
Abstract
The structures and intermolecular interactions in the halogen bonded complexes of six carbonyl bases with chlorotrifluoromethane have been studied by ab initio MP2 and CCSD(T) as well as density functional methods: B3LYP, B3LYP-D3 and wB97XD. The CCSD(T)/aug-cc-pVTZ calculated interaction energies of these complexes range between −1.22 and −2.62 kcal mol−1. The interaction energies are related to the ionization potential and proton affinity of the carbonyl bases. The intermolecular interaction results in a contraction of the C–Cl bond and an elongation of the C–F bonds accompanied by a relevant frequency shift of the corresponding stretching vibrations. The origin of the variation of the C–Cl and C–F bond lengths is discussed on the basis of an NBO analysis. It is suggested that the formation of these complexes can be considered as a two-step mechanism, involving the intermolecular charge transfer from the lone-pair orbitals on the oxygen atom to the antibonding σ*(C–Cl) orbital followed by a decrease of the intramolecular charge transfer from the lone-pair orbitals on the fluorine atoms to the antibonding σ*(C–Cl) orbital. A symmetry-adapted perturbation theory (SAPT) decomposition of the interaction energies has been performed.
Similar content being viewed by others
1 Introduction
Classical textbooks on molecular interactions mainly devoted to hydrogen bonds [1] and charge transfer complexes [2] have appeared in the years 1960–1970. It is only in the late 1990s that charge transfer complexes involving an X halogen atom in the ZX···Y system have been recalled halogen bonds. They involve an interaction of an electron-rich center Y with an electronegative halogen substituent in a ZX molecule. These bonds have been featured in an increasing numbers of areas. Their importance in the field of molecular recognition [3, 4] and crystal engineering [5] as well as in biological systems [6] also has been recognized. This wide range of applications has placed the halogen bonded interactions in a center of great attention, and many theoretical works have been focused on them [7–20].
In this work, we present the results of a theoretical investigation of the interaction between chlorotrifluoromethane (CClF3) and substituted carbonyl derivatives. Let us notice that the complexes between trifluoromethyl halides (CF3X) and guest molecules have been the subject of several theoretical investigations, as for example the interaction between CClF3 and NH3 [21, 22], H2O [23, 24], H2S [25], organic bases of different nature [26–29] or halide anions [30]. The influence of angular distortions on the energies of halogen bonds and hydrogen bonds has been recently discussed [31]. Very few experimental data on these interactions are available. The vibrational properties of trifluoromethyl halides complexed with dimethyl ether have been investigated by IR and Raman spectroscopies in liquid argon and in liquid krypton [32]. The halogen bond formed between CClF3 and dimethyl ether has been studied by rotational spectroscopy [33].
Although halogen atoms carry partial negative charges, there is a positive electrostatic potential at the head of the lone pair in the opposite direction of this lone pair. This region is called a σ-hole which accounts for the ability of halogens to accept an interaction with electronic rich atoms [34–36]. A σ-hole has been predicted in the Cl atom of CClF3 [34].
It has been shown that the properties of the hydrogen-bonded systems such as the thermodynamic or spectroscopic properties are intimately related to the nature of the proton acceptor (N, O, S) [37], and the influence of the basicity/acidity on these properties has been discussed in several works. Much less data are available for halogen bonds. In the present work, we want to discuss the influence of the basicity on the complexes formed between CClF3 and simple carbonyl derivatives (H2CO, HFCO, F2CO, CH3CHO, CH3CFO, (CH3)2CO) where different substitutions allow to modulate the electron donor ability. With the exception of the H2CO···ClCF3 complex [27, 28, 36], no theoretical data have been reported for these systems.
The present work is arranged as follows. In the first part, the structure of the complexes, the binding energies and the variation of the C–Cl and C–F bond lengths are discussed. The second part deals with the NBO (natural bond orbital) analysis, focusing mainly on the occupation of bonding and antibonding orbitals, the hybridization of the C atom and inter- and intramolecular stabilization energies. The results of SAPT calculations are presented as well. In the last part of our work, the frequency shifts of the ν(C–Cl) and ν(C–F) stretching vibrations induced by the interaction with ketones are discussed.
