
Abstract
This study reports the development of a sensitive magnetic bead-based enzyme-linked immunoassay (MELISA) for the pan-reactive detection of the Influenza A virus. The assay combines immunomagnetic beads and biotin-nanoparticle-based detection to quantify a highly conserved viral nucleoprotein in virus lysates. At the capture step, monoclonal antibody-coated magnetic microbeads were used to bind and concentrate the nucleoprotein in samples. The colorimetric detection signal was amplified using biotinylated silica nanoparticles (NP). These nanoparticles were functionalized on the surface with short DNA spacers bearing biotin groups by an automated supported synthesis method performed on nano-on-micro assemblies with a DNA/RNA synthesizer. A biotin-nanoparticle and immunomagnetic bead-based assay was developed. We succeeded in detecting Influenza A viruses directly in the lysis buffer supplemented with 10% saliva to simulate the clinical context. The biotin-nanoparticle amplification step enabled detection limits as low as 3 × 103 PFU mL−1 and 4 × 104 PFU mL−1 to be achieved for the H1N1 and H3N2 strains respectively. In contrast, a classical ELISA test based on the same antibody sandwich showed detection limit of 1.2 × 107 PFU mL−1 for H1N1. The new enhanced MELISA proved to be specific, as no cross-reactivity was found with a porcine respiratory virus (PRRSV).

Graphical abstract
Similar content being viewed by others

Introduction
Influenza viruses cause worldwide outbreaks and pandemics in humans and animals with high morbidity and mortality [1]. Of the Influenza A, B, C and D viruses, Influenza A viruses mutate the most rapidly leading to the continuous apparition of new strains. The high antigenic flexibility and hence greater virulence increase the severity of illnesses and death rates in the event of a pandemic [2]. The seasonal subtypes of circulating Influenza A viruses (H1N1 and H3N2) cause 3–5 million human severe infections and between 290,000 and 650,000 fatal cases annually, mostly in elderly and immunocompromised individuals and in the young children [3, 4]. There is an urgent need to improve diagnostic of H1N1 and H3N2 viruses to better control future outbreaks.
Rapid diagnostic tests for influenza, mostly based on antigen detection, facilitate early detection and then improve clinical decision-making. Furthermore, such tests can serve as tools for disease monitoring. Most of the assays dedicated to rapid detection of the currently circulating subtypes of Influenza A viruses use monoclonal antibodies (mAbs) against the nucleoprotein (NuP) encoded by Influenza A viruses. The NuP protein is highly abundant, notably during Influenza A virus infections [5, 6]. It is also a highly conserved protein among the different subtypes of Influenza A viruses with a > 90% amino acid sequence identity [7]. Thus, the approach targeting the nucleoprotein A is generic for a large panel of Influenza A strains. For instance, Yang et al. produced two monoclonal antibodies against the avian Influenza A nucleoprotein [8]. The immunoassays targeting NuP proved to be robust and specific for 15 subtypes of avian Influenza A viruses. In 2020, an antibody microarray was developed from a preselection of 50 mAbs [9]. The diagnostic system detected and differentiated six clinically important upper respiratory tract pathogen groups, including Influenza A and B viruses, by testing 25 mAbs against the nucleoprotein.
The first ELISA test based on immunocapture of the nucleoprotein was described by Chomel et al. [10]. This immunocapture sandwich assay was found to be specific for a large panel of human and animal influenza viruses and no cross-reaction with other respiratory viruses was observed. A minimal amount of 5 ng mL−1 of purified viral NuP was detected using highly sensitive radioactive detection [11]. In 2010, Chen et al. developed the Flu A DOT ELISA [12]. This test kit, which targets NuP in real samples of viruses, showed high sensitivity compared with the commercial kit QuickVue Flu A from Quidel Corp. and sufficient specificity for all the variants of the Influenza A virus considered (LoD: 1.6 × 103 TCID50 mL−1 for the H5N1 strain). A one-step immunoassay with interferometric measurement was developed for the real-time detection of Influenza A nucleoprotein in samples [13]. The technique enabled a level of detection of 1 μg mL−1 NuP. This work aimed at developing a rapid and portable sensor for in situ detection of Influenza A viruses. More recently, a highly sensitive fluorescent europium nanoparticle (NP)-based immunoassay, ENIA, was developed by Zhang et al. [14]. ENIA proved to be accurate in real samples infected with both Influenza A and B viruses. It demonstrated an analytical sensitivity of 2.5 × 102 EID50 mL−1 for the A/Brisbane/59/07 H1N1 strain. Leirs et al. screened commercial antibodies against the Influenza A nucleoprotein using a digital ELISA microscopic approach. This imaging technique on the microscale proved ultra-sensitive for detecting the recombinant Influenza A nucleoprotein spiked in buffer, reaching femtomolar detection limits. On the other hand, digital ELISA showed lower efficacy for purified viruses directly in the lysis buffer [15]. A horseradish peroxidase (HRP)-labeled lateral flow immunoassay (HRP-LFIA) was recently developed for simultaneous detection of Influenza A and B viruses [16]. Four monoclonal antibodies, recognizing specific epitopes in nucleoproteins A and B, were evaluated. Compared with the real-time RT-PCR kit for diagnosis, HRP-LFIA was found to be sensitive and specific for the detection of Influenza A and B viruses [16]. No cross-contamination was observed with other common viral pathogens. In another study, several europium detection-based LFIAs were evaluated and showed good performance in comparison to the RT-PCR test [17]. Although conventional LFIA is simple to perform and provide rapid tests at a low cost, most LFIAs produce only a qualitative result (positive or negative) and are not really sensitive. To improve the performance of the assay, a microfluidic-based LFIA was recently developed [18]. The device enabled a LoD of 0.1 ng mL−1 NuP but its use requires high technical skills.
