson M, Bettadapura J, Gahan ME, Mahalingam S (2011) Applications of animal models of infectious arthritis in drug discovery: a focus on alphaviral disease. Curr Drug Targets 12:1024–1036
Higashino A, Sakate R, Kameoka Y, Takahashi I, Hirata M, Tanuma R, Masui T, Yasutomi Y, Osada N (2012) Whole-genome sequencing and analysis of the Malaysian cynomolgus macaque (Macaca fascicularis) genome. Genome Biol 13:R58
Higgs S, Ziegler SA (2010) A nonhuman primate model of chikungunya disease. J Clin Invest 120:657–660
Jackson KJ, Liu Y, Roskin KM, Glanville J, Hoh RA, Seo K, Marshall EL, Gurley TC, Moody MA, Haynes BF, Walter EB, Liao HX, Albrecht RA, Garcia-Sastre A, Chaparro-Riggers J, Rajpal A, Pons J, Simen BB, Hanczaruk B, Dekker CL, Laserson J, Koller D, Davis MM, Fire AZ, Boyd SD (2014) Human responses to influenza vaccination show seroconversion signatures and convergent antibody rearrangements. Cell Host Microbe 16:105–114
Kapustin Y, Souvorov A, Tatusova T, Lipman D (2008) Splign: algorithms for computing spliced alignments with identification of paralogs. Biol Direct 3:20
Lavinder JJ, Wine Y, Giesecke C, Ippolito GC, Horton AP, Lungu OI, Hoi KH, DeKosky BJ, Murrin EM, Wirth MM, Ellington AD, Dorner T, Marcotte EM, Boutz DR, Georgiou G (2014) Identification and characterization of the constituent human serum antibodies elicited by vaccination. Proc Natl Acad Sci U S A 111:2259–2264
Lee A, Khiabanian H, Kugelman J, Elliott O, Nagle E, Yu GY, Warren T, Palacios G, Rabadan R (2014) Transcriptome reconstruction and annotation of cynomolgus and African green monkey. BMC Genomics 15:846
Lefranc MP (2014a) Immunoglobulin (IG) and T cell receptor genes (TR): IMGT(R) and the birth and rise of immunoinformatics. Front Immunol 5:22
Lefranc MP (2014b) Immunoglobulins: 25 years of immunoinformatics and IMGT-ONTOLOGY. Biomolecules 4:1102–1139
Lefranc MP, Giudicelli V, Duroux P, Jabado-Michaloud J, Folch D, Aouinti S, Carillon E, Duvergey H, Houles A, Paysan-Lafosse T, Hadi-Saljoqi S, Sasorith S, Lefranc G, Kossida S (2015) IMGT(R), the International ImMunoGeneTics information system (R) 25 years on. Nucleic Acids Res 43:D413–D422
Lefranc MP, Lefranc G (2001) The immunoglobulin facts book. Academic Press, London, pp. 1–458
Lefranc MP, Pommié C, Ruiz M, Giudicelli V, Foulquier E, Troung L, Thouvenin-Content V, Lefranc G (2003) IMGT unique numbering for immunoglobulin and T cell receptor variable domains and Ig superfamily V-like domains. Dev Comp Immunol 27:55–77
Li S, Lefranc MP, Miles JJ, Alamyar E, Giudicelli V, Duroux P, Freeman JD, Corbin VD, Scheerlinck JP, Frohman MA, Cameron PU, Plebanski M, Loveland B, Burrows SR, Papenfuss AT, Gowans EJ (2013) IMGT/HighV QUEST paradigm for T cell receptor IMGT clonotype diversity and next generation repertoire immunoprofiling. Nat Commun 4:2333
Li Z, Liu G, Tong Y, Zhang M, Xu Y, Qin L, Wang Z, Chen X, He J (2015) Comprehensive analysis of the T-cell receptor beta chain gene in rhesus monkey by high throughput sequencing. Sci Rep 5:10092
Link JM, Hellinger MA, Schroeder HW Jr (2002) The rhesus monkey immunoglobulin IGHD and IGHJ germline repertoire. Immunogenetics 54:240–250
Lucas AH (2003) Immunoglobulin gene construction: human. encyclopedia of life sciences. 8
Moraz ML, Kunz S (2011) Pathogenesis of arenavirus hemorrhagic fevers. Expert Rev Anti-Infect Ther 9(1):49–59
Nakayama E, Saijo M (2013) Animal models for Ebola and Marburg virus infections. Front Microbiol 4:267
Olivieri DN, Gambon-Deza F (2015) V genes in primates from whole genome sequencing data. Immunogenetics 67:211–228
Palermo RE, Tisoncik-Go J, Korth MJ, Katze MG (2013) Old world monkeys and new age science: the evolution of nonhuman primate systems virology. ILAR J 54:166–180
Parameswaran P, Liu Y, Roskin KM, Jackson KK, Dixit VP, Lee JY, Artiles KL, Zompi S, Vargas MJ, Simen BB, Hanczaruk B, McGowan KR, Tariq MA, Pourmand N, Koller D, Balmaseda A, Boyd SD, Harris E, Fire AZ (2013) Convergent antibody signatures in human dengue. Cell Host Microbe 13:691–700
Pena JC, Ho WZ (2015) Monkey models of tuberculosis: lessons learned. Infect Immun 83:852–862
Pripuzova NS, Gmyl LV, Romanova L, Tereshkina NV, Rogova YV, Terekhina LL, Kozlovskaya LI, Vorovitch MF, Grishina KG, Timofeev AV, Karganova GG (2013) Exploring of primate models of tick-borne flaviviruses infection for evaluation of vaccines and drugs efficacy. PLoS One 8:e61094
Reed DS, Larsen T, Sullivan LJ, Lind CM, Lackemeyer MG, Pratt WD, Parker MD (2005) Aerosol exposure to western equine encephalitis virus causes fever and encephalitis in cynomolgus macaques. J Infect Dis 192:1173–1182
