
Abstract
Archaeal communities from mercury and uranium-contaminated freshwater stream sediments were characterized and compared to archaeal communities present in an uncontaminated stream located in the vicinity of Oak Ridge, TN, USA. The distribution of the Archaea was determined by pyrosequencing analysis of the V4 region of 16S rRNA amplified from 12 streambed surface sediments. Crenarchaeota comprised 76% of the 1,670 archaeal sequences and the remaining 24% were from Euryarchaeota. Phylogenetic analysis further classified the Crenarchaeota as a Freshwater Group, Miscellaneous Crenarchaeota group, Group I3, Rice Cluster VI and IV, Marine Group I and Marine Benthic Group B; and the Euryarchaeota into Methanomicrobiales, Methanosarcinales, Methanobacteriales, Rice Cluster III, Marine Benthic Group D, Deep Sea Hydrothermal Vent Euryarchaeota 1 and Eury 5. All groups were previously described. Both hydrogen- and acetate-dependent methanogens were found in all samples. Most of the groups (with 60% of the sequences) described in this study were not similar to any cultivated isolates, making it difficult to discern their function in the freshwater microbial community. A significant decrease in the number of sequences, as well as in the diversity of archaeal communities was found in the contaminated sites. The Marine Group I, including the ammonia oxidizer Nitrosopumilus maritimus, was the dominant group in both mercury and uranium/nitrate-contaminated sites. The uranium-contaminated site also contained a high concentration of nitrate, thus Marine Group I may play a role in nitrogen cycle.
Similar content being viewed by others
Introduction
Archaeal communities from freshwater environments have been studied mainly from lake sediments [8, 10, 23, 25, 29, 34, 37, 58]. There are also a few studies of archaeal communities from river sediments, but they do not encompass archaeal communities from low order streams [26, 46]. In some cases, the research focused only on the methanogen communities from lake sediments [9, 18, 55]. These freshwater studies detected Crenarchaeota 16S rRNA gene sequences primarily from uncultivated groups such as Marine Group I [10, 29, 37, 58], Miscellaneous Crenarchaeota group [10, 23, 34, 46], Freshwater Group [25], Group I3 [8], Marine Benthic Groups B and C [23], and Rice Clusters IV and VI [26]. Most of the freshwater euryarchaeotal communities showed the presence of cultivated methanogens Methanomicrobiales and/or Methanosarcinales [9, 18, 23, 25, 26, 29, 37, 46, 55, 58]. In addition, the members of the uncultivated Euryarchaeota were found in freshwater samples: namely Group III or Rice Cluster III [10, 18, 25, 26], Marine Benthic Group D [23], Deep Hydrothermal Vent Euryarchaeota 1 [34], Eury 5 [29] and Rice Cluster V and LDS Cluster [18]. Members of the Rice Clusters and other groups were also isolated from different environments including rice paddy fields [36, 45], indicating the wide distribution of the archaeal communities.
The only study of the archaeal community from a mercury-contaminated site focused on an acidic hot spring environment [56]. In this environment, Crenarchaeota represented the majority of the microbial population, as identified by 16S rRNA phylogeny. In addition, all cultured archaeal isolates were closely related to Sulfolobus solfataricus, a hyperthermophile crenarchaeote isolated from acidic geothermal pools. A study reporting mercury resistance in S. solfataricus also demonstrated the function of the archaeal mercuric reductase and its regulator MerR in mercury resistance [53].
A large amount of mercury and uranium wastes have been discharged in the area surrounding the Oak Ridge Reservation, in Oak Ridge, TN, USA, over the past few decades [15, 21]. These contaminants still persist in the low order streams draining the watersheds. A complete analysis of 16S rRNA pyrosequencing from microbial communities in contaminated and uncontaminated surface sediments from four streams in the Oak Ridge area is under investigation (unpublished results) as part of a study of mercury contamination in the area. The goal of the present study was to leverage the sampling and chemical analysis done for the mercury project to characterize and analyze the diversity of the archaeal communities present in surface sediments at a number of impacted and control sites of low order streams near Oak Ridge, TN, USA. The present study is the first, to our knowledge, to investigate the impact of anthropogenic sources of heavy metal contaminants on the diversity of archaeal communities in surface streams.
Materials and Methods
Sample Collection and Chemical Analyses
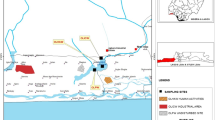
Twelve surface sediment samples from three contaminated streams; Bear Creek (BC), East Fork Poplar Creek (EF6, EF13 and EF23), White Oak Creek (WC); and one control stream, Hinds Creek (HC), were studied. All four low-order streams are located in the vicinity of Oak Ridge, TN, USA (Fig. 1). At each location, samples were collected from middle stream and bank points on one time point (July 2008). Sediment was collected by placing the wide-mouthed plastic bottle on the stream bottom and moving sediment and water into the bottle, bailing the mixture (1–2 L) into a bucket, pouring into a sterilized plastic container, and centrifuging to obtain the fine sediments. The sediments were mainly loose materials with a variety of grain size that varied among stations and samples.