In this paper, we present a sensitive and easy-to-use magnetic bead-based enzyme-linked immunoassay (MELISA) for detecting the Influenza A virus in a clinical setting (saliva). A magnetic bead-based immunoassay for detection of the Influenza A virus via the nucleoprotein A has already been described in the literature [19,20,21]. A fluorescence immunoassay (FIA) together with a magnetic concentration was integrated into a microfluidic module and proved highly sensitive and selective for detecting the Influenza A/H1 and Influenza A/H3 viruses [20]. The microfluidic system afforded a 1000-fold increase in sensitivity compared with a standard immunomagnetic-based FIA carried out according to a manual protocol. The LoD was 5 × 10−4 HAU in 45 μL, which corresponds approximately to 104 PFU mL−1 [22]. Although the assay demonstrated rapid and sensitive detection of the Influenza A infection, it was based on the use of a microsystem whose complex geometry makes it complicated and challenging to manufacture. HRP-LFIA associated with a preconcentration step of the NuP protein by magnetic beads improved 100-fold the sensitivity [21]. The LoD was 4.7 μg mL−1. Recently, Wu et al. developed a wash-free assay based on magnetic particle spectroscopy (MPS) and the self-assembly of magnetic NP to quantitatively detect H1N1 NuP [23, 24]. This rapid protocol reached the LoD of 0.25 μg mL−1 for NuP and 250 TCID50 mL−1 for Influenza A virus.
The biosensor sensitivity is of great importance to detect pathogens in environmental, agrifood, or biological samples because pathogens are typically present in very low concentrations. Therefore, various diagnostic assays using functionalized nanoparticles to amplify the signal and boost the sensitivity have been reported in recent years [25,26,27,28]. Herein, we propose to evaluate the sensitivity increase of MELISA for Influenza A detection via the nucleoprotein A, using immunomagnetic beads for the capture step and biotin-coated nanoparticles (biotin-NP) to enhance the signal at the detection step.
MELISA is based on a simple operating process that is easy to implement. It only requires magnetic beads and biologics to elaborate the different reagents. In addition, it achieves good sensitivities compared to other microsystems such as lateral flow assays, and the immunomagnetic extraction allows direct analysis of biological samples. We selected a couple of commercial anti-nucleoprotein A antibodies to construct the sandwich assay. The monoclonal antibody 9G8 is a mouse monoclonal antibody that recognizes the NuP of the Influenza A virus [29]. This antibody was used to functionalize magnetic beads. The monoclonal antibody 5D8 showed broad reactivity against Influenza A subtypes H1, H2, H3, H5, H7, and H9, and no cross-reactivity towards Influenza B and other respiratory viruses [30]. The latter was biotinylated to complete the sandwich test before HRP colorimetric detection.
The MELISA was carried out with a Streptavidin-HRP conjugate to detect purified NuP in buffer. We then used biotin-NP to amplify the signal for virus detection in complex matrices. Experiments were performed in the lysis buffer complemented with saliva to simulate clinical samples. Fifty-nanometre-diameter silica NP were functionalized via phosphoramidite chemistry, using a strategy of supported synthesis, adapting a protocol previously described by our group [31]. This well-controlled and reproducible method proved effective for NP functionalization with methylene blue-modified oligonucleotides [25] and proteins [32]. We used commercial biotin phosphoramidite for this NP functionalization. Then, the MELISA protocol with biotin-NP was optimized for nucleoprotein detection in virus samples. The LoD of MELISA was determined in a clinical setting.
Material and methods
Reagents
Bovine serum albumin (BSA), Bradford reagent, ELISA Colorimetric TMB (3,3′,5,5′-tertamethylbenzidine) reagent, Tween 20, Streptavidin-HRP, 1,8-Diazabicyclo[5.4.0]undec-7-ene (DBU), ELISA 5X Assay/Sample Diluent buffer B and high-quality grade chemicals were purchased from Sigma-Aldrich (Saint Quentin Fallavier, France). Phosphate-buffered saline (PBS) 10× was provided by Dominique Dutscher (Brumath, France). Sodium chloride was purchased from Laurylab (Brindas, France). Buffers were made with deionized water and sterilized by 0.22-μm filtration.
Dynabeads® M-280 Tosylactivated and DSB-XTM Biotin Protein Labelling Kit (D-20655) were obtained from Invitrogen and Molecular Probes, respectively (Thermo Fisher Scientific, Waltham, MA, USA). Influenza A NuP (9G8) and (5D8) antibodies were from Santa Cruz Biotechnology (St. Cruz, CA, USA). Rhodamine-coated silica nanoparticles were provided by Nano-H (Saint Quentin Fallavier, France). Oligonucleotides were synthetized using an Applied Biosystems 394 DNA/RNA synthesizer (Applied Biosystems, Foster City, CA). Phosphoramidite synthons: Biotin-TEG-phosphoramidite (1-dimethoxytrityloxy-3-O-(N-biotinyl-3-aminopropyl)-triethyleneglycolyl-glyceryl-2-O-(2-cyanoethyl)-(N,N-diisopropyl)-phosphoramidite); Spacer phosphoramidite 9 (9-O-dimethoxytrityl-triethylene glycol,1-[(2-cyanoethyl)-(N,N-diisopropyl)]-phosphoramidite); dT-CE phosphoramidite (5′-dimethoxytrityl-2′-deoxythymidine,3′-[(2-cyanoethyl)-(N,N-diisopropyl)]-phosphoramidite) and all synthesis reagents were purchased from Glen Research (Sterling, Virginia).
