
Abstract
A simple and efficient dispersive liquid-liquid microextraction technique (DLLME) was developed by using a mixture of two solvents: 40 μL of tetrachlorethylene (extraction solvent) and 1.0 mL of methanol (disperser solvent), which was rapidly injected with a syringe into 10 mL of water sample. Some important parameters affecting the extraction efficiency, such as type and volume of solvents, water sample volume, extraction time, temperature, pH adjustment and salt addition effect were investigated. Simultaneous determination of 34 commonly used pesticides was performed by using gas chromatography coupled with mass spectrometry (GC-MS). The procedure has been validated in order to obtain the highest efficiency at the lowest concentration levels of analytes to fulfill the requirements of regulations on maximum residue limits. Under the optimum conditions, the linearity range was within 0.0096–100 μg L−1. The limits of detection (LODs) of the developed DLLME-GC-MS methodology for all investigated pesticides were in the range of 0.0032 (endrin)–0.0174 (diazinon) μg L−1 and limits of quantification (LOQs) from 0.0096 to 0.052 μg L−1. At lower concentration of 1 μg L−1 for each pesticide, recoveries ranged between 84% (tebufenpyrad) and 108% (deltamethrin) with relative standard deviations (RSDs) (n = 7) from 1.1% (metconazole) to 11% (parathion-mehtyl). This methodology was successfully applied to check contamination of environmental samples. The procedure has proved to be selective, sensitive and precise for the simultaneous determination of various pesticides. The optimized analytical method is very simple and rapid (less than 5 min).

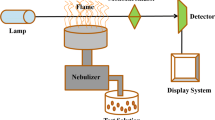
Analytical procedure for testing water samples consists of dispersive liquid-liquid microextraction (DLLME) and gas chromatography coupled with mass spectrometry (GC-MS).
Similar content being viewed by others


Introduction
Pesticides are a numerous and diverse group of chemical compounds. Nowadays, they are applied in many spheres of life, not only in agriculture [1, 2]. The range of applications is continually expanding; hence their consumption is ever increasing and more of them are getting into the environment [2,3,4]. As a result, the presence of pesticides, particularly those highly soluble and polar, in different type of environmental samples has been reported [5,6,7].
One of the basic ways of limiting the adverse effects of pesticides on human health is the continuous monitoring of these compounds [1]. The numerous regulations that have come into force, on both national and EU level, concerning permissible levels of pesticide residues, are driving the development of new analytical techniques and the improvement of existing ones [8,9,10,11]. However, pesticides monitoring in natural waters is very complex due to their low concentration levels, multiplicity in terms of physicochemical properties depending on their chemical structure (e.g. polarity, solubility in water and organic solvents, volatility, and acidic/base characteristics) and persistence in the environment [2, 6, 12,13,14,15].
The improvements of existing techniques are aimed at miniaturization, automation, simplification and the use of solvent-free techniques at the sample preparation stage, in order to make them consistent with ‘Green Chemistry’ principles [16].
Concerning the final determination of pesticides in environmental samples, the majority of these measurements are currently conducted by gas chromatography (GC) coupled with mass spectrometer (MS) detector (single or tandem), but liquid chromatography (LC) is also applicable, especially for polar and thermally unstable pesticides. Nevertheless, most of the GC methods tend to have long run times and to be very specific for a limited number of pesticides and sample matrices [17, 18].
Solvent microextraction, as an alternative for traditional liquid-liquid extraction (LLE), has become an increasingly used technique for sample preparation [2, 13, 17,18,19]. There are four main methods used in solvent microextraction: liquid-liquid microextraction (LLME), single-drop microextraction (SDME), hollow-fiber-protected microextraction (HFME) and dispersive liquid-liquid microextraction (DLLME) [20, 21]. DLLME was introduced in 2006 and has attracted great attention due to its wide range of applications [21,22,23,24,25,26]. In comparison with other solvent and sorptive microextraction techniques, DLLME does not require a long extraction time to obtain good extraction efficiency. The equilibrium is reached within a few seconds due to the very large surface area of the microdrops in the dispersed state. It allows for the analysis of small sample volumes and, because of small amounts of solvent used, is more environmentally friendly. Moreover, it is inexpensive, easy to operate, provides high recoveries and enrichment factors, and can be coupled with GC or HPLC. Nevertheless, this technique has some limitations, which are related mainly to the requirements posed for the extraction and disperser solvents. Only solvents slightly soluble in water and denser than water (mostly carbon disulfide and chlorinated solvents, for example: carbon tetrachloride, 1,1,1-trichloroethane, tetrachloroethylene or chlorobenzene) or high-melting liquids having densities lower than water (for instance, 1-undecanol, 1-dodecanol or hexadecane) can be used as extractants [27,28,29,30]. Regarding this second group of solvents, the sample needs additionally to be cooled and then the solidified drop of solvent is collected from the vial, melted and analyzed. This variant is called DLLME-Solidification of Floating Organic droplet (DLLME-SFO) [31, 32]. Special devices with narrow tube necks are required to facilitate the collection of floated extracts [33, 34]. Additionally, another solvent with higher density than water (an auxiliary solvent) can be added to adjust the density of the extraction solvent. Thus, through centrifugation, the extraction phase collects on the bottom of the conical vial [27, 35]. The use of an auxiliary solvent requires an additional step in the sample preparation procedure, which can be a source of an additional problems and errors.
As dispersive solvents, water-miscible and polar solvents, such as acetonitrile, acetone and methanol are routinely used [36]. The type and volume of disperser solvent can significantly affect the volume of the sedimented phase, which in turn influences the extraction efficiency. Moreover, the dispersive solvent allows for the extracting solvent to partition itself uniformly in the water sample and thus to achieve effective and rapid mass transfer. To accomplish this usually relatively large volumes of dispersive solvents are required (in the order of several milliliters), which decreases the partition coefficient of analytes into the extraction solvent [37,38,39]. This problem can be avoided by using ultrasonic stirring or a cationic surfactant in order to disperse the extraction solvent instead of dispersive solvent. In order to help the dispersion process, vortex agitation is also used [40,41,42]. Additionally, new solutions and improvements of the DLLME technique have recently been proposed. To overcome the non-selective characteristic of extraction solvents and their hazardous properties, new alternatives are being developed like the use of ionic liquids (ILs) and binary solvents (for example: mixture of tetrachloroethylene with tert-butyl methyl ether (4/6, v/v)) [43,44,45,46,47]. ILs have extremely low vapor pressures, possess good extractability and can dissolve a wide range of compounds. However, only ILs which are insoluble in water can be used in DLLME and the background peaks can interfere with analyte peaks [25]. In addition, ILs are relatively expensive, often need purification before application and their choice is based mainly on empirical experimentation due to the lack of precise physicochemical properties. Moreover, most ILs are incompatible with GC analysis [48].