Sampling locations in creeks near Oak Ridge, TN, USA. The location of DOE Y-12 National Security Complex (Y-12 NSC) and the sampling sites are indicated. The location coordinates for sampling sites are: Hinds Creek (HC) 36°08′N, 84°03′W; White Oak Creek (WC) 35°55′N, 84°18′W; Bear Creek (BC) 35°58′N, 84°16′W; East Fork Poplar Creek K23.4 (EF23): 35°59′N, 84°14′W; East Fork Poplar Creek K13.8 (EF13) 35°59′N, 84°18′W; East Fork Poplar Creek K6.3 (EF6) 35°57′N, 84°21′W
Chemical analyses were performed on water samples. Water samples were collected concurrently, centrifuged, and analyzed for major anions and cations by ion chromatography and inductively coupled plasma mass spectrometry (ICP-MS), respectively [20, 38]. Uranium was measured by either kinetic phosphorimetry analysis [5, 16] or ICP-MS [63]. In addition, total mercury was measured in sediments. The chemical measurements in the bank- and the mid-channel samples were similar at each location so the averages (Table 1, Supplemental Table 1 and Vishnivetskaya et al., unpublished data) are shown.
DNA Extraction and Pyrosequencing of Archaeal 16S rRNA Genes
The total community genomic DNA was extracted from approximately 0.5–1 g (wet weight) of the fine sediment using the PowerSoil™ DNA Isolation Kit (Mo Bio Labs, Inc., Carlsbad, CA, USA). To amplify the hypervariable V4 region (∼300 bp) of the 16S rRNA gene of Archaea, we used as forward primer oligonucleotides that contained the modified U519F primer [59] fused to variable 6-nucleotide key tags for multiplexing [12] and to the 454 FLX sequencing primer A (5′-GCCTCCCTCGCGCCATCAGxxxxxxCAGYMGCCRCGGKAAHACC, where the x region represents the various key tags and the 16S rRNA primer is bold). The reverse oligonucleotide primer represented a fusion of the 454 FLX sequencing primer B and a modified Arch806R primer [60] (5′-GCCTTGCCAGCCCGCTCAGGGACTACNSGGGTMTCTAAT, where the 16S rRNA region is bold). PCR reactions (50 μl) contained 1.5 μl of each forward and reverse primers (10 μM each), 1 μl template DNA (40–80 ng/μl), and 0.6 μl (2.5 U/μl) AccuPrime™ Pfx DNA polymerase (Invitrogen, Carlsbad, CA, USA). Samples were initially denatured at 95°C for 2 min; then amplified using 30 cycles of 95°C for 15 s, 55°C for 30 s, and 68°C for 45 s; with final extension of 3 min at 68°C. Negative control reactions without template were always performed. The PCR amplicons were purified using the Agencourt AMPure solid-phase paramagnetic bead technology (Agencourt Bioscience Corporation, Beverly, MA, USA) and the amplicon purity, concentration and size were estimated using DNA 1000 chip and Agilent 2100 Bioanalyzer (Agilent Technologies, Inc., Waldbronn, Germany). The sequencing reactions were performed on a 454 Life Sciences Genome Sequencer FLX (Roche Diagnostics, Indianapolis, IN, USA) using unidirection amplicon library sequencing protocol with emPCR Kit II (Roche). The Genome Sequencer FLX provides a single-read accuracy of >99.5% for each individual read of 200–300 bp. Raw 454 FLX data (∼30,000 kb) were initially processed through the RDP’s (Ribosomal Database Project) Pyrosequencing Pipeline (http://pyro.cme.msu.edu/index.jsp) [13]. During this process, the sequences were first sorted by tag sequence, then the 16S rRNA primers were removed and sequences under 200 bp or sequences containing Ns were filtered out. A total of 14,897 high-quality sequences of 200–250 bp were obtained for 12 samples.
Phylogenetic Analysis
Sequence alignment was performed using the fast RNA secondary structure similarity Infernal Software Package [40]. Aligned sequences were clustered by the complete-linkage clustering method using the RDP pyrosequencing tools (http://pyro.cme.msu.edu/index.jsp, [13]). Clustered files for all the Archaeal sequences at every site separately (midchannel and near bank locations combined) were used to calculate Shannon Index and Chao1 estimator of community diversity (RDP pyrosequencing tool). To get the similar number of sequences for each site, the datasets were randomly sampled from using Daisy Chopper (available from www.genomics.ceh.ac.uk/GeneSwytch/).
Representative sequences from 20 Crenarchaeota and from 52 Euryarchaeota clusters were used for BLASTN search in the National Center for Biotechnology Information (NCBI) database (http://blast.ncbi.nlm.nih.gov/Blast.cgi). Closely related sequences were included in phylogenetic analysis. Sequences affiliated with Crenarchaeota and Euryarchaeota were aligned separately with closely related sequences from the NCBI database, first using the ClustalW multiple alignments with the 1,000 bootstrap option included in the BioEdit program (version 7.0.9.0). Then, the V4 region was manually selected and its final alignment was manually improved using BioEdit. Neighbor-joining bootstrap trees (with 500 replicates) were constructed using MEGA (version 4.1). Sequences in clusters with 95% identity were grouped. Branches of the trees were further classified into previously reported groups.
Sequence Accession Numbers
Sequences from this study were deposited in the GenBank database under accession numbers GU080346–GU080354, GU080356–GU080415, GU080417–GU081588, GU081590–GU081594, GU081596–GU081606, GU081609–GU081611, GU081613 and GU081615–GU081619 for Crenarchaeota sequences and GU081620–GU082015 for Euryarchaeota sequences.
Statistical Analyses
One-way ANOVA analysis with Tukey’s Honestly Significant Difference (p < 0.05) was performed on each chemical and physical parameter to determine significant differences among the parameters by sampling site (SYSTAT 10). Pearson correlation coefficient determined colinearity among the environmental parameters (SYSTAT 10).