Saliva collection
Saliva samples were collected from healthy volunteers after thorough rinsing of the mouth with water no less than 1 h after a meal. Collection of saliva was carried using a 1.5-mL tube and diluted with PBS buffer. Saliva samples were stored at − 20 °C before use.
Scanning electron microscopy
Scanning electron microscopy (SEM) was performed on magnetic beads at an acceleration voltage of 5 kV using a VEGA TESCAN SEM. Before analysis, samples were metallized with 5 nm thickness of gold.
Transmission electron microscopy
Transmission electron microscopy (TEM) was performed on a Philips CM120 instrument operating at an acceleration voltage of 120 kV (Centre Technologique des Microstructures, Lyon). Silica NP were observed after deposition of 5 μL of diluted solution on a formvar-carbon-coated copper grid and evaporation to dryness.
Nucleoprotein expression and production
The full-length nucleoprotein (NuP) gene of the H1N1 (strain A/WSN/33) influenza virus with a 6-His-tag at its C-terminal was cloned in the Escherichia coli expression vector pET22 (Novagen) and expressed in the BL-21 Rosetta DE3 E. coli cells (Stratagene) [33]. Transformed cells were grown at 37 °C in 100 mL of Luria Bertani medium containing ampicillin to mid-exponential phase and then induced for 4 h by adding 1 mM isopropyl-β-d-thiogalactopyranoside at 37 °C under shaking. NuP purification was performed as previously described [33]. Briefly, after centrifugation, the bacterial pellet was resuspended in a lysis buffer (20 mM Tris at pH 7.4 with NaCl (50 mM or 300 mM), 5 mM imidazole, 1% Triton and 1 mg mL−1 lysozyme), sonicated and treated with 0.15 mg mL−1 RNAse A at 35 °C for 20 min in the presence of 10 mM Mg2+ ions. The protein was purified by affinity chromatography using Ni magnetic beads (PureProteome, Millipore), following the procedure recommended by the manufacturer. The NuP was then dialysed against Tris buffer (20 mM Tris, 300 mM NaCl, pH 7.5). After dialysis, the protein concentration was determined using a molar extinction coefficient ε of 56,200 M−1 cm−1 at 280 nm.
Viral strains
Influenza A/WSN/33 (H1N1), Influenza A/UDORN/72 (H3N2) and Porcine Reproductive and Respiratory Syndrome virus (PRRSV) Flanders13 (13V09) viruses inactivated by binary ethylenimine (BEI) were kindly provided by Dr. Nicolas Bertho (INRAE, France) and Dr. Fanny Blanc (INRAE, France). All inactivated viruses were stored at − 80 °C. Before inactivation, virus concentrations were determined in terms of MDCK cell infection. The initial titres of the viruses were 108 PFU mL−1 for H1N1 sample 1, 1.5 × 108 PFU mL−1 for H1N1 sample 2 and 108 PFU mL−1 for H2N3. The control virus PRRSV was titrated at 107 PFU mL−1.
Magnetic bead functionalization
Tosyl activated magnetic beads of 2.8 μm diameter (Dynabeads™ M-280, Invitrogen, USA) were coated with 10 μg of Influenza A NuP (9G8) antibody per milligram of beads for 1 h under gentle shaking, according to the protocol recommended by the manufacturer. Then, after 15 min passivation in BSA buffer (0.1 M sodium phosphate, 150 mM sodium chloride, 0.5% (w/v) BSA, pH 7.6), the magnetic beads were washed three times in 0.01 M sodium phosphate, 150 mM sodium chloride, 0.1% (w/v) BSA, pH 7.4, and stored at 4 °C until the assays.
Biotin coating on silica NP
Rhodamine-coated silica nanoparticles were functionalized by a supported synthesis strategy developed by our group [31]. The functionalization was achieved in a DNA/RNA synthesizer using phosphoramidite chemistry. First, dT10-PEG2-dT10 was synthesized via the 1 μM coupling program of the instrument, reaching an average phosphoramidite coupling yield per cycle of 98%; then, the biotin group was incorporated with a 10% coupling yield. Biotin incorporation was monitored at 498 nm using dimethoxytrityl quantification by a UV-visible spectrophotometer.
Nanoparticles were then released from the CPG by incubating the support in 1 mL of DBU (1,8-Diazabicyclo[5.4.0]undec-7-ene) 0.1% (w/v) in water-acetonitrile 1/1 (v/v). The DBU solution was stirred in a thermomixer at 22 °C for 1 h before recovering. A fresh DBU solution was added to the CPG suspension every hour. The released nanoparticles were washed with water (1× 4 mL then 3× 2 mL) and concentrated by centrifugation on a 30K Amicon Ultra filter (5000 g, 10 min). After the release kinetics, the nanoparticle concentration in each solution was quantified by UV-visible measurement at 560 nm (see Supplementary Information (ESM) Fig. S1A and B). Finally, the NP were characterized by DLS (ESM Fig. S2).
Biotin functionalization of Influenza A NuP (5D8) antibodies
The DSB-XTM Biotin Protein Labelling Kit (D-20655) was used. One hundred microlitres of 0.1 mg mL−1 5D8 antibody solution and 10 μL of 1 M sodium bicarbonate solution were transferred to a reaction tube. One microlitre of DSB-X biotin succinimidyl ester in DMSO solution was added. The reaction mixture was stirred for 90 min at RT. The biotinylated antibody was then purified on a purification resin by centrifugation.