2 Computational methods
The optimized geometries, vibrational harmonic frequencies and infrared intensities have been calculated for the following six carbonyl bases: H2C=O; HFC=O; F2C=O; CH3HC=O; CH3FC=O and (CH3)2C=O. Then, full geometry optimization and the calculations of the vibrational properties have been performed for the complexes of these bases with CClF3. In the calculations, we employed an ab initio MP2 method [38] combined with the cc-pVTZ basis set [39], as well as the B3LYP [40, 41], B3LYP-D3 (which is B3LYP corrected with the original D3 damping function) [42] and wB97XD [43] hybrid density functionals combined with the 6-311++G(d,p) basis set [44, 45]. The counterpoise CP-corrected gradient optimization has been used in the MP2 calculations [46].
The interaction energies of the studied complexes have been determined at the MP2/aug-cc-pVTZ, CCSD(T)/aug-cc-pVTZ, B3LYP/6-311++G(d,p), B3LYP-D3/6-311++G(d,p) and wB97XD/6-311++G(d,p) levels of theory. In the calculations of the interaction energies by two ab initio methods [MP2 and CCSD(T)] coupled with the aug-cc-pVTZ basis set, we have used the geometry of the complexes optimized at the MP2/cc-pVTZ level. All the interaction energies were corrected for the basis set superposition error (BSSE) computed by the CP method [46].
For all the complexes, the symmetry-adapted perturbation theory (SAPT) decomposition of the interaction energies has been performed at the MP2/cc-pVTZ level of theory. All the computations were carried out with the Gaussian 09 [47] or MOLPRO 2012 [48] programs.
A natural bond orbital (NBO) analysis provides a detailed insight into the nature of electronic structure and bonding in the molecules. In this work, the atomic charges, hybridization, occupation of orbitals and the second-order interaction energies were calculated by the DFT method using the 5.0 version of the NBO program [49, 50]. The atoms in molecules (AIM) analysis [51] was used in order to characterize the intermolecular interactions between the carbonyl bases and chlorotrifluoromethane investigated in this work.
3 Results and discussion
3.1 Structures of the complexes and interaction energies
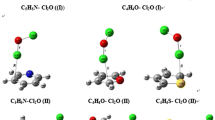
The structures of the interacting molecules optimized with the MP2 method are illustrated in Fig. 1. Intermolecular parameters (O···Cl distances, CO···Cl and CCl···O angles) are indicated in Table 1. In all the systems, the intermolecular O···Cl distances are all smaller than the sum of the van der Waals radii (3.27 Å).

Structures of CClF3 complexes with carbonyl bases optimized at the MP2/cc-pVTZ level
At any level of theory, the O···Cl distance is the longest in the F2C=O complex; these distances in the CH3CHO and (CH3)2CO complexes are nearly equal, being slightly longer in the first system when considering the MP2, B3LYP and B3LYP-D3 calculations. As in the case of the complexes between molecular chlorine and the same carbonyl bases, the asymmetrical ketones show two different structures where the C–Cl bond coincides approximately with the direction of the lone-pair orbitals of the oxygen atom (O LPs) [52, 53]. The C–Cl···O bonds calculated at the B3LYP level are nearly linear and show larger departures from linearity when calculated at other levels. It follows from the H···Cl distances that there is no bond formation between the C–H group of the ketones and the Cl atom of CClF3. However, a weak interaction between the H and Cl atoms cannot be ruled out in the CH3CFO (b) and (CH3)2CO complexes.
According to the AIM theory, two atoms are bonded to each other if their nuclei are linked by a line of maximal electron density named the bond path. Figure 2 demonstrates the presence of a bond critical point (BCP) in the H2CO···ClCF3 complex. The electron density (ρ), the Laplacian of the electron density (∇2 ρ) and the total electron energy (H) at the selected BCP in all the studied complexes are collected in Table 2.