In order to remove the centrifugation step, which is considered as one of the most time-consuming steps in this method, demulsified DLLME has been proposed [40,41,42]. After the introduction of the solvent mixture and consequently emulsion formation, another portion of dispersive solvent (serving as demulsifier) is introduced, leading to the breakup of the emulsion. This process is accomplished in two steps, flocculation and coalescence, and the solution is quickly cleared into two phases. However, the problem here is the selection of an appropriate extraction solvent, especially in the case of using low-density solvents. Due to their high-water solubility, resulting in good dispersion, the emulsion cannot be broken up by the demulsifier. Moreover, the commonly used demulsifiers are water miscible organic solvents, which has an adverse effect on the partition coefficients of analytes into the extraction solvent. Therefore, new alternatives are needed. To increase the dispersion of the extraction solvent into the aqueous solution, resulting in shorter equilibration time and enhancement the extraction efficiency, vortex agitation has been used [35]. For more complex matrices, such as honey, vegetable or soil samples, a two-step microextraction was proposed [49,50,51]. It is based on the combination of different extraction techniques, which are mostly DLLME and solid-phase extraction (SPE). This approach provides better analytical performance than its single-step counterpart, including high selectivity and sensitivity. On the other hand, it involves more operations in sample preparation, which can be a source of additional costs and problems.
All recently proposed improvements of the DLLME technique have their merits and drawbacks. All of them ensure higher enrichment factor, resulting in enhancement of the method sensitivity. Based on literature data, it can be concluded that halogenated extraction solvents, despite their drawbacks, are still the ones most frequently used for the extraction of various analytes [2, 17, 22, 23, 25, 26, 35,36,37,38,39]. DLLME has become a powerful tool for sample pretreatment due to its simplicity, rapidity (about 4–6 min), effectiveness and low cost. The biggest challenge has been the necessity of determining all contaminants (which have a broad spectrum of physicochemical properties) in a single analytical run. Thus, a solvent capable of extracting this group of diverse compounds is required. Such multi-contaminant analyses are currently recommended by European Union regulations and this makes DLLME a powerful tool in environmental science.
The purpose of this study was to develop and apply a multi-residue methodology to control and monitor the levels of 34 commonly used pesticides in aqueous samples. The pesticides selected for the study belong to various chemical groups with diverse types of action and some of them have not been measured previously and are considered difficult to extract. Table 1 presents the characteristics and physicochemical properties of the studied pesticides [52]. Different mixtures of extraction and disperser solvents were compared and evaluated for extraction of pesticides. Other parameters affecting extraction efficiency (solvents volume, aqueous sample volume, extraction time, temperature, salting out effect, centrifugation time, and sample pH) were systematically optimized. For the identification and quantitative determination, gas chromatography coupled with mass spectrometry (GC-MS) was used. The proposed methodology was validated and applied to environmental samples. The multi-residue procedure that was developed in this study reduces analysis time, solvent consumptions and the obtained values of limits of detection are adequate for trace analysis of these xenobiotics in aqueous samples.
Experimental
Chemicals and reagents
Certified individual pesticide standard solutions (100 mg L−1 in methanol and purities greater than 97%) of methiocarb, diazinon, γ-HCH (lindane), pirimicarb, carbendazim, fenpropidin, parathion-methyl, malathion, chlorpyrifos, tetraconazole, metazachlor, fipronil, alpha-endosulfan, flutolanil, flusilazole, endrin, cyproconazole, chlordecone (kepone), carfentrazone-ethyl, fluquinconazole, fenazaquin, tebufenpyrad, metconazole, phosalone, pyridaben, alpha-cypermethrin, quizalofop-ethyl, tau-fluvalinate, deltametrhin, azoxystrobin and dimethomorph, were obtained from Ultra Scientific, Kingstown (USA). Pesticide standard solutions (10 mg L−1 in acetonitrile, purity 98.5%) of fenoxaprop-ethyl, haloxyfop-R-methyl and prothioconazole were supplied by Dr. Ehrenstorfer GmbH (Germany). The internal standard (IS) of triphenylphosphate (TPP, 500 mg L−1 in methyl tert-butyl ether) was obtained from Supelco, Bellefonte (USA). The chemical formula, substance group, type of action, human health issues, molecular mass and physicochemical properties of the studied pesticides are presented in Table 1 [52].
Methanol, acetonitrile, carbon disulfide (CS2) and chlorobenzene (C6H5Cl) were supplied by Merck, Darmstadt (Germany). Tetrachloroethylene (C2Cl4), sodium chloride salt and potassium dihydrogen phosphate (purities 99+ %) were purchased from Sigma-Aldrich, Poole (UK). 1,1,1-trichloroethane (C2H3Cl3) was provided by Fisher Scientific, Loughborough (UK). Acetone and acetic acid were bought from POCH S.A., Gliwice (Poland). Chloroform (CHCl3) was supplied by CHEMPUR Company, Piekary Śląskie (Poland). Sodium hydroxide (pure to analysis) was purchased from STANLAB, Lublin (Poland). The reagents were analytical or higher grade and solvents were chromatography grade. The water used in this study was obtained from an ultrapure water purification system Milli-Q–Millipore Corporation, Bedford (USA).
A standard stock solution of 34 pesticides was prepared in acetonitrile at the concentration 100 μg L−1 and maintained at 4 °C. Individual standard solutions of 34 pesticides at 10 mg L−1 were made in acetonitrile and stored in freezer at − 20 °C. The working aqueous solutions were prepared daily by spiking purified water with the standard solutions at different concentration levels. These standards were used both for matrix spike, in order to optimize the extraction conditions and in the validation study in different concentration levels from 0.02 to 100 μg L−1. The calibration standards at concentrations of 0.02; 0.05; 0.1; 1.0; 10; 50 and 100 μg L−1 were prepared by dilution of the working standards directly into the matrix. Each aqueous solution (standard or environmental) was spiked by internal standard of TPP at the concentration of 10 μg L−1.
Water samples
In order to examine the applicability of the proposed multi-residue method, environmental water samples from various sources (with different values of the pH, salt content, solid particles, etc.) were tested. Natural samples from river, lake, sea, well, pond, canal and rain collected in northern Poland (Tri-city area: Gdańsk, Sopot and Gdynia) and northeast of Poland (central Masuria and Podlasie area) were selected for analysis. In total, samples from 34 locations situated in the study area were taken in cooperation with the Voivodship Inspectorate of Environmental Protection in Gdańsk (Poland) and the urban water supply Saur Neptun Gdańsk S.A.
Water samples were collected during the execution of agrochemical treatments (growing season of plants) into brown glass bottles with a capacity of 1 l, without a concave bottom. Before filling with water, the test bottles were rinsed with water and then filled with no headspace in order to eliminate the presence of air in the bottleneck. After collection, the samples were immediately placed in a cooler at 4 °C in the dark and transported to the laboratory, where they were analyzed within 24 h. Prior to the analysis, water samples were filtered through a 0.45-μm PTFE filter (Cronus, Gloucester, UK) and were stored at 4 °C. All samples were analyzed in triplicate using the DLLME optimized procedure and GC-MS.
