
Abstract
Several extraction methods are used to isolate natural compounds, and recent approaches utilize subcritical or supercritical extraction media. In this paper we compare extraction methods based on subcritical eluents, dimethyl ether (sC-DME) and n-butane (sC-nB), under mild conditions, using coffee beans and powder as an exemplary raw material. The parameters to be controlled to improve the extraction are considered, and the resulting data discussed. The results obtained display higher selectivity of sC-DME for caffeine (1.9%w/w sC-DME vs. 1.7%w/w sC-nB, on dry extract) and a good yield (0.479 mg/g of caffeine from green coffee beans) compared to, e.g., supercritical carbon dioxide (SC-CO2), which shows 0.32 mg/g of caffeine at higher pressure and temperature (25 MPa, 40 °C). We also discuss some technical implementations for optimizing the use of sub-critical eluents through proper combinations of pressure and temperature. We show that extraction processes based on sub-critical eluents are easy to operate and efficient, and can be easily automated.
Similar content being viewed by others


Introduction
Established extraction processes for natural compounds are contributing a very large collection of novel active substances for the pharmaceutical and nutraceutical industry. These include mechanical pressing, extraction with organic solvents or sub-critical water, and more recently, extraction by supercritical CO2 (SC-CO2). [1,2,3] The use of a mechanical press is energy-intensive and the extraction yields are low; however, this method (in particular in cold pressing) usually keeps the integrity of most molecules, resulting in the preservation of organoleptic and functional properties. Extraction with organic solvents refers mostly to n-hexane, which is the solvent of choice for most oils and oil-soluble compounds. A major drawback with n-hexane and higher organic molecules is the presence of residual solvent in the end products, which is harmful to humans, and also not environmentally sustainable [4]. Supercritical carbon dioxide methods are very interesting, because there is virtually no contamination of the products. However, the pressures and temperatures needed are energetically unfavorable. SC-CO2 is able to extract low molecular weight, non-polar compounds [5] but is a poor solvent for high molecular weight and polar compounds [6]. Typical extraction conditions for moderately polar molecules, e.g., caffeine, with subcritical water are 0.8–4 MPa/373–473 K, which cannot be considered as mild conditions and, especially at the higher temperatures, entail a risk of decomposition or transformation of the organic extracts [7, 8].
Extraction using liquefied (subcritical) solvents is rapidly growing in the field of extraction of active substances, and recent reports have described the extraction and characterization of flavoring molecules [9], edible oils [10], and coumarin and phenylpropanoid derivatives from Citrus spp. and Artemisia spp. [11, 12]. Liquefied gas eluents have been used as well for selective extraction, and it was shown that extraction selectivity depends strongly on the eluent used [13].
The extraction efficiency of organic eluents is a function of the polarity of the molecules to be extracted, which correlates directly to the dielectric constant and the polarity of the extraction medium. n-Butane (nB) and dimethyl ether (DME) are two organic eluents that display different polarities, and their use is proposed as a valid alternative for n-hexane [14,15,16,17,18].
Subcritical dimethyl ether (sC-DME) at 298 K and 0.1 MPa exists as a colorless gas with a characteristic etheric scent [19], and is classified as a flammable gas (EC Regulation No 1272/2008). Under the working conditions adopted in this work, sC-DME does not form peroxides [19,20,21,22] as they tend to form above 353 K. Even if this substance is considered flammable at room conditions, the standard industrial gas handling procedures are sufficiently established to use it safely [22, 23]. For the use proposed in this work, the favorable conditions that this molecule displays when liquefied (low viscosity, promotion of mixing, and penetration into the solid matrix) make it a perfect candidate as an eluent for extraction [24]. The extraction efficiency of sC-DME is increased on wet or fresh matrices [25,26,27]. DME is also considered a negligible contributor to the global warming [27,28,29,30].