ELISA protocol for Influenza A nucleoprotein detection
Fifty microlitres of 4 μg mL−1 solution of Influenza A NuP (9G8) antibody in PBS buffer was added to each well of a microtitration plate and incubated overnight at 4 °C. The wells were washed three times using Washing buffer 0.5 (PBS, 0.5% (v/v) Tween 20, pH 7.4). Three hundred microlitres of PBS buffer with 5% (w/w) milk was added to each well and incubated at 37 °C for 1 h under gentle shaking. Then, the wells were washed three times using Washing buffer 0.5. Fifty microlitres of Influenza A nucleoprotein in PBS buffer at different concentrations was added to each well, and the solutions were incubated at room temperature for 2 h under gentle shaking. Then, the wells were washed three times using Washing buffer 0.1 (PBS, 0.1% (v/v) Tween 20, pH 7.4). Fifty microlitres of 0.5 μg mL−1 biotinylated Influenza A NuP (5D8*) antibody in PBS with 0.1% Tween 20 and 0.1% BSA was added before incubation at RT for 2 h. Then, the wells were washed three times using Washing buffer 0.1. One hundred microlitres of 1 μg mL−1 Streptavidin-HRP revelation solution was added to the wells before incubating for 45 min at room temperature under gentle shaking. The wells were washed three times with Washing buffer 0.1 before adding 100 μL of TMB and incubating the plate in the dark for 30 min at RT. Then, 100 μL of 2 M sulfuric acid was added to the wells to stop the reaction. Absorbance was measured at 450 nm and 550 nm. The absorbance values retained were A450nm–A550nm to take into consideration the optical imperfections of the plate.
MELISA protocol for Influenza A nucleoprotein detection
1 × 106 magnetic beads previously coated with the Influenza A NP (9G8) antibody were first washed with PBS buffer three times. Nucleoprotein solutions were incubated with the beads under gentle shaking at room temperature for 1 h. The beads were then washed three times with Washing buffer 0.1 (PBS, 0.1% (v/v) Tween 20, pH 7.4). Conjugated biotin antibody 5D8* (0.5 μg mL−1) was incubated in a Capture buffer (PBS, 0.1% (v/v) Tween 20, 0.1% BSA (w/w), pH 7.4) for 1 h under gentle shaking. After three washing steps, Streptavidin-HRP revelation solution was added before incubation at room temperature for 30 min at 1000 rpm in a thermomixer. The beads were washed three times with Washing buffer 0.1 before adding 100 μL of TMB and incubating in the dark for 30 min at RT. Finally, the supernatant was diluted with 100 μL of 2 M sulfuric acid and absorbance was read at 450 nm in a microplate reader (Perkin Elmer). A450nm–A550nm gave the correct value after suppression of the background signal.
MELISA protocol for deactivated virus detection
1 × 106 magnetic beads previously coated with the Influenza A NP (9G8) antibody were first washed with PBS buffer three times. Deactivated virus solutions (PBS, 10% saliva) were incubated with the beads under gentle shaking at room temperature for 1 h. The beads were concentrated by means of a magnet and washed three times with Washing buffer 0.1. Conjugated biotin antibody 5D8* (0.5 μg mL−1) was added to the suspension in a Capture buffer (PBS, 0.1% Tween 20, 0.1% BSA) before shaking gently for 1 h. After three washing steps, Streptavidin-HRP revelation solution (100 μL, 1 μg mL−1) was added and the mixture was shaken at RT for 30 min at 1000 rpm in a thermomixer. The beads were washed three times with Washing buffer 0.1 before adding 100 μL of TMB and incubating in the dark for 30 min at RT. Finally, absorbance was read at 450 nm as previously described.
For the MELISA protocol using biotin-NP amplification, the same protocol was used for the antigen capture. Then, the two following steps were added for detection: biotin-NP (1 × 1011 NP mL−1 in PBS, 0.1% (v/v) Tween 20) was added to the magnetic bead suspension before incubation for 30 min at 1000 rpm in a thermomixer. After three washing steps, Streptavidin-HRP revelation solution (100 μL, 1 μg mL−1) was added before incubating the suspension at room temperature for 30 min at 1000 rpm in a thermomixer. The beads were washed three times with Washing buffer 0.1 before adding 100 μL of TMB and incubating in the dark for 5 min at room temperature. Finally, absorbance was read at 450 nm as previously described.
Results and discussion
We first compared the performance of a magnetic bead-based immunoassay called MELISA with that of a more conventional ELISA for specific detection of the Influenza A nucleoprotein. Figure 1 illustrates the two strategies.

Presentation of the ELISA and MELISA for NuP detection
ELISA detection of NuP protein
A sandwich ELISA method for Influenza A nucleoprotein detection was performed using Streptavidin-HRP colorimetric detection. The assay was carried out with (1) the anti-Influenza A NuP antibody (9G8) for microwell coating and (2) the anti-Influenza A NuP biotinylated antibody (5D8*) as second antibody for detection. The experiments were carried out within a range of NuP concentrations from 0 to 8 μg mL−1. 9G8 and 5D8* antibodies were kept constant at 4 μg mL−1 and 0.5 μg mL−1 respectively, concentrations which have been determined as suitable to obtain a measurable signal.
A positive control was performed in a well directly coated with 0.5 μg mL−1 of biotinylated antibody (5D8*). A strong colorimetric response was observed. A negative control with no 9G8 antibody coating on the bottom of the well and a blank corresponding to the full assay without the protein in the binding solution were also carried out.
The results are reported in Fig. 2. A detection threshold of 0.6 OD was determined, corresponding to the average of three blank values plus three times the standard deviation. A LoD of 0.25 μg mL−1 for NuP was obtained with this ELISA format.