Water from the river was used in this study to calculate the recovery of pesticides at two concentration levels (1 and 50 μg L−1) and to check the impact of different physicochemical properties of analyzed water samples on the results. The higher value of concentration was dictated by the upper limit of the linearity range for the studied pesticides. The pH values of the analyzed water samples were in the range of 4.6 (rain)–7.8 (lake).
DLLME procedure
A 10-mL water sample was transferred into a 12-mL screw cap glass test tube with conical bottom and spiked with standards at 1 mg L−1 for each pesticide, and 10 μg L−1 for TPP as internal standard. A mixture of 1 mL of methanol (as disperser solvent) and 40 μL of tetrachloroethylene (as extraction solvent) was rapidly injected into the sample solution by using a 5-mL syringe (Hamilton, Reno, NV, USA) and then the mixture was gently shaken for 30 s. A cloudy emulsion (water, methanol, and tetrachloroethylene) was formed in the test tube. The mixture was then centrifuged for 3 min at 2500 rpm, causing the dispersed fine droplets of the extraction phase to settle to the bottom of the conical test tube. The 30 μL of settled extraction phase was collected each time using a 50-μL microsyringe (Hamilton, Reno, USA) and transferred to the vials. Then, 2 μL of extract was injected into the GC-MS system.
GC-MS analysis
The analyses were carried out on an Agilent Technologies HP7890A gas chromatograph with an electronically controlled split/splitless injector coupled with an Agilent MSD 5975C mass spectrometry detector connected to MSD Chemstation software (Agilent Technologies). The temperature of the injection port was set to 250 °C while the sample injection was made in splitless mode with a purge flow 50 mL min−1 for 1 min. The injection volume of 2 μL was selected for all analyses. Chromatographic separation was performed by using a Zebron™ ZB-Multiresidue-1 fused silica column (30 m × 0.25 mm × 0.25 μm film thickness; phase specially designed for the separation of all types of pesticides) supplied by Phenomenex, Torrance, USA. The carrier gas was helium (purity 99.999%) maintained at a constant flow of 1.0 mL min−1.
The following temperature oven program was applied to separate pesticides effectively: 70 °C for 2 min, then an increase of 15 °C min−1 to 205 °C, held for 2 min, followed by an additional increase of 5 °C min−1 to 240 °C, then 10 °C min−1 up to 320 °C, held for 3 min. The total duration of the temperature program was 31 min. The transfer line temperature at 320 °C was maintained. The mass spectrometer was operated in electron ionization (EI) mode with the ion source temperature at 230 °C. The MS quad temperature was set at 150 °C. The electron energy was 70 eV.
The optimization of chromatographic resolution including: type and volume of injection, injector temperature, carrier gas flow, and temperature program were performed in the SCAN mode at the concentration level of 10 mg L−1 for each pesticide. Full-scan MS data were acquired in the range of m/z 45–450 to obtain the fragmentation spectra of pesticides. In order to quantify the analytes in water samples (standard or environmental), the mass spectrometer was operated in selected-ion-monitoring (SIM) mode with solvent delay of 5 min. Three specific ions were selected for each pesticide and used to identify the compound. Table 1 shows the relevant information on the selected ions, which were chosen for monitoring. The first ion, more intensive, was used for measurement and the other two for confirmation. Peak quantitation was done relative to the internal standard—TPP. For the pesticides of tau-fluvalinate, alpha-cypermethrin, which show stereoisomerism, two peaks were detected for each one, corresponding to the cis (Z) and trans (E) isomers.
Calculations and statistical analysis
In order to compare the importance of differences between the true value of concentration and the determined mean, Student’s t test (ƒ = n-1, α = 0.05, tcrit = 4.3029) has been applied. This test enables to examine whether the calculated average concentrations are not statistically different [53]. Limits of detection (LODs) were calculated from calibration functions using Eq. 1 [54]:
Where S is the residual standard deviation of the calibration function and b is the slope of the first linear function. The limits of quantitation (LOQs) were determined as 3 times the LOD values for each analyte.
Extraction efficiency, expressed as recovery, was defined as the percentage of total analyte amount expressed as a ratio of pesticide concentration in the sedimented phase (csed) with precisely specified volume for each time (Vsed; usually 30 μL of phase was obtained) to the initial concentration of pesticide (c0) in the sample solution with the volume of 10 mL at each time (V0) [21]:
The concentration of analyte in the sedimented phase was determined by comparing the peak areas obtained from direct injection of standard solution and injection of obtained sedimented phase.
In order to estimate the method suitability, the intra- and inter- day precisions of injection were evaluated by examining the retention times and peak areas of analytes [55]. Standard mixture solutions at three different concentration levels (low, medium and high) were injected thrice per day and on two different days. The obtained results indicated that the relative standard deviation (RSD) values of retention time and peak area were less than 5%.
Results and discussion
In this work, a DLLME-GC-MS methodology for routine determination of 34 different pesticides in environmental samples was applied. The biggest challenge was the optimization step of the analytical procedure, in order to obtain the highest efficiency at the lowest concentration levels of analytes to fulfill the requirements of regulations on maximum residue limits. Therefore, different mixtures of extraction and disperser solvents were compared and evaluated. In addition, the influences of other parameters, considered as critical, have been examined and optimized.
Based on the data presented in Table 1, it can be concluded that pesticides from various chemical classes have very different physicochemical properties, often extremely diverse. On the basis of solubility in water a wide range can be noticed, going from very soluble compounds such as pirimicarb (water solubility of 3100 mg L−1), to practically insoluble compounds like tau-fluvalinate or deltamethrin (with solubility of 1 mg L−1 and 0.2 mg L−1 respectively). Similarly, wide is the range of values of octanol-water partition coefficients at pH 7 and 20 °C. For example, for tau-fluvalinate is 7, while for carbendazim only 1.5. Such a large span values show that this group of compounds exhibits disparate properties, which cause problems in the selection of an appropriate analytical methodology for their qualitative and quantitative determination. Especially difficult was the optimization of the chromatographic analysis parameters to obtain complete separation of all analytes in the shortest period.
Selection of extraction and dispersive solvents
A fundamental step in the optimization of DLLME is the selection of the organic solvent mixture. Some of the previously mentioned properties need to be considered in this choice. A suitable extractant should exhibit affinity to pesticides and have a density greater than water density, and be compatible with the chromatographic system. On this basis, solvent extraction reagents were chosen. In this study, chlorobenzene (density 1.11 g mL−1), carbon disulfide (density 1.26 g mL−1), 1,1,1-trichloroethane (density 1.34 g mL−1) and tetrachloroethylene (density 1.62 g mL−1) were examined. Furthermore, when choosing a dispersive solvent, its ability to mix with the extractant and water sample should be taken into account. Additionally, the disperser should possess the capacity to decrease the interfacial tension of the extraction solvent in order to reduce droplet size, thus increasing the extraction efficiency. Methanol, acetonitrile and acetone were tested as dispersive solvents. Next, the extractions for each pair of extractant–dispersant mixture were conducted. In summary, 12 extractions for different combinations of organic solvent mixtures were examined, with three replications for each mixture. A series of 10 mL aqueous solution samples were studied by using 1.0 mL of methanol containing different extraction solvents to achieve every time 30 ± 1 μL volume of sedimented phase. The extraction efficiencies for different solvent mixtures are presented in Fig. 1. Figure 1 summarizes the results of chromatographic peak areas corrected to the IS, obtained for the extracts of water samples enriched with standard mixture at concentrations of 1 mg L−1 for each pesticide by DLLME-GC/MS. The graph was prepared using the Statistica 8 software package (StatSoft Inc., 2008; USA). In Fig. 1, the objects are determined pesticides (according to the numbering in Table 1) and variables are the combinations of organic solvent mixtures.