Reynaud JM, Horvat B (2013) Animal models for human herpesvirus 6 infection. Front Microbiol 4:174
Rice P, Longden I, Bleasby A (2000) EMBOSS: the European molecular biology open software suite. Trends Genet 16:276–277
Schroeder HW Jr, Cavacini L (2010) Structure and function of immunoglobulins. J Allergy Clin Immunol 125:S41–S52
Scinicariello F, Engleman CN, Jayashankar L, McClure HM, Attanasio R (2004) Rhesus macaque antibody molecules: sequences and heterogeneity of alpha and gamma constant regions. Immunology 111:66–74
Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Soding J, Thompson JD, Higgins DG (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7:539
Sundling C, Li Y, Huynh N, Poulsen C, Wilson R, O'Dell S, Feng Y, Mascola JR, Wyatt RT, Karlsson Hedestam GB (2012) High-resolution definition of vaccine-elicited B cell responses against the HIV primary receptor binding site. Science Transl Med 4
Thullier P, Chahboun S, Pelat T (2010) A comparison of human and macaque (Macaca mulatta) immunoglobulin germline V regions and its implications for antibody engineering. MAbs 2:528–538
Vallender EJ, Miller GM (2013) Nonhuman primate models in the genomic era: a paradigm shift. ILAR J 54:154–165
Watson CT, Steinberg KM, Huddleston J, Warren RL, Malig M, Schein J, Willsey AJ, Joy JB, Scott JK, Graves TA, Wilson RK, Holt RA, Eichler EE, Breden F (2013) Complete haplotype sequence of the human immunoglobulin heavy-chain variable, diversity, and joining genes and characterization of allelic and copy-number variation. Am J Hum Genet 92:530–546
Wiseman RW, Karl JA, Bohn PS, Nimityongskul FA, Starrett GJ, O'Connor DH (2013) Haplessly hoping: macaque major histocompatibility complex made easy. ILAR J 54:196–210
Yan G, Zhang G, Fang X, Zhang Y, Li C, Ling F, Cooper DN, Li Q, Li Y, van Gool AJ, Du H, Chen J, Chen R, Zhang P, Huang Z, Thompson JR, Meng Y, Bai Y, Wang J, Zhuo M, Wang T, Huang Y, Wei L, Li J, Wang Z, Hu H, Yang P, Le L, Stenson PD, Li B, Liu X, Ball EV, An N, Huang Q, Zhang Y, Fan W, Zhang X, Li Y, Wang W, Katze MG, Su B, Nielsen R, Yang H, Wang J, Wang X, Wang J (2011) Genome sequencing and comparison of two nonhuman primate animal models, the cynomolgus and Chinese rhesus macaques. Nat Biotechnol 29:1019–1023
Zimin AV, Cornish AS, Maudhoo MD, Gibbs RM, Zhang X, Pandey S, Meehan DT, Wipfler K, Bosinger SE, Johnson ZP, Tharp GK, Marcais G, Roberts M, Ferguson B, Fox HS, Treangen T, Salzberg SL, Yorke JA, Norgren RB Jr (2014) A new rhesus macaque assembly and annotation for next-generation sequencing analyses. Biol Direct 9:20
Acknowledgments
Special acknowledgments to the IMGT team for support and help with the IGH gene annotations. We would also like to thank Dr. John Trefry for providing study samples supporting this effort and Dr. Brett Beitzel for insightful discussions and thoughtful advice.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
This work was supported by the Defense Threat Reduction Agency under the US Army Medical Research Institute of Infectious Diseases project number 31741630.
The content of this publication does not necessarily reflect the views or policies of the US Department of the Army, the US Department of Defense, and the US Department of Health and Human Services or of the institutions and companies with which the authors are affiliated.
All procedures performed in studies involving animals were in accordance with the ethical standards of the institution or practice at which the studies were conducted. Research was conducted under an IACUC approved protocol in compliance with the Animal Welfare Act, PHS Policy, and other Federal statutes and regulations relating to animals and experiments involving animals. The facility where this research was conducted is accredited by the Association for Assessment and Accreditation of Laboratory Animal Care, International and adheres to principles stated in the Guide for the Care and Use of Laboratory Animals, National Research Council, 2011.
This article does not contain any studies with human participants performed by any of the authors.
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
ESM 1
(DOCX 35 kb)
Rights and permissions
About this article

Cite this article
Yu, GY., Mate, S., Garcia, K. et al. Cynomolgus macaque (Macaca fascicularis) immunoglobulin heavy chain locus description. Immunogenetics 68, 417–428 (2016). https://doi.org/10.1007/s00251-016-0921-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00251-016-0921-2