Constrained ordination techniques were utilized to identify patterns of variation in the distribution of archaeal gene sequences in streams and correlated the distribution with the environmental and physical descriptors. Sequence abundances for each cluster were converted into weight percentage values by dividing the total abundance for that sample; weight percentage values were natural log transformed (ln + 1). Detrended correspondence analysis, an indirect gradient analysis based on segment length, was performed to determine the modality of the sequence data. The analysis resulted in short (<1.5) segment lengths, indicating that the dataset was considered linear and suitable for direct gradient analysis. Redundancy analysis (RDA) was applied, with the response variables being the weighted sequence cluster data and the predictor variables were the measured environmental and physical parameters. Forward selection of the predictor variables followed by Monte Carlo permutation tests were used to prevent artificial inflation of variation due to autocorrelation in the constrained ordination model [33].
Results
Location and Chemical Properties of Sampling Sites
The BC site was the most unique site sampled as it was contaminated with high levels of uranium. In addition to the uranium, BC had elevated levels of co-contaminants: nitrate, barium, calcium, chloride, sodium, conductivity, DIC, iron, magnesium, sulfate, and strontium (Table 1 and Vishnivetskaya et al., unpublished data). The EF sites showed decreasing concentration of mercury in water and sediment samples with increasing the distance from the DOE Y-12 National Security Complex, NSC (i.e., sites from EF23 to EF6; Fig. 1). A high level of sulfate and pH of 8.8 characterized the WC site. The HC control site had the lowest levels of contaminants. Table 1 presents the total concentrations of mercury and uranium. In addition, the chemical equilibrium distributions of the mercury and uranium species were similar in all the samples (not shown).
Archaeal Sequences Classification
Only 1,838 sequences of the V4 region of 16S rRNA genes were identified as archaeal by the RDP classifier at the 80% confidence threshold. However, a total of 14,897 sequences were obtained from the 12 samples, including 11,172 bacterial sequences (Table 2). We had modified previously published oligonucleotides to increase the coverage of the Archaea domain in amplifying the V4 region. The original primer U519F [59] captured about 85% of bacteria but only a little more than 50% of the Archaea, missing groups like Nanoarchaeota, Korarchaeota and several groups within Euryarchaeota when no mismatches are allowed. The modified U519F primer was designed to capture 95% Archaea and only 1% Bacteria without mismatches. However, with one mismatch the primer became closer to the universal primer. When using the universal reverse primer under PCR conditions compatible for mismatches, a large fraction of the sequences amplified were bacterial in nature. The actual archaeal sequences were grouped into 252 clusters with a distance cut off of 0.03 using the RDP complete linkage clustering tool. At this level, only eight clusters matched the sequences of cultivated archaea. Clusters containing sequences from more than one sample as well as clusters with one or two sequences 97% identical to cultivated strains (M. bryantii, Thermococcus profundus, Methanolobus vulcani, Methanobacterium oryzae, Methanomethylovorans hollandica, and Methanobacterium subterraneum) were chosen for further analysis (a total of 109 clusters at 97% identity). Clusters containing sequences at 95% identity were grouped, resulting in 72 groups (Supplemental Table 2).
RDA indicated that there were relationships between archaeal community structure and chemical conditions at the site (Fig. 2). The analysis tested all the chemical parameters (Table 1 and July data in Vishnivetskaya et al., unpublished data). RDA is based on multivariate linear models relating principal axes to the observed environmental variables. RDA has been used for correlation of microbial communities with other parameters [1, 17]. The RDA axes 1 and 2 described 62.8% and 1.6% of the variation in the archaeal communities, respectively. Samples containing similar community composition clustered near to each other on the RDA biplot.

Redundancy analysis (RDA) of the archaeal sequences in correlation with chemical parameters. A total of 71 clusters at 95% sequence identity were used for analysis. RDA with clusters obtained grouped at 97% sequence identity showed similar distribution (not shown). Open symbols represent samples taken in mid-channel and closed symbols represent samples taken near the bank of stream. Correlations with SO 2−4 and temperature are indicated. The regression scores (R) for SO 2−4 is 0.4312 for axis 1 and −0.9024 for axis 2. The R scores for temperature are −0.9082 for axis 1 and −0.4185 for axis 2. In addition, pH showed positive correlation with SO 2−4 . Crenarchaeota clusters are indicated in bold and Euryarchaeota clusters are indicated in italic
Samples from the most contaminated sites, BC and EF23, and from sulfate-contained site WC had positive RDA axis 1 scores. The variation characterizing these samples was associated with Crenarchaeota sequences found in clusters 8, 9. The WC samples were also significantly correlated with relatively high SO 2−4 concentrations and pH levels of stream water.
Samples taken from EF6 and EF13 with moderate levels of Hg (Table 1) clustered together along the negative sides of RDA axis 1 and 2. The variations in the EF6 and EF13 samples were associated with the differences in water temperature (∼21°C), with the other samples being approximately 2°C colder (Table 1). Crenarchaeota and Euryarchaeota sequences from clusters 1, 2, 10, 16, 29, 30, 32, 42, and 48 were associated with variations described by the negative RDA scores. Samples taken from the control site HC had negative RDA 1 and positive RDA 2 scores. Euryarchaeota sequences from cluster 39 were associated with the positive RDA scores of axis 2. It is also noted that replicate samples taken from the middle of the stream and near to bank showed similar community composition (Fig. 2), therefore they were combined for phylogenetic analysis.