Comparison of different mixtures of organic solvents for the isolation of 34 pesticides from spiked water samples at the concentration level 1 mg L−1 for each pesticide by using DLLME–GC-MS. Variables: 1) methanol–chlorobenzene; 2) methanol–tetrachloroethylene; 3) methanol–1,1,1-trichloroethane; 4) methanol–carbon disulfide; 5) acetonitrile–chlorobenzene; 6) acetonitrile–tetrachloroethylene; 7) acetonitrile–1,1,1-trichloroethane; 8) acetonitrile–carbon disulfide; 9) acetone–chlorobenzene; 10) acetone–tetrachloroethylene; 11) acetone–1,1,1-trichloroethane; 12) acetone–carbon disulfide. The graph was plotted by using the Statistica 8 software package (StatSoft Inc., 2008; USA)
Based on the obtained results it can be concluded that the most efficient isolation of pesticides from water samples is provided by a mixture of methanol and tetrachloroethylene. Similar values of detector response were obtained for a mixture of acetonitrile and chlorobenzene. However, the shape of the obtained chromatographic peaks was not acceptable due to incomplete separation, dividing and blurring. Thereby, the values of peak areas were higher when compared to other mixtures of organic solvents. Narrowing the band of chromatographic peak by temperature was used. The difference between the peak of the first analyte and the initial temperature of the temperature program was at least 150 degrees of Celsius. Moreover, in the case of carbon disulfide the initial temperature of the temperature program was too high (higher than boiling point of this solvent) and no solvent film was arised on the forehead of the chromatographic column as it evaporated at once. For mixture of acetonitrile and tetrachloroethylene, only 19 pesticides gave satisfying detector response. Very low response was obtained for acetone and tetrachloroethylene. Cyproconazole, metconazole, fenoxaprop-ethyl, alpha-cypermethrin, tau-fluvalinate and deltametrhin were not detected. When a mixture of acetone and 1,1,1-trichloroethane was used, only flutolanil, flusilazole, metconazole and phosalone gave high detector response. The extraction efficiency for most of the studied pesticides was higher when methanol was used as the dispersive solvent, compared to acetonitrile and acetone. For mixtures of methanol and carbon disulfide, only malathion, chlorpyrifos and alpha-cypermethrin showed higher detector response when compared to use of a methanol and tetrachloroethylene mixture. Moreover, the cloudy state was stable only for a short period of time and sometimes phase separation was not obtained. Therefore, a mixture of methanol and tetrachloroethylene was chosen for this study.
Selection of organic solvent mixture volumes
The next step of the study was the selection of an appropriate volume of extracting and dispersing solvent mixture. The volume of extractant has a significant impact on the numerical value of the coefficient of enrichment. Increasing the volume of extractant enhances the settled phase volume created in the bottom of conical tube after centrifugation, which results in reduction of the enrichment factor. Therefore, an optimally chosen volume of extractant provides both a sufficient volume of settled phase for GC analysis and high enrichment factor. For this purpose, the effect of different volumes of tetrachloroethylene as extracting solvent has been examined: 10, 20, 40 and 60 μL, respectively. Furthermore, the volume of dispersing solvent significantly affects the formation of the cloudy emulsion, which influences the degree of dispersion of the extractant in the aqueous phase and consequently the volume of the sedimented phase. Therefore, the effect of different volumes of methanol as dispersant on the extraction efficiency was compared: 0.5, 1.0, 1.5 and 2 mL. For each volume, the extraction was performed three times from a sample of tap water spiked with a mixture of standards at concentrations of 1 mg L−1 for each pesticide by DLLME–GC-MS. The obtained results are presented in Figs. 2 and 3. The Y-axis represents a ratio between peak areas of analytes and internal standard. The error bars represent the standard error obtained for peak areas of pesticide series.

Effect of extraction solvent volume from 10 to 60 μL on extraction efficiency, concentration of each pesticide 1 mg L−1 (n = 3); dispersant solvent volume 1 mL; microextraction time 30 s; without adjustment of the pH and addition of NaCl salt; error bars represent the standard error obtained for peak areas of pesticide series

Effect of disperser solvent volume from 0.5 to 2.0 mL on extraction efficiency, concentration of each pesticide 1 mg L−1 (n = 3); extraction solvent volume 40 μL; microextraction time 30 s; without adjustment of the pH and addition of NaCl salt; error bars represent the standard error obtained for peak areas of pesticide series
Based on the obtained results, it can be unambiguously stated that the volume of 40 μL of tetrachloroethylene most effectively isolates pesticides from water samples. Only in the case of methiocarb and deltamethrin, a higher detector response was obtained when using a 60-μL extractant volume. In addition, for 10 μL of tetrachloroethylene, a very small drop of settled phase was observed and chromatographic peaks corresponding to carbendazim, fipronil, prothioconazole, chlordecone (kepone), azoxystrobin and dimethomorph were not detected. Therefore, a 40-μL volume of extracting solvent was chosen.
In the case of dispersant volume (Fig. 3), it can be stated that 1 mL of methanol ensures maximum efficiency of the extraction process. For most of the investigated pesticides an effective isolation from water samples was achieved. Exceptions are tetraconazole, flutolanil, flusilazole, cyproconazole, azoxystrobin and dimethomorph. For these compounds, higher detector response was obtained when 0.5 mL of dispersant was used for extraction. At the same time, such a volume of solvent decreased the volume of the extract which made collection of the extract for analysis difficult. Therefore, 1 mL of methanol as a dispersive solvent was chosen for the subsequent studies.
Extraction time
The microextraction time is defined as the time elapsed from the addition of solvent (extraction and dispersing) mixture to the start of centrifugation of the cloudy emulsion. Technically, it was a time of shaking, after the addition of solvent mixture to the water sample. This provided more reproducibility of the procedure, because the analyst can add the mixture at different speeds. The contact area at the interface between the extractant solvent and the aqueous phase is very large; therefore, the transport of analytes from aqueous to organic phase is rapid. Consequently, the equilibrium is quickly established. For this purpose, enriched water samples at 1 mg L−1 for each pesticide were placed in glass centrifuge tubes and extracted for 0.5; 1; 2 and 5 min. The influence of various microextraction times on its effectiveness is presented in Fig. 4.