In this paper, we focus on the extracting capacity of n-butane and dimethyl ether used as subcritical eluents to show that their extraction performance can be tuned by changing the design of the reactor and the pressure and temperature conditions. The toxicology of the eluent is of the utmost importance. According to the European Food Safety Authority (EFSA), DME residues present low toxicity because of low uptake and distribution into human tissues [31, 32], and the European Union permits its use as an extraction medium in a number of cases [33]. DME is also recognized as “Generally Recognized as Safe” (GRAS) by the FDA (Food and Drug Administration) [34], as by examining the residual levels in various extracted food products by gas chromatography–mass spectroscopy (GC–MS), it was found that DME levels are below the detection limit (2 ppm) [35].
The advantages of using subcritical n-butane (sC-nB) are a high solvation capacity for lipophilic compounds (i.e., oils) while working under milder conditions of temperature and pressure than SC-CO2 [36]. nB is more environmentally friendly than heavier organic solvents (e.g., n-hexane), and shares with SC-CO2 the absence of any residues in the final products. From the toxicological point of view, nB has been classified by the FDA as GRAS [37]. The EFSA has included nB in the list of the extraction solvents which can be used in the processing of food, food ingredients and raw materials according to good manufacturing practices [38].
Another advantage is that these subcritical eluents can be used at low pressures and ambient temperatures, which are suitable for eluent recycling. This allows to employ a small amount of gases compared to the conventional methods, and avoid the waste of non-recycling processes and eluents. The present work reports a comparison of extraction methods based on sC-DME and sC-nB. As an exemplary biomolecule we used caffeine, whose extraction methods are highly developed. Currently, the most used method is supercritical extraction using CO2. This procedure allows the extraction of almost all the caffeine from green beans, and the residual content in the biomass varies greatly depending on the temperature, extraction time, pressure [39], and the wetting and swelling of beans. It has been shown that water penetration and diffusion into the beans promote the dissolution of caffeine in water and the transfer from the inside to the surface of the beans' matrix [40].
The polarity of DME and nB varies significantly, as shown by their dielectric constants (1.8 for nB at 296 K, 4.3 for DME at 293 K and 80.4 for water at 293 K), suggesting that extraction selectivity might depend significantly on the polarity of the molecule to be extracted. In our case, the logP of caffeine is − 0.07 [41], which makes it a moderately polar molecule, and suggests that it should contract interactions with high dielectric constant solvents, thus allowing a good extraction without the need to pre-treat by wetting and swelling the beans. Industrially, caffeine extraction is performed mostly on green fresh beans. In the present work, extraction was also carried out on roasted beans and ground coffee powder.
Materials and methods
Materials
Caffeine standard was purchased from Sigma-Aldrich (Milan, Italy), while acetone, methanol, acetic acid, and ethanol were obtained from Carlo Erba Reagents (Milan, Italy). Dimethyl ether and n-butane were obtained from SOL S.p.A. (Chieti, Italy). The solid matrices (coffee beans and ground coffee, from Coffea arabica L.) were kindly donated by Mokambo S.p.A. (Chieti, Italy). The extraction thimbles were Whatman CELLULOSE (size: 19 mm × 90 mm), purchased from Sigma-Aldrich (Milan, Italy). All chemicals were of analytical-reagent grade or better.