The uranium/nitrate contaminated site (BC) had the lowest number of archaeal sequences. Most of the archaeal sequences were found in the mercury-contaminated EF sites (78%); with a decreasing amount toward the most contaminated site (EF23) in this creek (Table 2). WC and the control site (HC) had a similar number of archaeal sequences. The archaeal sequences found fell into both the Crenarchaeota (76%) and the Euryarchaeota (24%) phyla.
Diversity indices calculated at 0.03 distance level for each site (midchannel and near-bank locations combined) are shown in Table 3. Lower diversity was observed in the highly contaminated sites BC and EF23. In spite of the differences in sequence library sizes, the Shannon indices for less contaminated EF6, EF13, and WC and uncontaminated HC were in similar range (3.3–3.7). Diversity in high mercury-contaminated EF23 site characterized by 247 sequences was lower in comparison to the control site with 168 sequences.
Crenarchaeota Sequences Analysis
The Crenarchaeota sequences fell into seven described groups (Fig. 3a [22, 26, 28, 43, 62]). Only the Marine Group I include cultivated strains. Clear differences were observed in the distribution of the crenarchaeal sequences among the sampling sites (Fig. 4a). The sequences in groups Freshwater, Miscellaneous Crenarchaeota, Group I3, and Marine Benthic B were most abundance in the background site HC and the less contaminated sites EF6 and EF13. Members of the Marine Group I were found in all sites; however, they were dominant in the high mercury EF23 site (Fig. 4a). Rice Cluster VI was found mostly in EF23 and WC sites. Rice Cluster IV was found mostly in EF13 and EF23. The uranium/nitrate-contaminated site BC had the lowest number of Crenarchaeota (51 sequences) with the lowest variety: Marine Group I (74%), Miscellaneous Crenarchaeota (10%) and Group I3 (16%).


Neighbor-joining phylogenetic trees of crenarchaeotal (a) and euryarchaeotal (b) V4 region of 16S rRNA gene sequences obtained from all samples. Similar topology for both phylogenetic trees was obtained when only full length sequences of published 16S rRNA genes were included into analysis (not shown). Bootstrap values greater than 50% are indicated at the nodes. The scale bar indicates 0.05 nucleotide substitution per sequence position. Clusters obtained during this study are indicated in bold; the number of sequences in the corresponding cluster followed by the number of samples these sequences were found in (a total of 12 samples) is given in parentheses. Clusters represent a group of sequences with 95% identity. The complete list of sequences in each cluster is given in Supplemental Table 2. Accession numbers of published sequences are indicated in square brackets. Previously described major phylogenetic lineages are indicated. The sequences of Methanosarcina acetivorans C2A [AE010299] and Thermococcus kodakarensis NR [028216] serve as an out-group in A and Nitrosopumilus maritimus SCM1 [CP000866] and Thermoproteus neutrophilus [AB009618] were used for an out-group in B

Archaeal community structures within each sampling site. The sequences from samples collected at mid-channel and stream bank were combined for each location. The sequences were grouped by major phylogenetic lineages presented in Fig. 3. Only groups with more than five sequences were included. For the larger groups, the number of sequences is indicated. a Crenarchaeota community; b Euryarchaeota community
The majority of the archaeal sequences (798 sequences) fell into the Miscellaneous Crenarchaeota group (Fig. 3a). The members of the Miscellaneous Crenarchaeota group as well as the members of Group I3, Marine Benthic B and Freshwater group were isolated from a large variety of environments described in detail in Supplemental Table 3.
The second largest cluster, with 366 archaeal sequences, fell into the Marine Group I. This group includes the chemolithoautotrophic nitrifier Nitrosopumilus maritimus isolated from a tropical marine tank aquarium [30]. In addition, uncultivated sequences have been found in other environments (Supplemental Table 3).
Fewer sequences were found representing other groups. Rice Cluster IV and Rice Cluster VI members were found in fresh water, marine and agricultural environments (Supplemental Table 3).
Euryarchaeota Sequences Analysis
The Euryarchaeota sequences fell into nine described groups (Fig. 3b [11, 23, 27, 47, 48, 54]). In addition, seven clusters (52, 75, 37, 38, 31, 46, and 65) were not included in any known group. Most of the euryarchaeotal sequences (83%) were obtained from the background (HC) and the less contaminated sites (EF6 and EF13; Table 2). A few low-diversity Euryarchaeota sequences were detected in the mercury-contaminated site EF23 and uranium-contaminated site BC (Fig. 4b). Despite this, Fig. 4b clearly showed the differences between the BC and EF23 communities. In addition, these Euryarchaeota sequences included many related to cultivated strains.
The H2-dependent methanogens Methanomicrobiales and the Rice Cluster III, with 30% and 18% of the euryarchaeota sequences, respectively, were found in all sites (Fig. 4b). Methanomicrobiales sequences were closely related to an acidiphilic methanogen Methanoregula boonei [3]; Methanolinea tarda, a methanogen isolated from an anaerobic propionate-degradation enrichment culture and to Methanosphaerula palustris, a mesophilic methanogen isolated from minerotrophic fen peatlands [7]. Members of the Rice Cluster III were isolated from oil contaminated soil and groundwater; and rice fields (Supplemental Table 4). Additionally isolated was a heterotrophic, anaerobic, mesophilic, and neutrophilic member of Rice Cluster III, related to the Thermoplasmatales [27].