Contour line diagram of the Laplacian of the electron density of the H2CO···ClCF3 complex (in the plane passing through the O, Cl and C atoms marked on the figure). The solid (blue) lines represent negative values of the Laplacian, while the dashed (red) lines represent the positive values. The electron density description was obtained at the MP2/cc-pVTZ level
As indicated in Table 2, in the case of two complexes: CH3CFO···ClCF3 (b) and (CH3)2CO···ClCF3, the AMI calculations suggest an additional stabilization interaction between the chlorine and hydrogen atoms (H···Cl). According to the Popelier’s criteria for hydrogen bonds, the electron density at the BCP ranges from 0.002 to 0.035 au, and the Laplacian of the electron density ranges from 0.024 to 0.139 au [54, 55]. As follows from Table 2, these two H···Cl interactions do not fulfill the second criterion. Therefore, they cannot be classified as hydrogen bonds but as weak van der Waals interactions. It is worth to mention that in both the CH3CFO···ClCF3 (b) and (CH3)2CO···ClCF3 complexes, the H···Cl distances are longer than the sum of the corresponding van der Waals radii by 0.29 and 0.20 Å, respectively.
When considering only the O···Cl interactions, we have found the following correlations between the B3LYP-D3 binding energies and the AMI parameters (ρ and ∇2 ρ):
The interaction energies calculated at different levels are reported in Table 3. The values of the energies calculated with the wB97XD and B3LYP methods are lower than the ones calculated by the other methods. The values obtained at the B3LYP-D3, MP2 and CCSD(T) levels of theory are similar. Let us notice that the interaction energy for the H2CO···ClCF3 system calculated in Ref. [29] at the MP2(full)/6-311++G(3df, 3pd) level is equal to −1.90 kcal mol−1, which is by 0.3 kcal mol−1 lower than our B3LYP-D3 calculated value.
At each level of theory, larger intermolecular O···Cl distances result in lower interaction energies, and in the further discussion, we will consider the B3LYP-D3 interaction energies.
As indicated in Table 3, the electron-releasing CH3 group strengthens the interaction and the electron-attracting substituents weaken the interaction. This prompted us to compare the interaction energies with the basic properties of the ketones. The proton affinities (PA) calculated at the B3LYP-D3/6-311++G(d,p) level as well as the calculated and experimental ionization potentials (IP) of the ketones [56–61] are reported in Table 4.
From the B3LYP-D3 energies, we have calculated the following correlations:
The correlation 3 is illustrated in Fig. 3. The slopes of these two correlations are much lower than those recently reported for the stronger complexes between the same carbonyl bases and molecular chlorine (−0.76 and 0.055, respectively) [52].

Interaction energy –ΔE (kcal mol−1) as a function of the ionization potential IP(eV)
Comparison with the literature values is difficult. The reported interaction energy in the NH3···ClCF3 system is equal to −2.37 kcal mol−1 (MP2/aug-cc-pVDZ) [21, 22]. The PA of NH3 is 204 kcal mol−1, about the same as that of (CH3)2CO, 203.5 kcal mol−1 (see Table 4), and the ΔE of the (CH3)2CO···ClCF3 complex (calculated at the MP2/aug-cc-pVTZ level of theory) is −2.63 kcal mol−1, as shown in Table 3. The interaction energy in the H2O···ClCF3 system is equal to −1.46 kcal mol−1 (MP2/cc-pVTZ) [26] or −1.76 kcal mol−1 [MP2/6-311++G(d,p)] [24], the PA of H2O being equal to 166.5 kcal mol−1. In the H2CO···ClCF3 complex, the interaction energy is larger (−2.07 kcal mol−1), and this is consistent with the fact that the value of the PA of H2CO (177.5 kcal mol−1, Table 4) is larger than that of H2O by 11 kcal mol−1.
As outlined in several works [21, 24, 25, 28] when CClF3 interacts with electron donors of medium strength such as NH3, H2O or H2S, the C–Cl bond is contracted by moderate amounts. In the F3CCl···NH3 system, the C–Cl bond is contracted by 6.6 mÅ [24] or 6.3 mÅ [21]. In the F3CCl···OH− system, the C–Cl bond is contracted by 7 mÅ [24], and in the weaker F3CCl···SH2 complex, the C–Cl bond is contracted by only 2.9 mÅ [25].
The values of the C–Cl bond contraction for the present systems are reported in Table 5 which also indicates the elongation of the three CF bonds. These results show that the contraction of the C–Cl bond is moderate, ranging from 4.7 mÅ in F2CO to 9.4 mÅ in (CH3)2CO. As in general, bond contractions are connected with blue shifts of the corresponding stretching frequency, the present complexes can be categorized as blue-shifting complexes. Further, the elongation of the C–F bonds is comprised between 1.6 and 5.1 mÅ and is also the largest in the complexed (CH3)2CO system.