Effect of microextraction time from 0.5 min (30 s) to 5 min. at ambient temperature on extraction efficiency, concentration of each pesticide 1 mg L−1 (n = 3); without adjustment of the pH and addition of NaCl salt; error bars represent the standard error obtained for peak areas of pesticide series
Based on the obtained results it can be observed that the extraction efficiency decreases with the extraction time. The longer the extraction time, the less effective analytes isolation from the water sample is. Furthermore, it should be noted that prolonged agitation negatively affects the sedimented phase volume. For most of the investigated pesticides, 30 s of solution agitation after addition of the organic solvents mixture gave the most efficient and repeatable results with regards to analytes isolation from the water samples. Therefore, a 30-s agitation time was chosen as optimal.
Influence of temperature, the pH of solution and addition of NaCl salt on the efficiency of the DLLME process
The next step in selecting the optimum conditions for the microextraction process was to investigate the effect of temperature on its efficiency. For this purpose, experiments were carried out at different temperatures: about 20 ± 5 °C (ambient), 35 °C and 50 °C. At higher temperatures, the microextraction was carried out from enriched water sample previously heated in a heating mantle to suitable temperature controlled electronically. Shaking for 30 s was done outside of a heating jacket. Each step was performed in triplicate. The effect of temperature on the extraction efficiency is presented in Fig. 5.

Effect of extraction temperature on its effectiveness, concentration of each pesticide 1 mg L−1 (n = 3); without adjustment of the pH and addition of NaCl salt; error bars represent the standard error obtained for peak areas of pesticide series
Based on the conducted tests, it can be unequivocally stated, that the increase of temperature negatively influenced the efficiency of pesticides isolation from water samples. Moreover, due to increase of solubility of the extraction solvent in aqueous solution at higher temperatures, the volume of the sedimented phase decreases. It was observed that at ambient temperature the efficiency of the process was highest, which confirms the values of chromatographic peak areas for the determined compounds. Only for methiocarb, flusilazole and flutolanil extraction efficiency was higher at 50 °C. However, at this temperature other analytes exhibited a much lower efficiency of isolation. Thus, room temperature (20 ± 5 °C) was chosen as the optimum for further research.
The next step in optimization of microextraction conditions was to investigate the effect of the pH of the water sample solution on extraction efficiency. Some of the studied pesticides, for example carbamates, can degrade under alkaline condition. At low pH value, they tend to form neutral molecules, thus increasing the affinity for nonpolar solvents, resulting in improved extraction efficiency. On the other hand, at acidic pH values, dispersion capability can decrease to some extent. Therefore, selection of an appropriate pH value of water samples is crucial in order to obtain the highest extraction efficiency. This applies in particular to multi-residue methods due to diverse physicochemical properties of analytes. For this purpose, experiments for enriched water samples at different pH values (3, 6 and 11) were carried out. The pH values were adjusted by adding hydrochloric acid or phosphate buffer, respectively. The influence of sample solution pH on microextraction efficiency is presented in Fig. 6.

Effect of the pH of sample solution on extraction efficiency, concentration of each pesticide 1 mg L−1 (n = 3); without addition of NaCl salt; error bars represent the standard error obtained for peak areas of pesticide series
Based on the obtained results, it was observed that changing the pH value of the water sample negatively affects the extraction efficiency. After the addition of an acid solution (0.1 mol L−1 HCl) to reduce the pH value of the sample, sometimes, the sedimentation phase was not formed as a solvent layer and therefore no further analysis was possible. When the pH value increased up to 11, the extraction efficiency decreased significantly compared to the water sample without adjusting the pH value. For fenoxaprop-ethyl, alpha-cypermethrin, tau-fluvalinate, deltametrhin and malathion the detector responses (areas) were not obtained. Consequently, the pH of the water sample was not adjusted at subsequent stages of the analytical procedure, and its value in all extracts was constant about 6.0 ± 0.5.
The final step of the DLLME procedure development was to investigate the effect of NaCl salt addition on extraction efficiency (salting out). NaCl addition increases the ionic strength of the solution, which accelerates the process of pesticides isolation from water samples. Hydration spheres are formed around salt ions by water molecules, resulting in a reduction of available water to dissolve the analyte molecules, thus enhancing their transportation into the organic phase. This effect is observed mainly for polar compounds. In this study, pesticides with various polarity were examined. For this purpose, experiments for enriched water samples containing different quantities of inorganic salt (without addition, 5%, 10% and 15% w/v – weight to volume ratio) were performed. The effect of NaCl salt addition on microextraction efficiency is presented in Fig. 7.

Effect of addition of inorganic salt to the sample solution on extraction efficiency, concentration of each pesticide 1 mg L−1 (n = 3); error bars represent the standard error obtained for peak areas of pesticide series
Based on the results of experiments, it can generally be concluded, that addition of NaCl salt did not significantly affect the pesticides isolation from water samples. For most of the studied compounds, the extraction yield was the highest without addition of inorganic salt to the water samples, as evidenced by the measured values. Only for fenpropidin, tetraconazole and flusilazole increased efficiency of isolation from samples containing 5% of NaCl salt (w/v) have been noticed. Whereas, for parathion-methyl, metazachlor, flutolanil and cyproconazole the effectiveness was higher when the water sample contained 15% (w/v) of salt. At the same time a decrease of extraction efficiency of other compounds was observed, in particular of phosalone for which no chromatographic peak was obtained. The results for samples containing the 5% (w/v) of NaCl were better, but not for all studied pesticides. The Y-axis represents the summary of peak areas; therefore, the detector responses were higher in this case. This improvement was not so significant and at the same time is associate with an additional step in the procedure. Therefore, NaCl salt addition was excluded and not used in further studies.
Method validation
In order to validate the analytical procedure, enriched water samples at different concentration levels were prepared: 0.02 μg L−1, 0.05 μg L−1, 0.1 μg L−1, 1 μg L−1, 10 μg L−1 and 100 μg L−1, respectively. A suitable internal standard volume (TPP) was added to all water samples, so that a concentration of 10 μg L−1 was reached in each solution. The concentration of TPP was always constant. For each level of pesticides concentration, the extraction was performed four times. Based on the obtained results, calibration curves were plotted separately for each pesticide, and the limits of detection and quantification were calculated. Basic validation data obtained for each analyte by using the DLLME–GC-MS procedure is presented in Table 2.
The values of LODs were in the range of 0.0032 μg L−1 (endrin)–0.017 μg L−1 (diazinon) and are at least 100 times lower than those established in legislative regulations concerning the maximum residue levels of pesticides in environmental samples [9,10,11]. Therefore, the developed methodology is more sensitive, allowing for the identification of the compounds of interest in water samples at a concentration level far lower than required, and meets the expectations for multi-residue methods. The selected optimized parameters, affecting extraction efficiency, qualitative and quantitative determination, have ensured reproducible and reliable measurement results, as demonstrated by the coefficient of variation which was in the range of 2.4% (chlorpyrifos)–9.6% (fluquinconazole).