Extraction system
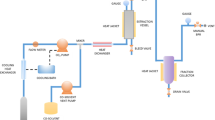
The in-house designed and assembled experimental apparatus is shown in Fig. 1. The apparatus consists of an evaporation chamber (250 mL), condenser column (500 mL), tubing, and extraction chamber (30 mL), all being made of AISI 316L steel. Both liquids and solids can be processed in this apparatus. The system is entirely controlled by an Advanced Process Controller/Programmer (Eurotherm 2604, Schneider Electric, Italy) which allows precise control on the pressure and temperature (accuracy > 0.002 MPa; > 0.01 K, respectively). The operating conditions are measured using temperature probes [RTD-PT100 class 1/3 DIN purchased from OMEGA Engineering, Inc. (USA)] positioned both on the evaporator chamber and the condenser column, and a 4−20 mA pressure transducer (Gems Sensors and Controls, UK) with a measuring range of 0−10 MPa. Temperature control of all components is carried out by two chiller/heater units with a cooling power of 1000 W. The evaporation chamber has an operating pressure range of 0.101–10 MPa. The solvent flux is controlled by a flowmeter (Bronkhorst EL-Flow, NL) and observed visually through a quartz window positioned between the condenser and the extraction chamber. The tubing is insulated with 5 cm of foam rubber. The liquefied gas influx is measured both by the weight of the input gas and by flowmeter measurement.

Extraction apparatus
Extraction methods and conditions
The extraction of caffeine was carried out using two different conditions. The setup of extraction with DME is as follows: 287 K for the condenser column, 318 K for the evaporation chamber, and a liquefied DME flux of 9–10 mL/min. The setup of extraction with nB is as follows: 283 K for the condensation column, 318 K for evaporation chamber and the flux of liquefied nB (8–9 mL/min). The sample (ca. 3.5–3.7 g of whole beans or powder) was put into the extraction chamber using a cellulose thimble as physical support and filter. Then, a vacuum (ca. 5 × 10–3 MPa) was applied to evacuate the air inside the tubing. The liquefied gas eluent was injected when the temperatures reached the setpoints. In each run, 40.0 g of eluent (nB or DME) was loaded, and the experiments were conducted for 30 min, 1 h and 2.5 h, every time loading fresh samples. At the end of the extraction, the liquefied gas was removed from the sample by gasification of the eluent into a recovery chamber. The extract was kept under nitrogen atmosphere at 275 K. After each experiment, the tubing was disassembled and accurately cleaned using polar and nonpolar solvents before assembling for the next run.
Quantification of caffeine in the residue
Considering that even the most efficient extraction methods leave a residual amount of caffeine inside coffee beans, we needed to quantify the residual amounts after extraction with nB and DME [25]. With this purpose, we used a traditional maceration in ethanol, which is considered an exhaustive extraction method [17, 42]. The maceration was performed by keeping the product for 48 h at 298 K with a material/solvent ratio of 0.05 w/w. The quantification of caffeine in fresh samples, and residual caffeine in extracted matrices was determined using HPLC–PDA [43].
The analysis of caffeine extracts was performed on a Waters system consisting of a model 600 pump and a UV–Vis photodiode array detector model 2996 (Waters, Milford, MA, USA). A Rheodyne model 7125i injector (Rheodyne, Cotati, CA, USA) equipped with 20 µL loop and a degasser system model DG-4400 (Phenomenex, Torrance, CA, USA) was used. Chromatographic separation was achieved using a Kinetex-XB C18 (150 × 4.6 mm I.D. 5 μm particle size) column protected by a disposable Security Guard precolumn (3.0 × 4.0 mm) (Phenomenex, Torrance, CA, USA) maintained at 298 ± 1 K, using a thermostatically controlled column heater. An XS104 Mettler Toledo analytical balance was used to weigh the analytes for the preparation of the stock solution and calibration standard. Water HPLC-grade water obtained by passage through an Elix 3 and Milli-Q Academic water purification system (18 mΩ/cm, TOC < 5 ppb) (Millipore, Bedford, MA, USA). The mobile phase consisted of 10 mM ammonium acetate buffer adjusted to pH 3.5 with acetic acid (phase A) and methanol (phase B). To perform the best separation and detection of caffeine, a linear gradient elution program was used. The mobile phase composition was 78% and 22% of phases A and B, respectively, from 0 to 8 min. Next, within 17 min the composition of the eluting mixture was turned to 5% and 95% of phases A and B, respectively. Then, the mobile phase composition remained 5% and 95% of phases A and B, respectively, between 25 and 30 min. Finally, the mobile phase composition returned to the original ratio between 30 and 33 min, followed by 12 min of re-equilibration of the column to the initial condition. The flow rate was set at 1.0 mL/min. The solvents were filtered before use through a 0.45 µm WTP membrane, while ammonium acetate solution was filtered through a WCN 0.5 µm membrane (Whatman, Maidstone, UK). Detection was carried out at 271 nm. Empower v.2 Software (Waters Spa, Milford, MA, USA) was used for setting up the analysis and for data management. All experiments were performed in triplicate.