3.2 NBO analysis
In this section, we want to discuss the results of a NBO analysis, focusing mainly on the NBO charges, the occupation of bonding and antibonding orbitals, the hybridization of the C atom along with the second-order inter- and intramolecular stabilization energies.
Let us remember that halogen bonds have been compared to hydrogen bonds and have been shown to be similar in many aspects [63–67]. Briefly summarizing, let us mention that the contraction of a Z–H bond in a hydrogen-bonded system ZH···Y has been explained mainly by a variation of the σ*(C–H) occupation and a change in the hybridization of the Z(H) atom [68–74]. These parameters can also be discussed for the blue-shifting halogen bonds. The results of our NBO analysis for the present systems are presented in Tables 6, 7 and 8.
The variation of the NBO charges on the C and Cl atoms of CClF3 induced by the interaction with the ketones is indicated in Table 6. This table also reports the variation of hybridization of the C atom in the C(Cl) and C(F) bonds along with the global charge transfer (CT) occurring from the ketones to CClF3. Table 7 reports the second-order stabilization energies E (2) → σ*(C–Cl), the occupation of the σ*(C–Cl) antibonding and σ(C–Cl) bonding orbitals. In Table 8, the intramolecular charge transfer energies from the lone-pair orbitals (LPs) of the three F atoms to the σ*(C–Cl) orbitals and the variation of the occupation of the σ*(C–F) orbitals are indicated.
The results of Table 6 show that the charge transfer from the ketones to CClF3 is rather low, ranging from 0.5 me (F2CO) to 4.9 me (CH3CHO). Let us notice that for the systems involving H2CO, F2CO and HFCO, there is also a CT from the LPs of the O atom to the Rydberg orbitals of the Cl atom. The corresponding second-order stabilization energies are comprised between 0.39 kcal mol−1 (F2CO) and 0.44 kcal mol−1 (H2CO). It must be mentioned that these values are of the same order of magnitude as the E 2 (LPO → σ*(C–Cl) stabilization energies which range between 0.3 and 0.58 kcal mol−1 (Table 7). This effect is rather unusual and illustrates the electrophilic feature of the halogen. For the other systems, the charge transfer energy to the Rydberg orbitals of Cl is smaller (between 0.07 and 0.18 kcal mol−1).
In a first step, we will discuss the origin of the variation of the C–Cl and C–F bond lengths. The interaction with ketones results in an increased polarization of the C–Cl bond. The variations of the charges on the C and Cl atoms are +6.2 and −14.5 me for the weakest F2CO complex. The largest changes of the charges on the C atom (+16.9 me) and on the Cl atom (−31.6 me) are predicted for the (CH3)2CO···ClCF3 system. For this system, the CT to the three F atoms is also the largest (19 me). Let us notice that for CClF3 complexed with stronger electron donors such as Br−, the CT is equal to 15.4 me [24].
As indicated in Table 7, the second-order stabilization energies E 2 → σ*(C–Cl) increase from F2CO (0.3 kcal mol−1) to CH3CHO (1.24 kcal mol−1). Despite this charge transfer, the occupation of the σ*(C–Cl) orbital decreases, its decrease being slightly larger for the (CH3)2CO system (12.7 me) than for the F2CO one (11.4 me). Our calculations also predict a small decrease of the σ(C–Cl) occupation between 0.7 and 1.1 me. Further, as indicated in Table 6, the interaction results in an increase of the s-character of the C(Cl) atom, smaller for the weaker complexes (F2CO = 0.37 %) than for the stronger ones [(CH3)2CO = 0.92 %]. It must be pointed out that a variation of hybridization of the C(Cl) atom of CClF3 is a known effect which has already been predicted for CClF3 complexed with molecules such as NH3 [24], H2O and H2S [25].