The Methanosarcinales and the Eury 5 groups (23.5% of the total Euryarchaeota sequences) were found in all sites except the BC site (Fig. 4b) and are closely related to the mesophilic acetotrophic Methanosarcina acetivorans [57]; the methanol and methylamines dependent Methanolobus profundi [39] (Supplemental Table 4); M. hollandica, isolated from fresh water sediments containing dimethyl sulfide [35]; and Methanothrix soehngenii [44]. The Eury 5 group [48]; also named Pedant-33 [2], UEG-2 [61] or SM1 [49]; formed a deeply divergent, but well-supported branch in the Euryarchaeota phylogenetic tree (Fig. 3b). Members of the Eury 5 group were found in agricultural and marine environments (Supplemental Table 4).
Sequences affiliated with H2-dependent methanogen Methanobacteriales and Cluster 31 were found in all the sites except the high mercury site EF23 (Fig. 4b). M. oryzae, M. bryantii, M. subterraneum, and Methanobacterium thermoautotrophicum [19, 24, 31] were included in Cluster 25 (Fig. 3b, Supplemental Table 4). Cluster 31 fell into a separate branch of the phylogenetic tree (Fig. 3b), homologs to this cluster were found in surface water of a freshwater forest lake and also fell in a separate phylogenetic group [25].
The Marine Benthic Group D and Deep Sea Hydrothermal Vent Euryarchaeota 1; including a total of 22 sequences; were found in EF sites and the WC site (Fig. 4b). Members of these two groups were not cultivated and were found in minerotropic fen peatland; lake sediments; and water from a cave rich in hydrogen sulfide, carbon dioxide and methane (Supplemental Table 4).
Few sequences from clusters 31 and 65 (Fig. 3b) were found mostly in the WC site. Cluster 31 contained four sequences in WC and cluster 65 contained two sequences, one in WC and one in HC. Homologs to these sequences were isolated from marine environments (Supplemental Table 4).
Discussion
The size and diversity of the archaeal community in the surface freshwater sediments studied were related to the contamination present in the streams (Table 3). Differences in the archaeal community structure along the East Fork Poplar Creek (EF sites) were related to increasing concentration of mercury. Except for mercury (Table 1 and Vishnivetskaya et al., unpublished data), most of the chemical parameters except nitrate and uranium showed minimal differences among the EF sites. The most evident effect of the chemistry on the archaeal community was found in BC, with the lowest number of archaeal sequences and lowest diversity (Tables 2 and 3) and highest nitrate and uranium (Table 1). In another study, the archaeal community was reduced in heavy-metal (Cd, Cu, Ni, and Zn) contaminated soil [50].
The amount of Archaea, quantified by real-time PCR, was approximately 1% or between 1.3% and 1.7% of the total microbial community in freshwater lake sediments [55] or in soils [6, 42, 50], respectively. During this study, no real-time PCR was performed; however, it was noticed that the number of archaeal sequences was low in comparison to the number of bacterial sequences obtained from the same samples, using the same archaeal primers (Table 2) or bacterial primers (Vishnivetskaya et al., unpublished data). In independent studies using the same pyrosequencing approach (and primers) on samples isolated from hot springs in Yellowstone Natural Park, the archaeal sequences exceeded 90% of total (unpublished results). The current study clearly showed that Crenarchaeota dominated over Euryarchaeota in the freshwater stream sediments. Similar results have been obtained for river sediments where 70% of the clones were affiliated with Crenarchaeota and 30% belonged to Euryarchaeota [46]. Thus, the composition of archaeal community in low-order streams was consistent at the highest taxonomic level with that from larger river systems.
The results from pyrosequencing may artificially increase the diversity of the community due to sequencing errors [32]. In this study, a large number of clusters (252) at 97% sequence identity were obtained; including 112 clusters with one sequence only. In order to address this concern, only the sequences found in at least two samples were analyzed, reducing the number of clusters by more than half. The number of clusters with more than one sequence in one site was 34 (counting mid-channel and bank point separately). However, based on the high homology of the pyrosequences analyzed to previously isolated clones sequenced by the dideoxy chain-termination method [52] (Supplemental Tables 3 and 4), the diversity obtained in the current study appears to be real.
Marine Group I was found to be the dominant group in the contaminated sites EF23 and BC (Fig. 4a). This group included the ammonia oxidizer N. maritimus [30] which is capable of converting ammonia to nitrite. The BC site contained high concentrations of nitrate (Table 1). In addition, these samples contained a number of uncultivated bacteria similar to the Myxococcales group (Vishnivetskaya et al., unpublished data). Previous studies detected Anaeromyxobacter sp. in uranium contaminated sediments from the Y-12 NSC, Oak Ridge, TN, USA [41]. Some of the Myxococcales, like Anaeromyxobacter dehalogenans, can use nitrate as an electron acceptor and reduce it to ammonia [51]. In the BC site 0.01% of the bacterial sequences were related to Nitrospira sp. and to γ-Proteobacteria affiliated with Nitrococcus sp. (Vishnivetskaya et al., unpublished data); these bacteria could convert nitrite into nitrate. Then the hypothesis is that members of Marine Group I could have a role in oxidizing ammonia previously produced by the Myxococcales. The reason for Marine Group I to be dominant in the mercury-rich site EF23 is unclear.
The methanogens were the third largest group of Archaea found in all samples. Although these samples were obtained from oxygen-saturated sediments, micro-niches with low redox conditions may exist in the stream sediments to allow these anaerobic groups to grow or be maintained. Methanogens, especially members of the orders Methanomicrobiales and Methanosarcinales, were described in freshwater lake and river sediments [4, 9, 18, 25, 26, 29, 46, 55]. In this study, hydrogenotrophic methanogens (methane production from H2 and CO2) were the dominant group. Acetoclastic methanogens (methane production from acetate) were also detected in large numbers. The occurrence of methanogens indicates the possibility of complete degradation of organic biomass in freshwater sediments; then the hydrogen and CO2 or acetate produced by other microbes is converted to methane.