Influenza A NuP detection by ELISA. Absorbance (Abs 450 nm–Abs 550 nm) versus 0–8 μg mL−1 of Influenza A nucleoprotein in PBS buffer, pH 7.4. Values are mean ± SD of three independent experiments
MELISA detection of NuP protein
The first step in the MELISA sandwich assay consisted of grafting the 9G8 antibody onto magnetic beads. Then, passivation of the bead surface was performed with BSA to reduce the signal-to-noise ratio. After grafting, the number of beads used for the assay was calculated considering the concentration of 9G8 antibody grafted to the beads, so a volume of beads containing the same amount of the 9G8 antibody as that used to coat the wells of the ELISA plate was used for each assay. 1 × 106 magnetic beads were therefore used at the capture step for each test. BSA was added to PBS to improve the washing steps and obtain the best signal-to-noise ratio.
The MELISAs were carried out by varying the Influenza A NuP concentration from 0 to 4 μg mL−1 and the obtained data were compiled and averaged (Fig. 3). The capture step was performed with magnetic beads previously functionalized with the monoclonal antibody (9G8). After NuP capture, the detection step consisted of a second binding operation with biotinylated 5D8* mAb and Streptavidin-HRP.

MELISA detection of Influenza A NuP. Absorbance (Abs 450 nm–Abs 550 nm) versus 0–4 μg mL−1 of Influenza A nucleoprotein in PBS buffer, pH 7.4. Values are mean ± SD of three independent experiments
MELISA shows a strong response correlated with NuP detection. In the protocol, the beads were incubated with NuP protein solutions for 60 min. A plateau was reached at 0.5 μg mL−1 of protein. A detection threshold of 1.4 OD was determined, corresponding to the average of the three blank values plus three times the standard deviation. The results with MELISA show a NuP detection limit of 0.063 μg mL−1, which is 4 times better than with ELISA. A range of linearity was observed between 0.063 and 1 μg mL−1 of NuP. The detection limit of MELISA is also better than that obtained by Wu et al. using magnetic nanoparticles and direct measurement by spectroscopy (LoD of 0.25 μg mL−1) [23] .
To optimize the time required to carry out the complete MELISA, we also investigated MELISA with different NuP and antibody incubation times. Incubation times of 30 min and 60 min were investigated to bind the capture antibody (9G8) with the NuP and the NuP with the second antibody (5D8*). The results are shown in Fig. S3 (see ESM). With a detection threshold calculated at 1.4 OD, the LoD reached by the faster test (incubation time of 30 min) was the same as that previously obtained (0.063 μg mL−1). However, the saturation plateau was reached more rapidly. The test response after 30-min incubation was more like a yes/no answer than a NuP concentration-dependent response. No progressive increase of the signal as a function of protein concentration was observed. This can be explained by a steric hindrance effect that impacted the kinetics. The binding reaction that occurs on the magnetic bead surface takes longer than that in solution. Considering these results, we preferred to maintain an incubation time of 60 min for the following experiments with detection of whole Influenza viruses.
MELISA detection of H1N1 and H2N3 viruses
To determine MELISA performance for Influenza A virus detection, two different strains, H1N1 and H3N2, were studied, together with PRRSV, a porcine virus, whose nucleoprotein is not recognized by the antibodies.
ELISA was first performed with the H1N1 sample 1, following the protocol used for Influenza A NuP detection. Different dilutions of the mother solution of the virus were tested to estimate the lowest virus concentration that could be detected. The blank run with PBS instead of the virus gave a signal threshold (average of three blanks plus three times the standard deviation) of 0.5 OD. As shown in Fig. 4, ELISA obtained a LoD of 1.2 × 107 PFU mL−1 for H1N1 sample 1.

MELISA and ELISA with whole viruses of different strains (H1N1 sample 1, H3N2, PRRSV). Absorbance (Abs 450 nm–Abs 550 nm) for different concentrations of viruses (1 × 104–1 × 108 PFU mL−1) in PBS buffer, pH 7.4
The same sample was then analysed by MELISA (green curve, Fig. 4). The protocol was the same as for Influenza NuP detection. The detection threshold of 0.5 OD was determined as the average value of three blanks plus three times the standard deviation. MELISA shows a LoD of 2 × 105 PFU mL−1 for H1N1, which is approximatively 60 times more sensitive than with ELISA. As for the assays with purified NuP, MELISA increases the sensitivity of the assays run with whole viruses. MELISA was also carried out with different concentrations of H3N2 (blue curve, Fig. 4). As expected, a positive response was observed above a detection limit of 3 × 106 PFU mL−1. MELISA shows good sensitivity for H1N1 and H3N2 detection but lacks sensitivity compared with the other immunomagnetic bead-based assays described in the literature [20, 34, 35]. The negative responses obtained for non-related PRRSV at different concentrations indicate the selectivity of MELISA for Influenza A viruses (Fig. 4).
Biotin-NP MELISA for Influenza A virus sensitive detection
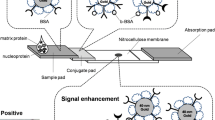
To enhance MELISA’s performance and push detection limits even further, we developed biotin-NP to amplify the positive signal at the detection step, as illustrated on Fig. 5.