Both the increase of the s-character of the C(Cl) atom and the decrease of the σ*(C–Cl) occupation contribute to the contraction of the C–Cl bond. The variation of the σ(C–Cl) occupation is very small, but it has been considered in our calculations. From the present results, we have deduced the following dual expression [Δr(C–Cl) in mÅ, Δσ*(C–Cl) in me]:
which illustrates the predominance of the hybridization effect over the σ*(C–Cl) and σ(C–Cl) occupations. The values of Δr(C–Cl) calculated by Eq. (5) and by the B3LYP method are compared in Fig. 4 (r 2 = 0.971).

Δr (Cl–Cl) (mÅ) calculated by Eq. (5) as a function of Δr (Cl–Cl) calculated at the B3LYP/6-311++G(d,p) level
In order to test the validity of this equation, we have calculated the Δr(C–Cl) value for the F3CCl···Br− system from the parameters reported in Ref. [24]. This complex is strong, the interaction energy calculated by MP2(full)/6-311++G(d,p) being −7.27 kcal mol−1. The parameters of Eq. (5) are: Δ%s (C–Cl) = +2.68, Δσ*(C–Cl) = −4.1 me, Δσ(C–Cl) = +1.2 me. Note that in contrast to the present systems, there is an increase of the σ(C–Cl) occupation. From these parameters, a Δr(C–Cl) value of −22.5 mÅ has been calculated from Eq. (5) which is very close to the value of −22.1 mÅ predicted by MP2 calculation in Ref. [24].
In a similar way, the elongation of the C–F bond can be evaluated from the same parameters, the variation of the occupation of the σ(C–F) orbitals being negligible:
Our calculations predict an increase of the polarity of the C–Cl bond along with an increase of the s-character of the C(Cl) atom. These results are in agreement with the Bent’s rule. Let us briefly remember that the Bent’s rule states that atoms maximize their s-character in hybrid orbitals aimed toward electropositive substituents and the atoms maximize their p-character toward electronegative substituents. This rule has been discussed in detail for systems involving CClF3 complexed with Lewis acids and Lewis bases [28]. Let us notice that in the present systems, an increase of the s-character is also reflected in the variation of the intramolecular angle in CClF3. Indeed, in isolated CClF3, the CClF and FCF angles are equal to 110.3° and 108.6°; in the complex with (CH3)2CO, these angles take values of 110.8° and 108.1°.
For the present systems, the correlation between the charges on the Cl atom and the % s-character in C orbital of the C–Cl bond can be written as:
Interestingly, for CClF3 complexed with stronger electron donors (OH−, Cl−), we have found a very similar correlationFootnote 1 from the data reported in Ref. [28].
Let us now discuss more in detail the changes occurring in the C–F bonds of CClF3. The interaction with ketones results in a decrease of the s-character in C orbital of the C–F bond (in order to preserve the total s-character of the C atom). Further, as indicated in Table 6, the interaction with ketones results in an increase of the charges on the external F atoms. The sum of these charges increases is larger than the variation of charges on the C atoms, being, for example, 8.9 me for the F2CO system and 19.4 me for the (CH3)2CO system. These variations can be explained by the fact that in isolated CClF3 there is a strong intramolecular delocalization, taking place from one of the LP of the F atoms to the σ*(C–Cl) orbital. For each of the F atoms, this delocalization is equal to 12.37 kcal mol−1, giving a sum of 37.11 kcal mol−1 in the isolated molecule. Complex formation results in a decrease of this delocalization, in the range between 0.93 kcal mol−1 (F2CO) and 2.01 kcal mol−1 (CH3CHO). It must be also mentioned that in isolated CClF3, there are other intramolecular delocalizations which are not very sensitive to complex formation. This is the case of the energy of the LPF1 → σ*(CF2) delocalization equal to 30.30 kcal mol−1 in the isolated molecule and decreasing by only 0.39 kcal mol−1 in the CH3CHO complex. Further, the weak delocalization from σ(C–Cl) to σ*(C–Cl) decreases by ca 0.15 kcal mol−1 by complex formation. These delocalizations will no more be considered hereafter.
Let us mention that a nice effect of electron transfer from the LPs of the halogen atoms to the remote part of the complex has been predicted in the H3N-CCl4 system characterized by the N···ClCCl3 interaction. In this system, the three LPs of the bonded Cl atom are delocalized to the three external C–Cl bonds, resulting in an increase of the σ*(C–Cl) occupation in these three external bonds and a contraction of the C–Cl bond involved in the interaction [64].