Recently, the Rice Cluster III group has been enriched from anoxic soils and characterized [27]. For the enrichment, Kemnitz et al. used yeast extract, observing acetate as a major fermentation product and methane produced as a final compound. The following quantification of the enrichment showed that the mixture contained both Bacteria and Archaea. Within the Archaea, 60% were members of Rice Cluster III and the rest were found to be methanogens [27]. The authors concluded that the enriched Rice Cluster III archaeon was a heterotrophic anaerobic, mesophilic, and neutrophilic microorganism utilizing oligopeptides for growth. In the present study, this Euryarchaeota group was distributed in all samples.
The remaining groups identified in this study (with 60% of the sequences) included Freshwater Group, Miscellaneous Crenarchaeota group, Group I3, Rice Cluster VI and IV, Marine Benthic Group B and D, Deep Sea Hydrothermal Vent Euryarchaeota 1, and Eury 5. These groups do not include any cultured representative strains and it is difficult to assign putative roles for these microorganisms in the absence of an isolate whose physiology has been characterized. In addition, the names of the groups, in many cases, indicate the original discovery of their members; for example, marine and rice paddy field environments. However, as shown in Supplemental Tables 3 and 4, the members of uncultivated Crenarchaeota and Euryarchaeota are found in many other locations.
In comparison with bacterial ecology and physiology, the community ecology and functional role of Archaea in mesophilic environments remain poorly characterized [14, 47]. Molecular and phylogenetic studies have demonstrated the diversity of the archaeal communities in many environments and raised the need for cultivation attempts of the unknown groups [54]. Our study associates some of the groups with particular contaminants and may give some indication of their roles in the environment.
References
Bourne D, Iida Y, Uthicke S, Smith-Keune C (2008) Changes in coral-associated microbial communities during a bleaching event. ISME J 2:350–363
Bowman JP, McCuaig RD (2003) Biodiversity, community structural shifts, and biogeography of prokaryotes within Antarctic continental shelf sediment. Appl Environ Microbiol 69:2463–2483
Brauer SL, Cadillo-Quiroz H, Yashiro E, Yavitt JB, Zinder SH (2006) Isolation of a novel acidiphilic methanogen from an acidic peat bog. Nature 442:192–194
Briee C, Moreira D, Lopez-Garcia P (2007) Archaeal and bacterial community composition of sediment and plankton from a suboxic freshwater pond. Res Microbiol 158:213–227
Brina R, Miller AG (1992) Direct detection of trace levels of uranium by laser-induced kinetic phosphorimetry. Anal Chem 64:1413–1418
Buckley DH, Graber JR, Schmidt TM (1998) Phylogenetic analysis of nonthermophilic members of the kingdom crenarchaeota and their diversity and abundance in soils. Appl Environ Microbiol 64:4333–4339
Cadillo-Quiroz H, Yavitt JB, Zinder SH (2009) Methanosphaerula palustris gen. nov., sp. nov., a hydrogenotrophic methanogen isolated from a minerotrophic fen peatland. Int J Syst Evol Microbiol 59:928–935
Casamayor EO, Schafer H, Baneras L, Pedros-Alio C, Muyzer G (2000) Identification of and spatio-temporal differences between microbial assemblages from two neighboring sulfurous lakes: comparison by microscopy and denaturing gradient gel electrophoresis. Appl Environ Microbiol 66:499–508
Chan OC, Claus P, Casper P, Ulrich A, Lueders T, Conrad R (2005) Vertical distribution of structure and function of the methanogenic archaeal community in Lake Dagow sediment. Environ Microbiol 7:1139–1149
Chaudhary A, Haack SK, Duris JW, Marsh TL (2009) Bacterial and archaeal phylogenetic diversity of a cold sulfur-rich spring on the shoreline of Lake Erie, Michigan. Appl Environ Microbiol 75:5025–5036
Chen Y, Wu L, Boden R, Hillebrand A, Kumaresan D, Moussard H, Baciu M, Lu Y, Colin Murrell J (2009) Life without light: microbial diversity and evidence of sulfur- and ammonium-based chemolithotrophy in Movile Cave. ISME J 3:1093–1104
Cole C, Sobala A, Lu C, Thatcher SR, Bowman A, Brown JW, Green PJ, Barton GJ, Hutvagner G (2009) Filtering of deep sequencing data reveals the existence of abundant Dicer-dependent small RNAs derived from tRNAs. RNA 15:2147–2160
Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ, Kulam-Syed-Mohideen AS, McGarrell DM, Marsh T, Garrity GM, Tiedje JM (2009) The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res 37:D141–D145
DeLong EF (1998) Everything in moderation: archaea as ‘non-extremophiles’. Curr Opin Genet Dev 8:649–654
DOE Report (1997) Field characterization report on Phase 1 of the Bear Creek Valley treatability study, Oak Ridge Y-12 Plant, Oak Ridge, Tennessee http://www.osti.gov/bridge/product.biblio.jsp?query_id=0&page=0&osti_id=468549