Results
In the present work, we compared the extraction of caffeine as an exemplary biomolecule using sC-nB and sC-DME as eluents to elucidate the conditions that control the selectivity and yield of the extraction.
The experiments were conducted at set temperatures (283–288 K for condenser column and 313 K for evaporation chamber) and vapor tension pressures of sC-nB and sC-DME, and lasted 0.5, 1 and 2.5 h (see Material and Methods). The mean extraction yield was measured by HPLC on dry extract. The results show a higher affinity of caffeine for sC-DME vs. sC-nB, where extracted amounts at 2.5 h for roasted ground coffee were 0.85 mg/g for sC-nB and 3.52 mg/g for sC-DME. An interesting finding is the residual amount of caffeine after the extraction. Figure 2 reports the caffeine extraction efficiencies for the two eluents and three starting matrices, where DME shows the best performances for all starting materials. Results have a coefficient of variation at 2% or less.

Chromatographic profiles of extracted caffeine
Table 1 reports the amounts of extracted caffeine expressed as milligrams of caffeine per gram of matrix at different extraction times for sC-nB and sC-DME, respectively. The extraction efficiency of sC-DME is more dependent on the extraction time as compared to sC-nB.
Table 2 reports residual caffeine in the extracted material after the exhaustive maceration process in ethanol. In agreement with the higher efficiency of sC-DME on coffee beans extraction, the residual caffeine from coffee maceration after sC-DME extraction is from two to three times lower than with sC-nB.
Table 3 reports data of exhaustive caffeine extraction with sC-DME after 24 h. The results show higher yields of extraction, which are obviously due to the increased extraction time. These yields are not high enough to render an increase of extraction time commercially viable from an industrial standpoint. It should be noted, however, that reported data refer to a percolation system in which the sample is not submerged in the eluent, and its performance can be improved by a modification of the extraction chamber that allows the accumulation of the liquefied eluent.
Table 4 shows comparative data for the performance of different eluents. The sample used is the fresh whole beans, which are the most used raw material for industrial applications. Most importantly, sC-DME displays higher extraction yields than SC-CO2 at 313 K and 25 MPa, possibly because of a higher chemical affinity with the fresh matrix.
Discussion
The results obtained using sub-critical n-Bu and DME are shown in Fig. 2 and Table 1. The plots highlight the high caffeine extraction levels achieved with DME as compared to nB. As mentioned above, this selectivity depends significantly on the polarity of caffeine (logP = − 0.07) whose extraction requires a moderately polar eluent, such as sC-DME. In addition, the higher extraction found with sC-DME on fresh vs. roasted beans may be due to a stronger interaction of this extractant with a wet or fresh starting material. In other words, sC-DME should have a higher permeability into fresh beans as compared to sC-nB. Another finding, based on the HPLC analysis of dry extract, is that sC-DME is more selective to caffeine than sC-Bu, with 1.9%w/w and 1.7%w/w, respectively, this percentage referring to the total amount of dry extract (i.e., 1.9%w/w means that 1.9% of the total mass of the dry extract is composed of caffeine). This aspect is very important in food processing, where the extraction of unwanted components should be highly selective to avoid the concurrent extraction of other desired molecules. The selectivity of extraction depends on the processing status of the starting material, i.e., roasted vs. fresh coffee.