In order to calculate the values of analyte recovery and check the soundness of the developed analytical procedure, extractions from environmental water samples, enriched at two concentration levels: 1 μg L−1 and 50 μg L−1 were carried out. The higher concentration value in this experiment was chosen due to the upper limit of the linearity range, which for all compounds was 100 μg L−1. Conversely, the lower selected concentration was based on the limits of quantification for each pesticide in order to examine two extremely diverse concentration levels within the linearity range. At this stage, samples of river water were selected for the study. Firstly, the water sample itself was analyzed to check for the influence of the matrix effect on pesticides separation and the presence of interfering substances. Then, the sample, enriched with a mixture of pesticide standards, was examined seven times for each concentration level. The chromatogram obtained for the extract from enriched river sample at 1 μg L−1 for each pesticide by using DLLME-GC/MS is presented in Fig. 8.

Chromatogram obtained for spiked tap water at the concentration level 1 μg L−1 for each pesticide by using DLLME–GC-MS: 1) methiocarb, 2) diazinon, 3) γ-HCH (lindane), 4) pirimicarb, 5) carbendazim, 6) fenpropidin, 7) parathion-methyl, 8) malathion, 9) chlorpyrifos, 10) tetraconazole, 11) metazachlor, 12) fipronil, 13) haloxyfop-R-methyl, 14) alpha-endosulfan, 15) flutolanil, 16) flusilazole, 17) prothioconazole, 18) endrin, 19) cyproconazole, 20) kepone, 21) carfentrazone-ethyl, IS (internal standard) triphenylphosphate (TPP), 22) fluquinconazole, 23) fenazaquin, 24) tebufenpyrad, 25) metconazole, 26) phosalone, 27) fenoxaprop-ethyl, 28) pyridaben, 29) alpha-cypermethrin, 30) quizalofop-ethyl, 31) tau-fluvalinate, 32) deltametrhin, 33) azoxystrobin, 34) dimethomorph
On the basis of the obtained results, analyte recoveries and relative standard deviation values were calculated, which is a measure of reproducibility of the developed methodology. Collective measurement results are presented in Table 3.
The obtained pesticide recovery values are satisfactory and meet the regulatory requirements. They were in the range of 84% (tebufenpyrad)–108% (deltamethrin) at the concentration levels of 1 μg L−1 for each pesticide with RSD values (n = 7) in the range of 1.1% (metconazole)–11% (parathion-methyl). Recoveries at higher concentration of 50 μg L−1 were in the range of 86% (prothioconazole)–115% (tau-fluvalinate) with RSD values (n = 7) between 1.9% for cyproconazole and 14% for carbendazim. In summary, the obtained RSD values at different concentrations are less than 20%, which, according to the SANTE/11945/2015 guidelines, indicates a good repeatability of the analytical procedure [56].
Environmental samples
In order to check the suitability of the developed methodology for testing natural water samples, cooperation with Saur Neptun Gdańsk (water supply company) and the Voivodship Inspectorate of Environmental Protection in Gdańsk has been established, in order to provide the necessary environmental samples. Surface water samples were collected at the measuring points, as defined in the Water Directive [8, 10], in cooperation with the Environmental Inspectorate and samples of tap water at various stages of treatment were provided by the Saur Neptun company. In addition, water samples were collected independently in northern and northeastern Poland (see Electronic Supplementary Material (ESM) Fig. S1). Samples were collected during agro-chemical treatments at different times of the year. The study area is dominated by large areas of forests, lakes and access to the Baltic Sea. This gives the possibility of dynamic development of agritourism. Agricultural production in the area is mainly focused on milk production, pig herding, cereals, rape, maize, potatoes, sugar beets and horticultural production. Water samples were taken with a telescopic scoop in locations where bridges were available at least 1 m from the shore. Samples were collected in the morning into brown glass bottles with a capacity of 1 l. In the analyzed water samples, four pesticides have been detected in concentrations above the maximum permissible levels (see ESM Fig. S2):
-
chlorpyrifos 0.350 ± 0.012 μg L−1 (lake sample from Klasztorne Duże in Kartuzy),
-
alpha-cypermethrin 9.50 ± 0.21 μg L−1 (river sample from Wierzyca in Stara Kiszewa),
-
metconazole 5.10 ± 0.67 μg L−1 (river sample from Wietcisa in Lubieszyn),
-
malathion 0.750 ± 0.014 μg L−1 (river sample from Radunia in Kolbudy).
These values exceed the maximum permissible content of pesticides in waters established by the European Union, which is 0.1 μg L−1 for individual compounds [9,10,11]. The chromatogram for the extract from the sample obtained at the Klasztorne Duże lake in Kartuzy is presented in Fig. 9.

Chromatogram obtained for environmental sample from lake by using DLLME–GC-MS; 1) chlorpyrifos, IS (internal standard) TPP
The results have confirmed the relevance of the method development that has been undertaken. The developed procedure allows for the determination of the groups of compounds used in plant protection that may cause pollution of the aquatic environment. Pesticides introduced into the environment are an important problem as their consumption and application area is constantly increasing. Water samples, in which pesticides were detected, were collected in rural areas, near agricultural crops, where pollution can be attributed to fertilization of farmland or animal husbandry. There is also a high probability that pesticides found in water samples may be present due to mistakes made in formulation application or improper handling of equipment used for spraying with plant protection products on nearby crop fields. The information obtained on the state of the aquatic environment makes it possible to assess the exposure to pesticides associated with contamination of surface waters and drinking water. This is an innovative topic, covering an extremely important issue of estimating the risk to life and health not only of people, but all living organisms. In addition, the obtained results may be the basis for improvement or upgrading of water treatment technologies, wastewater treatment and proper waste management.
Conclusion
Increasing consumption of pesticides and their use in many areas of life leads to ever larger quantities entering the environment. Presence of pesticides in rivers and lakes is particularly dangerous because they are a source of drinking water for human consumption. In accordance with “Green Chemistry” principles, nowadays the aim is to develop fast and cost-effective analytical methodologies that enable the identification of compounds with various physicochemical properties in a single analytical run. Therefore, an attempt was made to develop an analytical procedure for the determination of the largest possible number of contemporary pesticides from different chemical groups in water samples. In addition, the procedure can be expanded with other analytes as needed. The optimized and validated methodology can be successfully implemented for routine analysis. In addition, it fulfills all the legal requirements for analytical procedures for the determination of pesticide residues in environmental samples. This method has proved to be selective, sensitive and precise for the simultaneous determination of various pesticides as well as being simpler, having a lower cost and requiring a less labor-intensive sample preparation procedure than conventional methods.
By reduction the amounts of solvents used, some of the inconveniences limiting the technique’s application in the liquid-liquid system are eliminated. The DLLME technique can be coupled with GC and HPLC, provided that no ionic liquids are used as extraction solvents. This extraction technique requires only 1040 μL of solvents used, which makes it more environmentally friendly. Moreover, any special devices or sorbents are not needed making this procedure exceptionally cheap and easily accessible. Also, any conditioning steps before extraction are not required. With regard to our previous studies [6] on the development of a new analytical procedure by using solid-phase microextraction coupled with GC-MS, this methodology allows simultaneous determination of significantly more pesticides at concentrations of at least 100 times lower.