Presentation of MELISA (a) and Biot-NP MELISA (b) assays for Influenza A virus detection
Biotin-NP development
Silica nanoparticles of 50 nm diameter were functionalized on surface with an oligonucleotidic strand bearing a biotin group at its extremity. The method used for NP functionalization was based on a solid-phase synthesis technology previously reported by our group [31]. Nanoparticles (NP) were first immobilized on a control pore glass (CPG) support. The resulting assembled nano-on-micro (NOM) support was characterized by scanning electron microscopy (SEM) as shown in Fig. S4 (see ESM). The images show that the NP are grafted densely and homogeneously on the surface of CPG. Then, the NOM material was used in a DNA/RNA synthesizer to perform NP functionalization via phosphoramidite chemistry. This strategy provided highly functionalized nanoparticles. An oligonucleotidic spacer (ODN sequence: dT10-PEG2-dT10) was synthesized on the NP in order to distance the biotins from the NP surface. Around 5000 ODNs per NP were estimated by measuring UV absorbance at 260 nm and 560 nm and quantifying the ODN per NP ratio in the suspension, a protocol described by Bonnet et al. [25]. Then, the biotin group was incorporated. We decided to control the synthesis program on the instrument to obtain a 10% coupling yield for biotin incorporation. Using this strategy, we incorporated around 50 biotins per NP. This amount was optimized to achieve efficient binding of the biotin group during the MELISA. After NP release from the CPG support by DBU treatment and washing in water, the number of NP in the resulting suspension was estimated from the fluorescent NP quantification curve (ESM Fig. S1C, D). The NP structure after release was controlled by TEM (Fig. 6a). This observation showed that NP morphology remained stable during synthesis.

(a) TEM images of Biot-NP; SEM images of magnetic beads after the Biot-NP MELISA test (b) assay with 0.25 μg mL−1 nucleoprotein; (c) blank assay with PBS
We also confirmed the accessibility and reactivity of the biotins grafted onto the NP surface by SPR, via the biotin/avidin interaction. After immobilization of avidin on a SPR chip coated with a polyelectrolyte multilayer, we managed to observe the specific capture of the biotinylated nanoparticles (ESM Fig. S5A). The NP specific anchoring on the avidin-functionalized SPR chip was also confirmed by fluorescence optical microscopy (surface in red on the picture, see ESM Fig. S5B).
Biotin-NP display by electronic microscopy
The magnetic beads used during the biotin-nanoparticle and immunomagnetic bead-based assay (Biot-NP MELISA) were analysed by SEM.
For this experiment, the complete assay was carried out in the presence of nucleoprotein (0.25 μg mL−1) at the capture step (Fig. 6b). For comparison, a control was performed with PBS at the capture step (Fig. 6c). The images clearly show nanoparticles of the expected size of 50 nm decorating the magnetic bead surface in the positive assay carried out with the target protein. This confirms the selectivity of the assay, with efficient binding of the biotin-NP on the magnetic beads through the NuP present in the sample.
Influenza A virus detection in a clinical setting
The biotin-nanoparticle and immunomagnetic bead-based assay (Biot-NP MELISA) was investigated for detection of H1N1 (samples 1 and 2), H3N2 and PRRSV viruses (Fig. 7).

Biot-NP MELISA with whole viruses of different strains (H1N1 sample 1, H1N1 sample 2, H3N2, PRRSV). Absorbance (Abs 450 nm–Abs 550 nm) for different concentrations of viruses (2 × 103–8 × 106 PFU mL−1) in PBS buffer, pH 7.4 + 10% saliva
To mimic clinical samples, the viral solutions were diluted in lysis buffer supplemented with 10% saliva. After the sandwich part of the assay with viruses, the detection step was carried out using highly biotinylated nanoparticles to increase the anchoring efficiency of the Streptavidin-HRP. This strategy provided a signal increase in the case of a positive response (Fig. 7). This improved sensitivity demonstrated the efficiency of the amplification strategy. Detection limits of 3 × 103 PFU mL−1, 6 × 104 PFU mL−1 and 4 × 104 PFU mL−1 were calculated for H1N1 sample 1, H1N1 sample 2, and H3N2, respectively. Use of biotin-NP at the detection step improved MELISA’s sensitivity by a factor of 65 to 75. Detection limits below 104 PFU mL−1 and 105 PFU mL−1 were achieved for the H1N1 and H3N2 strains respectively, in the presence of 10% saliva to simulate a clinical setting. Good selectivity of the strains was also confirmed by the negative responses observed at different concentrations of PRRSV (Fig. 7).
The biotin-functionalized silica nanoparticles used in this work allowed a strong detection signal enhancement in MELISA immunological tests, which could fulfill the detection requirements for Influenza A virus in saliva. The excellent reproducibility, accuracy and practicability of the proposed assay obtained in samples comparable to clinical indicate the considerable potential of the present strategy. Biotin highly functionalized silica nanoparticles could be easily adapted to be a component of a variety of kits for detection of different analytes (DNA, enzyme, IgG, biomarkers). In addition, biotin-NP can be additionally functionalized to enable single step detection. For instance, this can be obtained with attaching organic dye-decorated antibodies or streptavidin-functionalized quantum dots conjugated to biotinylated antibodies. Overall, this technology is not limited to streptavidin directly labeled with HRP but could also work with labeled fluorophores via the streptavidin-biotin recognition.
Conclusions
In this study, a sensitive immunomagnetic bead-based assay (MELISA) was developed for Influenza A virus pan-reactive detection. The test was first compared with the standard ELISA and showed a significant improvement in sensitivity thanks to the magnetic capture and concentration of the target nucleoprotein. Then, we developed a new MELISA (Biot-NP MELISA) based on the use of biotin nanoparticles for signal amplification at the detection step and succeeded in detecting Influenza A/H1 and A/H3 viruses in a clinical setting. We demonstrated that biotinylated nanoparticles further increased sensitivity by a factor of 65–75. Detection limits of 3 × 103 PFU mL−1 and 4 × 104 PFU mL−1 were achieved for the H1N1 and H3N2 strains respectively. Biot-NP MELISA sensitivity appears competitive compared with other immunoassays developed with magnetic concentration at the capture step [20]. Our strategy allows capture of the viral nucleoprotein directly in solutions mimicking clinical samples, which is a huge advantage when developing assays with a fast time-to-result.