Ground coffee powder is the best matrix for solid–liquid extraction, due to its much higher surface area as compared to beans. This is also apparent from the results in Fig. 2. However, powder volatility represents a problem in the processing steps as it tends to be entrained by the extraction solvent, thereby clogging parts of the processing units. This is one of the reasons why processing companies tend to employ beans as the extraction matrix. In the present work, the yields for roasted and green beans are similar for each solvent (Fig. 2), but with an approximately threefold increase for sC-DME, reflecting the higher penetration ability of this extractant into the beans as compared to nB. In general, extraction yields are always lower with nB under all conditions tested.
As relates to the kinetics of extraction (Tables 1A and 2), we observed that yields for sC-nB on ground coffee at 1 h and 2.5 h are similar, whereas an approximately 60% increase was obtained on both fresh and roasted beans. This behaviour seems to suggest that sC-nB performs most of its extracting action within 1 h on ground powder, reaching a kind of plateau afterward. By contrast, sC-DME continues to extract caffeine with the elapse of time, and does not reach a plateau at 2.5 h (Table 1B).
Another interesting difference between sC-DME and sC-nB can be found when considering the residual caffeine remaining in the “spent” starting material, i.e., coffee ground powder or beans. Table 2 shows the relative amount of caffeine remaining in the extracted material as a weight percentage compared to the amount obtained after exhaustive extraction of untreated material (powder, fresh beans, roasted beans) by maceration in ethanol for 48 h. Data show that most of the caffeine remains in the starting material after extraction with sC-nB, whereas only ca. 30–40% is found as a residue when processing with sC-DME. The fact that sC-DME is more effective as an extractant for a moderately polar compound (caffeine) than sC-nB is not surprising at a first glance, but, interestingly, similar efficiencies were observed when processing a ground powder and a whole (roasted or fresh) bean. This behavior can be explained by the tendency of sC-DME to penetrate through whole natural matrices [16], such as green beans.
Extraction time is an important parameter for establishing the extraction yields. A moderate increase in the amount of extracted caffeine was predictably observed for sC-DME when increasing the treatment times up to 24 h (Table 3). Extraction yields might be enhanced by simple optimization of the extraction chamber and process, such as to increase the amount of liquefied eluent in contact with the raw material. With this purpose, the extractor can be modified by inserting a second tubing positioned on the top, to create the classical Soxhlet extractor scheme that can operate both at moderate pressures and under vacuum conditions to increase the yield of the extraction. Another strategy intrinsically present in the proposed system is the possibility to switch among several solvents and solvent mixtures with different polarities. In this way, the choice of the liquefied gas solvent will be based on the physico-chemical properties of the compounds of interest, to improve the extraction yields and, more importantly, increase the selectivity. Finally, comparison of sC-DME with SC-CO2 (Table 4) shows that weight ratios of extracted caffeine are higher for sC-DME under much milder conditions than those used with SC-CO2.
In conclusion, subcritical solvents may represent a low environmental impact alternative to higher hydrocarbon solvents (e.g., n-hexane) as extraction media for biomolecules. The peculiarities of sub-critical solvents such as sC-DME and sC-nB to behave both as liquids and gases in an extraction process, depending on the pressure and temperature conditions, allow them to be suited for finely tuning the extraction efficiency and selectivity. For example, the time and extent of contact between the subcritical solvent and the biological matrix can be tailored by changing the temperatures and pressures in the different stages of the extraction apparatus, i.e., the evaporation chamber, condenser column, tubing, and extraction chamber. The residual amount of subcritical solvent into the extracted product can be easily reduced to non-detectable amounts by simple vaporization under mild pressure and temperature conditions. The selectivity of extraction may also be improved using mixtures of sub-critical solvents, whose liquid/gas transitions are tailored by changing P/T values. Those eluents work under milder temperature conditions than, e.g., supercritical solvents, thereby any potential risk of degradation of labile actives may be reduced [44]. Finally, the industrial scale-up should not involve engineering challenges, as it does not require complex configurations due to low pressures (below 10 bar instead of 70–400 for SC-CO2) and temperatures below 40 °C. For the same reason, a large scale application would be much less energy-intensive as compared to supercritical processes.