References
Kim KH, Kabir E, Jahan SA. Exposure to pesticides and the associated human health effects. Sci Total Environ. 2017;575:525–35.
Biziuk M, Fenik J, Kosikowska M, Tankiewicz M. Pesticides. In: Jorgensen SE, editor. Encyclopedia of environmental management. Boca Raton: CRC Press, Taylor & Francis; 2013. p. 1963–82.
Lefrancq M, Jadas-Hécart A, La Jeunesse I, Landry D, Payraudeau S. High frequency monitoring of pesticides in runoff water to improve understanding of their transport and environmental impacts. Sci Total Environ. 2017;587-588:75–86.
Gonzalez-Rey M, Tapie N, Le Menach K, Dévier MH, Budzinski H, Bebianno MJ. Occurrence of pharmaceutical compounds and pesticides in aquatic systems. Mar Pollut Bull. 2015;96:384–400.
Masiá A, Campo J, Vázquez-Roig P, Blasco C, Picó Y. Screening of currently used pesticides in water, sediments and biota of the Guadalquivir River Basin (Spain). J Hazard Mater. 2013;263P:95–104.
Tankiewicz M, Morrison C, Biziuk M. Multi-residue method for the determination of 16 recently used pesticides from various chemical groups in aqueous samples by using DI-SPME coupled with GC-MS. Talanta. 2013;107:1–10.
Liu H, Qi S, Xing X, Yang D, Hu Y, Qu C. Currently used organochlorine pesticides in Mianzhu-Aba prefecture transect, eastern of the Tibetan Plateau, western China. J Geochem Explor. 2015;150:115–24.
Voulvoulis N, Arpon KD, Giakoumis T. The EU water framework directive: from great expectations to problems with implementation. Sci Total Environ. 2017;575:358–66.
Directive 2008/105/EC of the European Parliament and of the Council on environmental quality standards in the field of water policy, amending and subsequently repealing Council Directives 82/176/EEC, 83/513/EEC, 84/156/EEC, 84/491/EEC, 86/280/EEC and amending Directive 2000/60/EC of the European Parliament and of the Council. Official Journal of the European Union. 2008; 348: 84–97.
European Commission. Introduction to the New EU water framework directive. (Available at: http://ec.europa.eu/environment/water/water-framework/info/intro_en.htm, Accessed 23 Aug 2017).
Directive 98/83/EC on the quality of water intended for human consumption. Official Journal of the European Communities. 1998; L330: 32–54.
Kaufmann A. Validation of multiresidue methods for veterinary drug residues; related problems and possible solutions. Anal Chim Acta. 2009;637:144–55.
Caldas SS, Rombaldi C, de Oliveira Arias JL, Marube LC, Primel EG. Multi-residue method for determination of 58 pesticides, pharmaceuticals and personal care products in water using solvent demulsification dispersive liquid-liquid microextraction combined with liquid chromatography-tandem mass spectrometry. Talanta. 2016;146:676–88.
Primel EG, Caldas SS, Escarrone ALV. Multi-residue analytical methods for the determination of pesticides and PPCPs in water by LC-MS/MS: a review. Cent Eur J Chem. 2012;10:876–99.
Cotton J, Leroux F, Broudin S, Poirel M, Corman B, Junot C, et al. Development and validation of a multiresidue method for the analysis of more than 500 pesticides and drugs in water based on on-line and liquid chromatography coupled to high resolution mass spectrometry. Water Res. 2016;104:20–7.
Yusa V, Millet M, Coscolla C, Roca M. Analytical methods for human biomonitoring of pesticides. A review. Anal Chim Acta. 2015;891:15–31.
Kokosa JM. Advances in solvent-microextraction techniques. TrAC Trends Anal Chem. 2013;43:2–13.
Pinto MI, Sontag G, Bernardino RJ, Noronha JP. Pesticides in water and the performance of the liquid-phase microextraction based techniques. Microchem J. 2010;96:225–37.
Khalili-Zanjani MR, Yamini Y, Yazdanfar N, Shariati S. Extraction and determination of organophosphorus pesticides in water samples by a new liquid phase microextraction-gas chromatography-flame photometric detection. Anal Chim Acta. 2008;606:202–8.
Kujawski MW, Pinteaux E, Namieśnik J. Application of dispersive liquid-liquid microextraction for the determination of selected organochlorine pesticides in honey by gas chromatography-mass spectrometry. Eur Food Res Technol. 2012;234:223–30.
Rezaee M, Assadi Y, Milani Hosseini MR, Aghaee E, Ahmadi F, Berijani S. Determination of organic compounds in water using dispersive liquid-liquid microextraction. J Chromatogr A 2006; 1116: 1–9.
Zang X, Wang J, Wang O, Wang M, Ma J, Xi G, et al. Analysis of captan, folpet, and captafol in apples by dispersive liquid-liquid microextraction combined with gas chromatography. Anal Bioanal Chem. 2008;392:749–54.
Rezaee M, Yamini Y, Faraji M. Evolution of dispersive liquid-liquid microextraction method. J Chromatogr A. 2010;1217:2342–57.
Cheng J, Zhou Y, Zuo M, Dai L, Guo X. Application of dispersive liquid-liquid microextraction and reversed phase-high performance liquid chromatography for the determination of two fungicides in environmental water samples. Int J Environ Anal Chem. 2010;90:845–55.
Zgoła-Grześkowiak A, Grześkowiak T. Dispersive liquid-liquid microextraction. TrAC Trends Anal Chem. 2011;30:1382–99.
Herrera-Herrera AV, Asensio-Ramos M, Hernández-Borges J, Rodríguez-Delgado MÁ. Dispersive liquid-liquid microextraction for determination of organic analytes. TrAC Trend Anal Chem. 2010;29:728–51.
Chen H, Chen R, Li S. Low-density extraction solvent-based solvent terminated dispersive liquid-liquid microextraction combined with gas chromatography-tandem mass spectrometry for the determination of carbamate pesticides in water samples. J Chromatogr A. 2010;1217:1244–8.
Cruz-Vera M, Lucena R, Cárdenas S, Valcárcel M. Sample treatments based on dispersive (micro) extraction. Anal Methods. 2011;31:719–1728.
Kocúrová L, Balogh IS, Šandrejová J, Andruch V. Recent advances in dispersive liquid-liquid microextraction using organic solvents lighter than water. A review. Microchem J. 2012;102:11–7.
Moreno-González D, Gámiz-Gracia L, Bosque-Sendra JM, García-Campaña AM. Dispersive liquid-liquid microextraction using a low density extraction solvent for the determination of 17 N-methylcarbamates by micellar electrokinetic chromatography-electrospray-mass spectrometry employing a volatile surfactant. J Chromatogr A. 2012;1247:26–34.
Sanagi MM, Abbas HH, Wan Ibrahim WA, Aboul-Enien HY. Dispersive liquid-liquid microextraction method based on solidification of floating organic droplet for the determination of triazine herbicides in water and sugarcane samples. Food Chem. 2012;133:557–62.