References
Miller MA, Viboud C, Balinska M, Simonsen L. The signature features of influenza pandemics — implications for policy. N Engl J Med. 2009;360(25):2595–8. https://doi.org/10.1056/NEJMp0903906.
Taubenberger JK, Morens DM. 1918 Influenza: the mother of all pandemics. Rev Biomed. 2006;17(1):69–79.
World Health Organisation (WHO). Available from https://www.who.int/news-room/fact-sheets/detail/influenza-(seasonal). 2018.
Khanna M, Kumar P, Choudhary K, Kumar B, Vijayan VK. Emerging influenza virus: a global threat. J Biosci. 2008;33(4):475–82. https://doi.org/10.1007/s12038-008-0066-z.
Moeller A, Kirchdoerfer RN, Potter CS, Carragher B, Wilson IA. Organization of the influenza virus replication machinery. Science. 2012;338(6114):1631–4. https://doi.org/10.1126/science.1227270.
Albo C, Valencia A, Portela A. Identification of an RNA binding region within the N-terminal third of the influenza A virus nucleoprotein. J Virol. 1995;69(6):3799–806.
Shu LL, Bean WJ, Webster RG. Analysis of the evolution and variation of the human influenza A virus nucleoprotein gene from 1933 to 1990. J Virol. 1993;67(5):2723–9.
Yang M, Berhane Y, Salo T, Li M, Hole K, Clavijo A. Development and application of monoclonal antibodies against avian influenza virus nucleoprotein. J Virol Methods. 2008;147(2):265–74. https://doi.org/10.1016/j.jviromet.2007.09.016.
Plotnikova MA, Klotchenko SA, Lebedev KI, Lozhkov AA, Taraskin AS, Gyulikhandanova NE, et al. Antibody microarray immunoassay for screening and differential diagnosis of upper respiratory tract viral pathogens. J Immunol Methods. 2020;478:112712. https://doi.org/10.1016/j.jim.2019.112712.
Chomel JJ, Thouvenot D, Onno M, Kaiser C, Gourreau JM, Aymard M. Rapid diagnosis of influenza infection of NP antigen using an immunocapture ELISA test. J Virol Methods. 1989;25(1):81–91. https://doi.org/10.1016/0166-0934(89)90102-X.
Coonrod JD, Betts RF, Linnemann CC, Hsu LC. Etiological diagnosis of influenza A virus by enzymatic radioimmunoassay. J Clin Microbiol. 1984;19(3):361–5.
Chen Y, Xu F, Gui X, Yang K, Wu X, Zheng Q, et al. A rapid test for the detection of influenza A virus including pandemic influenza A/H1N1 2009. J Virol Methods. 2010;167(1):100–2. https://doi.org/10.1016/j.jviromet.2010.02.001.
Farris LR, Wu N, Wang W, Clarizia L-JA, Wang X, McDonald MJ. Immuno-interferometric sensor for the detection of influenza A nucleoprotein. Anal Bioanal Chem. 2010;396(2):667–74. https://doi.org/10.1007/s00216-009-3235-5.
Zhang P, Vemula SV, Zhao J, Du B, Mohan H, Liu J, et al. A highly sensitive europium nanoparticle-based immunoassay for detection of influenza A/B virus antigen in clinical specimens. J Clin Microbiol. 2014;52(12):4385–7. https://doi.org/10.1128/jcm.02635-14.
Leirs K, Tewari Kumar P, Decrop D, Pérez-Ruiz E, Leblebici P, Van Kelst B, et al. Bioassay development for ultrasensitive detection of influenza A nucleoprotein using digital ELISA. Anal Chem. 2016;88(17):8450–8. https://doi.org/10.1021/acs.analchem.6b00502.
Zhang J, Gui X, Zheng Q, Chen Y, Ge S, Zhang J, et al. An HRP-labeled lateral flow immunoassay for rapid simultaneous detection and differentiation of influenza A and B viruses. J Med Virol. 2019;91(3):503–7. https://doi.org/10.1002/jmv.25322.
Yoo SJ, Shim HS, Yoon S, Moon H-W. Evaluation of high-throughput digital lateral flow immunoassays for the detection of influenza A/B viruses from clinical swab samples. J Med Virol. 2019;92(8):1040–6. 1–7. https://doi.org/10.1002/jmv.25626.
Kim J, Hong K, Kim H, Seo J, Jeong J, Bae PK, et al. Microfluidic immunoassay for point-of-care testing using simple fluid vent control. Sensors Actuators B Chem. 2020;316:128094. https://doi.org/10.1016/j.snb.2020.128094.
Hung L-Y, Chang J-C, Tsai Y-C, Huang C-C, Chang C-P, Yeh C-S, et al. Magnetic nanoparticle-based immunoassay for rapid detection of influenza infections by using an integrated microfluidic system. Nanomedicine. 2014;10(4):819–29. https://doi.org/10.1016/j.nano.2013.11.009.
Lien K-Y, Hung L-Y, Huang T-B, Tsai Y-C, Lei H-Y, Lee G-B. Rapid detection of influenza A virus infection utilizing an immunomagnetic bead-based microfluidic system. Biosens Bioelectron. 2011;26(9):3900–7. https://doi.org/10.1016/j.bios.2011.03.006.
Noh JY, Yoon S-W, Kim Y, Lo TV, Ahn M-J, Jung M-C, et al. Pipetting-based immunoassay for point-of-care testing: application for detection of the influenza A virus. Sci Rep. 2019;9(1):16661. https://doi.org/10.1038/s41598-019-53083-8.