References
Patra A, Abdullah S, Pradhan RC (2022) Review on the extraction of bioactive compounds and characterization of fruit industry by-products. Bioresour Bioprocess 9:14. https://doi.org/10.1186/s40643-022-00498-3
Xu DP, Li Y, Meng X et al (2017) Natural antioxidants in foods and medicinal plants: extraction, assessment and resources. Int J Mol Sci 18:96. https://doi.org/10.3390/ijms18010096
Altemimi A, Lakhssassi N, Baharlouei A et al (2017) Phytochemicals: extraction, isolation, and identification of bioactive compounds from plant extracts. Plants 6:42. https://doi.org/10.3390/plants6040042
Gu LB, Pang HL, Lu KK et al (2017) Process optimization and characterization of fragrant oil from red pepper (Capsicum annuum L.) seed extracted by subcritical butane extraction. J Sci Food Agric 97:1894–1903. https://doi.org/10.1002/jsfa.7992
Shi Y, Ma Y, Zhang R et al (2015) Preparation and characterization of foxtail millet bran oil using subcritical propane and supercritical carbon dioxide extraction. J Food Sci Technol 52:3099–3104. https://doi.org/10.1007/s13197-014-1311-0
Goto M, Kanda H, Wahyudiono MS (2015) Extraction of carotenoids and lipids from algae by supercritical CO2 and subcritical dimethyl ether. J Supercrit Fluids 96:245–251. https://doi.org/10.1016/j.supflu.2014.10.003
He C, Du H, Tan C et al (2018) Semi-continuous pressurized hot water extraction of black tea. J Food Eng 227:30–41. https://doi.org/10.1016/j.jfoodeng.2018.02.001
Shalmashi A, Abedi M, Golmohammad F, Eikani MH (2010) Isolation of caffeine from tea waste using subcritical water extraction. J Food Process Eng 33:701–711. https://doi.org/10.1111/j.1745-4530.2008.00297.x
Khuwijitjaru P, Sayputikasikorn N, Samuhasaneetoo S et al (2012) Subcritical water extraction of flavoring and phenolic compounds from cinnamon bark (Cinnamomum zeylanicum). J Oleo Sci 61:349–355. https://doi.org/10.5650/jos.61.349
Pereira MG, Hamerski F, Andrade EF et al (2017) Assessment of subcritical propane, ultrasound-assisted and Soxhlet extraction of oil from sweet passion fruit (Passiflora alata Curtis) seeds. J Supercrit Fluids 128:338–348. https://doi.org/10.1016/j.supflu.2017.03.021
Fiorito S, Genovese S, Palumbo L et al (2020) Umbelliprenin as a novel component of the phytochemical pool from Artemisia spp. J Pharm Biomed Anal 184:113205. https://doi.org/10.1016/j.jpba.2020.113205
Genovese S, Epifano F, Preziuso F et al (2020) A novel and efficient subcritical butane extraction method and UHPLC analysis of oxyprenylated phenylpropanoids from grapefruits peels. J Pharm Biomed Anal 184:113185. https://doi.org/10.1016/j.jpba.2020.113185
Moreira I, Scheel GL, Hatumura PH, Scarminio IS (2014) Efeito do solvente na extração de ácidos clorogênicos, cafeína e trigonelina em coffea arabica. Quim Nova 37:39–43. https://doi.org/10.1590/S0100-40422014000100008
Yang C, Teo KC, Xu YR (2004) Butane extraction of model organic pollutants from water. J Hazard Mater 108:77–83. https://doi.org/10.1016/j.jhazmat.2003.12.007
Rapinel V, Santerre C, Hanaei F et al (2018) Potentialities of using liquefied gases as alternative solvents to substitute hexane for the extraction of aromas from fresh and dry natural products. Comptes Rendus Chim 21:590–605. https://doi.org/10.1016/j.crci.2018.04.006