Xiong J, Guan Z, Zhou G, Tang X, Lv Y, Wang H. Determination of chlorpyrifos in environmental water samples by dispersive liquid-liquid microextraction with solidification of a floating organic drop followed by gas chromatography with flame photometry detection. Anal Methods. 2012;4:3246–50.
Seebunrueng K, Santaladchaiyakit Y, Srijaranai S. Vortex-assisted low density solvent based demulsified dispersive liquid-liquid microextraction and high-performance liquid chromatography for the determination of organophosphorus pesticides in water samples. Chemosphere. 2014;103:51–8.
Farajzadeh MA, Djozan D, Khorram P. Development of a new dispersive liquid-liquid microextraction method in a narrow-bore tube for preconcentration of triazole pesticides from aqueous samples. Anal Chim Acta. 2012;713:70–8.
Ahmad W, Al-Sibaai AA, Bashammakh AS, Alwael H, El-Shahawi MS. Recent advances in dispersive liquid-liquid microextraction for pesticide analysis. TrAC Trends Anal Chem. 2015;72:181–92.
Ma J, Lu W, Chen L. Recent advances in dispersive liquid-liquid microextraction for organic compounds analysis in environmental water. Curr Anal Chem. 2012;8:78–90.
Yan H, Wang H. Recent development and applications of dispersive liquid-liquid microextraction. J Chromatogr A. 2013;1295:1–15.
Leong MI, Fuh MR, Huang SD. Beyond dispersive liquid-liquid microextraction. J Chromatogr A. 2014;1335:2–14.
Saraji M, Boroujeni MK. Recent developments in dispersive liquid-liquid microextraction. Anal Bioanal Chem. 2014;406:2027–66.
Zacharis CK, Tzanavaras PD, Roubos K, Dhima K. Solvent-based deemulsification dispersive liquid-liquid microextraction combined with gas chromatography-mass spectrometry for determination of trace organochlorine pesticides in environmental water samples. J Chromatogr A. 2010;1217:5896–900.
Cinelli G, Avino P, Notardonato I, Russo MV. Ultrasound-vortex-assisted dispersive liquid-liquid microextraction coupled with gas chromatography with a nitrogen-phosphorus detector for simultaneous and rapid determination of organophosphorus pesticides and triazines in wine. Anal Methods. 2014;6:782–90.
Wu Q, Chang Q, Wu C, Rao H, Zeng X, Wang C, et al. Ultrasound-assisted surfactant-enhanced emulsification microextraction for the determination of carbamate pesticides in water samples by high performance liquid chromatography. J Chromatogr A. 2010;1217:1773–8.
Asensio-Ramos M, Hernández-Borges J, Borges-Miquel TM, Rodríguez-Delgado MÁ. Ionic liquid-dispersive liquid-liquid microextraction for the simultaneous determination of pesticides and metabolites in soils using high-performance liquid chromatography and fluorescence detection. J Chromatogr A. 2011;1218:4808–16.
Wang S, Xiang B, Tang Q. Trace determination of dichlorvos in environmental samples by room temperature ionic liquid-based dispersive liquid-phase microextraction combined with HPLC. J Chromatogr Sci. 2012;50:702–8.
Wang X, Xu QC, Cheng CG, Zhao RS. Rapid determination of DDT and its metabolites in environmental water samples with mixed ionic liquids dispersive liquid-liquid microextraction prior to HPLC-UV. Chromatographia. 2012;75:1081–5.
Zhang J, Li M, Yang M, Peng B, Li Y, Zhou W, et al. Magnetic retrieval of ionic liquids: fast dispersive liquid-liquid microextraction for the determination of benzoylurea insecticides in environmental water samples. J Chromatogr A. 2012;1254:23–9.
Tsai WC, Huang S. Dispersive liquid-liquid microextraction with little solvent consumption combined with gas chromatography-mass spectrometry for the pretreatment of organochlorine pesticides in aqueous samples. J Chromatogr A. 2009;1216:5171–5.
Kroon MC, Buijs W, Peters CJ, Witkamp GJ. Quantum chemical aided prediction of the thermal decomposition mechanisms and temperatures of ionic liquids. Thermochim Acta. 2007;465:40–7.
Farajzadeh MA, Djozan D, Nouri N, Bamorowat M, Shalamzari MS. Coupling stir bar sorptive extraction-dispersive liquid-liquid microextraction for preconcentration of triazole pesticides from aqueous samples followed by GC-FID and GC-MS determinations. J Sep Sci. 2010;33:1816–28.
Samadi S, Sereshti H, Assadi Y. Ultra-preconcentration and determination of thirteen organophosphorus pesticides in water samples using solid-phase extraction followed by dispersive liquid-liquid microextraction and gas chromatography with flame photometric detection. J Chromatogr A. 2012;1219:61–5.
Moreda-Piñeiro J, Moreda-Piñeiro A. Recent advances in combining microextraction techniques for sample pre-treatment. TrAC Trends Anal Chem. 2015;71:265–74.
The PPDB Pesticide Properties Database. Agriculture & Environment Research Unit (AERU) at the University of Hertfordshire. (Available at: http://sitem.herts.ac.uk/aeru/ppdb/en/index.htm, Accessed 23 Aug 2017).
Mechlińska A, Wolska L, Namieśnik J. The effect of adding a standard on the result of determination of polychlorinated biphenyls in bottom sediment samples. Talanta. 2010;82:627–30.
Konieczka P, Namieśnik J. Quality assurance and quality control in the analytical chemical laboratory—a practical approach. In: Lochmüller ChH, editor. Boca Raton: CRC Press, Taylor & Francis; 2009. pp. 131–216.
Margoum C, Guillemain C, Yang X, Coquery M. Stir bar sorptive extraction coupled to liquid chromatography-tandem mass spectrometry for the determination of pesticides in water samples: method validation and measurement uncertainty. Talanta. 2013;116:1–7.
SANTE/11945/2015. Guidance document on analytical quality control and method validation procedures for pesticides residues analysis in food and feed. European Commission. (Available at: https://ec.europa.eu/food/sites/food/files/plant/docs/pesticides_mrl_guidelines_wrkdoc_11945.pdf, Accessed 23 Aug 2017).
Acknowledgements
This study was supported by the National Science Centre in Poland (decision number: DEC-2011/01/N/ST4/01977) and the Project for Young Scientists (MN 01-0212/08/322) funded by the Ministry of Science and Higher Education in Poland.
The authors would like to thank Mr. Emil Salim Parhimpunan Siregar for technical assistance, and are grateful to Mr. Goran Gržinić for assistance with proof reading and constructive comments.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
ESM 1
(PDF 375 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Tankiewicz, M., Biziuk, M. Fast, sensitive and reliable multi-residue method for routine determination of 34 pesticides from various chemical groups in water samples by using dispersive liquid–liquid microextraction coupled with gas chromatography–mass spectrometry. Anal Bioanal Chem 410, 1533–1550 (2018). https://doi.org/10.1007/s00216-017-0798-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-017-0798-4