Killian ML. (2008) Hemagglutination assay for the avian influenza virus. In: Spackman E, editor. Avian Influenza Virus. Methods in Molecular Biology™, vol 436: Humana Press. p. 47.
Wu K, Liu J, Saha R, Su D, Krishna VD, Cheeran MCJ, et al. Magnetic particle spectroscopy for detection of influenza a virus subtype H1N1. ACS Appl Mater Interfaces. 2020;12(12):13686–97. https://doi.org/10.1021/acsami.0c00815.
Su D, Wu K, Krishna VD, Klein T, Liu J, Feng Y, et al. Detection of influenza a virus in swine nasal swab samples with a wash-free magnetic bioassay and a handheld Giant Magnetoresistance sensing system. Front Microbiol. 2019;10(1077):01077. https://doi.org/10.3389/fmicb.2019.01077.
Bonnet R, Farre C, Valera L, Vossier L, Léon F, Dagland T, et al. Highly labeled methylene blue-ds DNA silica nanoparticles for signal enhancement of immunoassays: application to the sensitive detection of bacteria in human platelet concentrates. Analyst. 2018;143(10):2293–303. https://doi.org/10.1039/C8AN00165K.
Dutta Chowdhury A, Ganganboina AB, Nasrin F, Takemura K, Doong R-A, Utomo DIS et al. Femtomolar detection of dengue virus DNA with serotype identification ability. Anal Chem 2018;90(21):12464–12474. doi:https://doi.org/10.1021/acs.analchem.8b01802.
Afonso AS, Pérez-López B, Faria RC, Mattoso LHC, Hernández-Herrero M, Roig-Sagués AX, et al. Electrochemical detection of Salmonella using gold nanoparticles. Biosens Bioelectron. 2013;40(1):121–6. https://doi.org/10.1016/j.bios.2012.06.054.
Sena-Torralba A, Ngo DB, Parolo C, Hu L, Álvarez-Diduk R, Bergua JF, et al. Lateral flow assay modified with time-delay wax barriers as a sensitivity and signal enhancement strategy. Biosens Bioelectron. 2020;168:112559. https://doi.org/10.1016/j.bios.2020.112559.
Zu M, Yang F, Zhou W, Liu A, Du G, Zheng L. In vitro anti-influenza virus and anti-inflammatory activities of theaflavin derivatives. Antivir Res. 2012;94(3):217–24. https://doi.org/10.1016/j.antiviral.2012.04.001.
Liu J, Zhao J, Petrochenko P, Zheng J, Hewlett I. Sensitive detection of influenza viruses with europium nanoparticles on an epoxy silica sol-gel functionalized polycarbonate-polydimethylsiloxane hybrid microchip. Biosens Bioelectron. 2016;86:150–5. https://doi.org/10.1016/j.bios.2016.06.044.
De Crozals G, Farre C, Hantier G, Léonard D, Marquette CA, Mandon CA, et al. Oligonucleotide solid-phase synthesis on fluorescent nanoparticles grafted on controlled pore glass. RSC Adv. 2012;2:11858–66. https://doi.org/10.1039/C2RA22077F.
De Crozals G, Kryza D, Sánchez GJ, Roux S, Mathé D, Taleb J, et al. Granulocyte colony-stimulating factor nanocarriers for stimulation of the immune system (part I): synthesis and biodistribution studies. Bioconjug Chem. 2018;29(3):795–803. https://doi.org/10.1021/acs.bioconjchem.7b00605.
Tarus B, Chevalier C, Richard C-A, Delmas B, Di Primo C, Slama-Schwok A. Molecular dynamics studies of the nucleoprotein of influenza A virus: role of the protein flexibility in RNA binding. PLoS One. 2012;7(1):e30038. https://doi.org/10.1371/journal.pone.0030038.
Sayhi M, Ouerghi O, Belgacem K, Arbi M, Tepeli Y, Ghram A, et al. Electrochemical detection of influenza virus H9N2 based on both immunomagnetic extraction and gold catalysis using an immobilization-free screen printed carbon microelectrode. Biosens Bioelectron. 2018;107:170–7. https://doi.org/10.1016/j.bios.2018.02.018.
Luo F, Long C, Wu Z, Xiong H, Chen M, Zhang X, et al. Functional silica nanospheres for sensitive detection of H9N2 avian influenza virus based on immunomagnetic separation. Sensors Actuators B Chem. 2020;310:127831. https://doi.org/10.1016/j.snb.2020.127831.
Acknowledgements
This work was supported by the ERASMUS program in the form of internships for Sara Viezzi and Alice Wright. The authors would like to express their gratitude to Sylvain Minot and François Bessueille for electronic microscopy (SEM) imaging. Thanks are also extended to Dr. Nicolas Bertho (INRA, France) and Dr. Fanny Blanc (INRA, France) for providing us with viral strains. We thank Dr. Anny Slama-Schwok for initiating the production of recombinant NuP. Finally, the authors thank the Centre Technologique des Microstructures of Lyon 1 University for providing access to its TEM facilities.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The saliva samples used in this work were blind samples, with no identification or any information required or registered. The informed consent was obtained from all volunteer participants included in the study. There was no association with a clinical trial.
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Published in the topical collection celebrating ABCs 20th Anniversary.
Supplementary information
ESM 1
(PDF 2379 kb)
Rights and permissions
About this article

Cite this article
Farre, C., Viezzi, S., Wright, A. et al. Specific and sensitive detection of Influenza A virus using a biotin-coated nanoparticle enhanced immunomagnetic assay. Anal Bioanal Chem 414, 265–276 (2022). https://doi.org/10.1007/s00216-020-03081-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-020-03081-x