Dong W, Brooks SC (2006) Determination of the formation constants of ternary complexes of uranyl and carbonate with alkaline earth metals (Mg2+, Ca2+, Sr2+, and Ba2+) using anion exchange method. Environ Sci Technol 40:4689–4695
Edwards IP, Burgmann H, Miniaci C, Zeyer J (2006) Variation in microbial community composition and culturability in the rhizosphere of Leucanthemopsis alpina (L.) Heywood and adjacent bare soil along an alpine chronosequence. Microb Ecol 52:679–692
Glissman K, Chin KJ, Casper P, Conrad R (2004) Methanogenic pathway and archaeal community structure in the sediment of eutrophic Lake Dagow: effect of temperature. Microb Ecol 48:389–399
Godsy EM (1980) Isolation of Methanobacterium bryantii from a deep aquifer by using a novel broth-antibiotic disk method. Appl Environ Microbiol 39:1074–1075
Gu B, Brooks SC, Roh Y, Jardine PM (2003) Geochemical reactions and dynamics during titration of a contaminated groundwater with high uranium, aluminum, and calcium. Geochim Cosmochim Acta 67:2749–2761
Han FX, Su Y, Monts DL, Waggoner CA, Plodinec MJ (2006) Binding, distribution, and plant uptake of mercury in a soil from Oak Ridge, Tennessee, USA. Sci Total Environ 368:753–768
Inagaki F, Suzuki M, Takai K, Oida H, Sakamoto T, Aoki K, Nealson KH, Horikoshi K (2003) Microbial communities associated with geological horizons in coastal subseafloor sediments from the sea of Okhotsk. Appl Environ Microbiol 69:7224–7235
Jiang H, Dong H, Yu B, Ye Q, Shen J, Rowe H, Zhang C (2008) Dominance of putative marine benthic Archaea in Qinghai Lake, north-western China. Environ Microbiol 10:2355–2367
Joulian C, Patel BK, Ollivier B, Garcia JL, Roger PA (2000) Methanobacterium oryzae sp. nov., a novel methanogenic rod isolated from a Philippines ricefield. Int J Syst Evol Microbiol 50 (Pt 2): 525–528
Jurgens G, Glockner F, Amann R, Saano A, Montonen L, Likolammi M, Munster U (2000) Identification of novel Archaea in bacterioplankton of a boreal forest lake by phylogenetic analysis and fluorescent in situ hybridization (1). FEMS Microbiol Ecol 34:45–56
Kemnitz D, Chin KJ, Bodelier P, Conrad R (2004) Community analysis of methanogenic archaea within a riparian flooding gradient. Environ Microbiol 6:449–461
Kemnitz D, Kolb S, Conrad R (2005) Phenotypic characterization of Rice Cluster III archaea without prior isolation by applying quantitative polymerase chain reaction to an enrichment culture. Environ Microbiol 7:553–565
Koch M, Rudolph C, Moissl C, Huber R (2006) A cold-loving crenarchaeon is a substantial part of a novel microbial community in cold sulphidic marsh water. FEMS Microbiol Ecol 57:55–66
Koizumi Y, Takii S, Fukui M (2004) Depth-related change in archaeal community structure in a freshwater lake sediment as determined with denaturing gradient gel electrophoresis of amplified 16S rRNA genes and reversely transcribed rRNA fragments. FEMS Microbiol Ecol 48:285–292
Konneke M, Bernhard AE, de la Torre JR, Walker CB, Waterbury JB, Stahl DA (2005) Isolation of an autotrophic ammonia-oxidizing marine archaeon. Nature 437:543–546
Kotelnikova S, Macario AJ, Pedersen K (1998) Methanobacterium subterraneum sp. nov., a new alkaliphilic, eurythermic and halotolerant methanogen isolated from deep granitic groundwater. Int J Syst Bacteriol 48 Pt 2: 357–367
Kunin V, Engelbrektson A, Ochman H, Hugenholtz P (2009) Wrinkles in the rare biosphere: pyrosequencing errors lead to artificial inflation of diversity estimates. Environ Microbiol 12:118–123
Leps J, Smilauer P (2003) Multivariate Analysis of Ecological Data Using CANOCO. Cambridge University Press, Cambridge, UK
Lliros M, Casamayor EO, Borrego C (2008) High archaeal richness in the water column of a freshwater sulfurous karstic lake along an interannual study. FEMS Microbiol Ecol 66:331–342
Lomans BP, Maas R, Luderer R, Op den Camp HJ, Pol A, van der Drift C, Vogels GD (1999) Isolation and characterization of Methanomethylovorans hollandica gen. nov., sp. nov., isolated from freshwater sediment, a methylotrophic methanogen able to grow on dimethyl sulfide and methanethiol. Appl Environ Microbiol 65:3641–3650
Lueders T, Friedrich M (2000) Archaeal population dynamics during sequential reduction processes in rice field soil. Appl Environ Microbiol 66:2732–2742
MacGregor BJ, Moser DP, Alm EW, Nealson KH, Stahl DA (1997) Crenarchaeota in Lake Michigan sediment. Appl Environ Microbiol 63:1178–1181
Madden AS, Palumbo AV, Ravel B, Vishnivetskaya TA, Phelps TJ, Schadt CW, Brandt CC (2009) Donor-dependent extent of uranium reduction for bioremediation of contaminated sediment microcosms. J Environ Qual 38:53–60
Mochimaru H, Tamaki H, Hanada S, Imachi H, Nakamura K, Sakata S, Kamagata Y (2009) Methanolobus profundi sp. nov., a methylotrophic methanogen isolated from deep subsurface sediments in a natural gas field. Int J Syst Evol Microbiol 59:714–718
Nawrocki EP, Eddy SR (2007) Query-dependent banding (QDB) for faster RNA similarity searches. PLoS Comput Biol 3:e56