Bauer MC, Kruse A (2019) The use of dimethyl ether as an organic extraction solvent for biomass applications in future biorefineries: A user-oriented review. Fuel 254:115703. https://doi.org/10.1016/j.fuel.2019.115703
Chemat F, Abert Vian M, Fabiano-Tixier AS et al (2020) A review of sustainable and intensified techniques for extraction of food and natural products. Green Chem 22:2325–2353
Rapinel V, Rombaut N, Rakotomanomana N et al (2017) An original approach for lipophilic natural products extraction: Use of liquefied n-butane as alternative solvent to n-hexane. LWT Food Sci Technol 85:524–533. https://doi.org/10.1016/j.lwt.2016.10.003
Ihmels EC, Lemmon EW (2007) Experimental densities, vapor pressures, and critical point, and a fundamental equation of state for dimethyl ether. Fluid Phase Equilib 260:36–48. https://doi.org/10.1016/j.fluid.2006.09.016
Naito M, Radcliffe C, Wada Y et al (2005) A comparative study on the autoxidation of dimethyl ether (DME) comparison with diethyl ether (DEE) and diisopropyl ether (DIPE). J Loss Prev Process Ind 18:469–473. https://doi.org/10.1016/j.jlp.2005.07.001
Ladera R, Finocchio E, Rojas S et al (2012) Supported niobium catalysts for methanol dehydration to dimethyl ether: FTIR studies of acid properties. Catal Today 192:136–143. https://doi.org/10.1016/j.cattod.2012.01.025
Tallon S, Fenton K (2010) The solubility of water in mixtures of dimethyl ether and carbon dioxide. Fluid Phase Equilib 298:60–66. https://doi.org/10.1016/j.fluid.2010.07.009
Cardea S, Reverchon E (2019) Supercritical fluid processing of polymers. Polymers (Basel) 11:1551. https://doi.org/10.3390/polym11101551
Herrero M, Castro-Puyana M, Mendiola JA, Ibañez E (2013) Compressed fluids for the extraction of bioactive compounds. TrAC Trends Anal Chem 43:67–83. https://doi.org/10.1016/j.trac.2012.12.008
Kanda H, Kamo Y, Machmudah S et al (2014) Extraction of fucoxanthin from raw macroalgae excluding drying and cell wall disruption by liquefied dimethyl ether. Mar Drugs 12:2383–2396. https://doi.org/10.3390/md12052383
Catchpole O, Tallon S, Dyer P et al (2012) Integrated supercritical fluid extraction and bioprocessing. Am J Biochem Biotechnol 8:263–287. https://doi.org/10.3844/ajbbsp.2012.263.287
Semelsberger TA, Borup RL, Greene HL (2006) Dimethyl ether (DME) as an alternative fuel. J Power Sources 156:497–511. https://doi.org/10.1016/j.jpowsour.2005.05.082
Azizi Z, Rezaeimanesh M, Tohidian T, Rahimpour MR (2014) Dimethyl ether: a review of technologies and production challenges. Chem Eng Process Process Intensif 82:150–172
Wu J, Yin J (2008) Vapor pressure measurements of dimethyl ether from (213 to 393) K. J Chem Eng Data 53:2247–2249. https://doi.org/10.1021/je800375t
Good DA, Francisco JS, Jain AK, Wuebbles DJ (1998) Lifetimes and global warming potentials for dimethyl ether and for fluorinated ethers: CH3OCF3 (E143a), CHF2OCHF2 (E134), CHF2OCF3 (E125). J Geophys Res Atmos 103:28181–28186. https://doi.org/10.1029/98JD01880
European Food Safety Authority (EFSA) (2009) Safety in use of dimethyl ether as an extraction solvent. EFSA J. https://doi.org/10.2903/j.efsa.2009.984
European Food Safety Authority (EFSA) (2015) Scientific Opinion on the safety of use of dimethyl ether as an extraction solvent under the intended conditions of use and the proposed maximum residual limits. EFSA J. https://doi.org/10.2903/j.efsa.2015.4174
Commission E (2016) Commission Directive (EU) 2016/1855 of 19 October 2016 amending Directive 2009/32/EC of the European Parliament and of the Council on the approximation of the laws of the Member States on extraction solvents used in the production of foodstuffs and food i. Off J Eur Union 284:19–20
Food and Drug Administration (2017) GRAS Notice for the use of dimethyl ether as an extraction solvent. GRAS Notice No. GRN 000741
Adams MS (2017) GRAS Notice for the use of dimethyl ether as an extraction solvent. https://www.fda.gov/media/115219/download. Accessed 31 Aug 2022
Zanqui AB, De Morais DR, Da Silva CM et al (2015) Subcritical extraction of flaxseed oil with n-propane: Composition and purity. Food Chem 188:452–458. https://doi.org/10.1016/j.foodchem.2015.05.033
Food and Drug Administration (2021) Title 21—Food and Drugs. 21CFR184.1165
The European Food Safety Authority (2009) Directive 2009/32/EC of the European Parliament and of the Council of 23 April 2009 on the approximation of the laws of the Member States on extraction solvents used in the production of foodstuffs and food ingredients
Menzio J, Binello A, Barge A, Cravotto G (2020) Highly-efficient caffeine recovery from green coffee beans under ultrasound-assisted SC–CO2 Extraction. Processes 8:1062. https://doi.org/10.3390/pr8091062
MacHmudah S, Kitada K, Sasaki M et al (2011) Simultaneous extraction and separation process for coffee beans with supercritical CO2 and water. Ind Eng Chem Res 50:2227–2235. https://doi.org/10.1021/ie101252w
International Labour Organization; World Health Organization (2017) Icsc 0405 - Caffeine. https://www.ilo.org/dyn/icsc/showcard.display?p_version=2&p_card_id=0405%0Ahttp://www.ilo.org/dyn/icsc/showcard.display?p_version=2&p_card_id=0405. Accessed 3 Feb 2022
Renard CMGC (2018) Extraction of bioactives from fruit and vegetables: State of the art and perspectives. Lwt 93:390–395. https://doi.org/10.1016/j.lwt.2018.03.063
Yashin A, Yashin Y, Xia X, Nemzer B (2017) Chromatographic methods for coffee analysis: a review. J Food Res 6:60. https://doi.org/10.5539/jfr.v6n4p60
Chaves JO, de Souza MC, da Silva LC, et al (2020) Extraction of flavonoids from natural sources using modern techniques. Front Chem 8
Acknowledgements
We would like to thank Dr. Caterina Sergiacomo for experimental support.
Funding
Open access funding provided by Università degli Studi G. D'Annunzio Chieti Pescara within the CRUI-CARE Agreement. This research received no external funding.
Author information
Authors and Affiliations
Contributions
Conceptualization, VC and PDP; methodology, VC, MC and RDW; software, RDW; validation, VC, MC and RDW; formal analysis, VF; investigation, GS; resources, AF; data curation, VF; NDA; writing—original draft preparation, PDP; writing—review and editing, VC, RDW; visualization, GS; supervision, PDP; project administration, AF; funding acquisition, PDP All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Compliance with Ethics requirements
As the corresponding author, I declare that the subject article does not contain any studies with human or animal subjects.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ciulla, M., Canale, V., Wolicki, R.D. et al. Comparison of extraction methods for active biomolecules using sub-critical dimethyl ether and n-butane. Eur Food Res Technol 249, 367–374 (2023). https://doi.org/10.1007/s00217-022-04122-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00217-022-04122-8