North NN, Dollhopf SL, Petrie L, Istok JD, Balkwill DL, Kostka JE (2004) Change in bacterial community structure during in situ biostimulation of subsurface sediment cocontaminated with uranium and nitrate. Appl Environ Microbiol 70:4911–4920
Ochsenreiter T, Selezi D, Quaiser A, Bonch-Osmolovskaya L, Schleper C (2003) Diversity and abundance of Crenarchaeota in terrestrial habitats studied by 16S RNA surveys and real time PCR. Environ Microbiol 5:787–797
Park SJ, Park BJ, Rhee SK (2008) Comparative analysis of archaeal 16S rRNA and amoA genes to estimate the abundance and diversity of ammonia-oxidizing archaea in marine sediments. Extremophiles 12:605–615
Patel GB, Sprott GD (1990) Methanosaeta concilii gen. nov., sp. nov. ("Methanothrix concilii") and Methanosaeta thermoacetophila nom. rev., comb. nov. Int J Syst Bacteriol 40:79–82
Peng J, Lu Z, Rui J, Lu Y (2008) Dynamics of the methanogenic archaeal community during plant residue decomposition in an anoxic rice field soil. Appl Environ Microbiol 74:2894–2901
Rastogi G, Sani RK, Peyton BM, Moberly JG, Ginn TR (2009) Molecular studies on the microbial diversity associated with mining-impacted Coeur d’Alene River sediments. Microb Ecol 58:129–139
Robertson CE, Harris JK, Spear JR, Pace NR (2005) Phylogenetic diversity and ecology of environmental Archaea. Curr Opin Microbiol 8:638–642
Robertson CE, Spear JR, Harris JK, Pace NR (2009) Diversity and stratification of archaea in a hypersaline microbial mat. Appl Environ Microbiol 75:1801–1810
Rudolph C, Moissl C, Henneberger R, Huber R (2004) Ecology and microbial structures of archaeal/bacterial strings-of-pearls communities and archaeal relatives thriving in cold sulfidic springs. FEMS Microbiol Ecol 50:1–11
Sandaa RA, Enger O, Torsvik V (1999) Abundance and diversity of Archaea in heavy metal-contaminated soils. Appl Environ Microbiol 65:3293–3297
Sanford RA, Cole JR, Tiedje JM (2002) Characterization and description of Anaeromyxobacter dehalogenans gen. nov., sp. nov., an aryl-halorespiring facultative anaerobic myxobacterium. Appl Environ Microbiol 68:893–900
Sanger F, Nicklen S, Coulson AR (1977) DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci USA 74:5463–5467
Schelert J, Dixit V, Hoang V, Simbahan J, Drozda M, Blum P (2004) Occurrence and characterization of mercury resistance in the hyperthermophilic archaeon Sulfolobus solfataricus by use of gene disruption. J Bacteriol 186:427–437
Schleper C, Jurgens G, Jonuscheit M (2005) Genomic studies of uncultivated archaea. Nat Rev Microbiol 3:479–488
Schwarz JI, Eckert W, Conrad R (2007) Community structure of archaea and bacteria in a profundal lake sediment Lake Kinneret (Israel). Syst Appl Microbiol 30:239–254
Simbahan J, Kurth E, Schelert J, Dillman A, Moriyama E, Jovanovich S, Blum P (2005) Community analysis of a mercury hot spring supports occurrence of domain-specific forms of mercuric reductase. Appl Environ Microbiol 71:8836–8845
Sowers KR, Baron SF, Ferry JG (1984) Methanosarcina acetivorans sp. nov., an acetotrophic methane-producing bacterium isolated from marine sediments. Appl Environ Microbiol 47:971–978
Stein LY, La Duc MT, Grundl TJ, Nealson KH (2001) Bacterial and archaeal populations associated with freshwater ferromanganous micronodules and sediments. Environ Microbiol 3:10–18
Suzuki MT, Giovannoni SJ (1996) Bias caused by template annealing in the amplification of mixtures of 16S rRNA genes by PCR. Appl Environ Microbiol 62:625–630
Takai K, Horikoshi K (2000) Rapid detection and quantification of members of the archaeal community by quantitative PCR using fluorogenic probes. Appl Environ Microbiol 66:5066–5072
Walsh DA, Papke RT, Doolittle WF (2005) Archaeal diversity along a soil salinity gradient prone to disturbance. Environ Microbiol 7:1655–1666
Wust PK, Horn MA, Drake HL (2009) Trophic links between fermenters and methanogens in a moderately acidic fen soil. Environ Microbiol 11:1395–1409
Zhou P, Gu B (2005) Extraction of oxidized and reduced forms of uranium from contaminated soils: effects of carbonate concentration and pH. Environ Sci Technol 39:4435–4440
Acknowledgments
We thank Lisa Fagan, Miguel Rodriguez Jr., and Marilyn Kerley of ORNL for their help with sample collection and analysis. We thank Baohua Gu of ORNL for providing assistance with chemical analyses.
This work was supported by the US Department of Energy’s Office of Science Biological and Environmental Research, Environmental Remediation Sciences Program. Oak Ridge National Laboratory is managed by UT-Battelle, LLC, for the US Department of Energy under contract DE-AC05-00OR22725.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Porat, I., Vishnivetskaya, T.A., Mosher, J.J. et al. Characterization of Archaeal Community in Contaminated and Uncontaminated Surface Stream Sediments. Microb Ecol 60, 784–795 (2010). https://doi.org/10.1007/s00248-010-9734-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00248-010-9734-2