It follows from these considerations that the formation of the present complexes can be considered as originating from a two-step mechanism: the intermolecular charge transfer from the LPsO to σ*(C–Cl) followed by a decrease of the intramolecular charge transfer from LPF to σ*(C–Cl). Both effects are connected as illustrated by the following correlation:
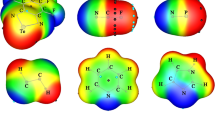
showing the predominance of the intramolecular effect over the intermolecular one. Figure 5 illustrates selected orbitals involved in the above-mentioned two-step mechanism. It must be mentioned that a similar effect has been demonstrated for the CHF3 molecule hydrogen-bonded to proton acceptor of medium strength [75]. In the isolated CHF3 molecule, the intramolecular hyperconjugation energy occurring from the three F atoms to the σ*(CH) bond is equal to 26.40 kcal mol−1. This value is similar to the hyperconjugation energy from the three F atoms to σ*(C–Cl) deduced in the present work for isolated CClF3. In the F3CH···OH2 complex, this hyperconjugation energy decreases by 3.8 kcal mol−1 and predominates over the intermolecular effect. As a consequence, the C–H bond is contracted by a small amount. It should be noted that the interaction between CHF3 and stronger proton acceptors such as F− or Cl− results in a net increase of the σ*(C–H) occupation, a decrease of the σ*(C–F) occupation and an elongation of the C–H bond. This is in strong contrast to the halogen bonds, the interaction between CClF3 and Br− or OH− resulting in a contraction of the C–Cl bond, as previously mentioned.

The donor–acceptor orbitals involved in the two-step mechanism: first step, LP1O → σ*(C–Cl) and LP2O → σ*(C–Cl); second step, LP(F) → σ*(C–Cl) interaction
As for CClF3, CBrF3, CBrCl3 complexed with F−, Cl− and Br− [30], in the present systems there is also a correlation between the binding energies and the second-order interaction energies E 2 LPsO → σ*(C–Cl). The best fit is found for the following logarithmic equation:
It should be mentioned that a unique correlation between the –ΔE and E 2 values was proposed in Ref. [30] for the different systems. The lower correlation coefficient of 0.819 probably results from the fact that each system is characterized by another correlation depending on the specific features of the halomethanes. This shows the advantage to consider closely related systems.
3.3 SAPT decomposition of the energies
To evaluate the physically meaningful components of the interaction energy in the complexes studied, a SAPT analysis has been performed at the MP2/cc-pVTZ level of theory. The results are collected in Table 9. The SAPT interaction energies vary between −1.13 and −2.32 kcal mol−1 for the complexes investigated. As follows from these results, the electrostatic term E(elec) is the dominant attraction component representing about 60 % of the total attraction forces. With regard to the induction E(ind) and dispersion E(disp) components, they account for about 10 and 30 % of the total attraction forces, respectively. The value of the repulsive exchange energy E(exch) is lower than the absolute value of the electrostatic term E(elec). Similar results have been found for the halogen bonded complexes between carbonyl bases and molecular chlorine calculated at the same level of theory [52].
3.4 Frequencies of the ν(C–Cl) and ν(C–F) vibrations
The calculated frequencies of the ν(C–Cl) and ν(C–F) vibrations in isolated and complexed CClF3 are indicated in Table 10. For isolated CClF3 the ν(C–Cl) stretching vibration is predicted at 1073.5 cm−1 in good agreement with the value reported in Ref. [30]. The ν(C–F) vibration is predicted at 1181.3 cm−1. These values are in good agreement with the respective values of 1099.4 and 1208.0 cm−1 observed in liquid argon at 89 K [19]. The interaction with ketones results in an increase of the ν(C–Cl) frequency from 4.3 cm−1 (F2CO) to 9.5 cm−1 [(CH3)2CO]. The degenerate ν(C–F) vibrations are split into two components, and their frequencies decrease from 7.8 to 19.3 cm−1.
As demonstrated in numerous hydrogen-bonded systems, the interaction energies and the frequency shifts of the stretching vibration of the bond involved in the interaction are linearly related. This correlation was originally referred as the Badger–Bauer correlation [76, 77].
For the present systems, there is only a very crude correlation between the interaction energies and the frequency shifts:
This can be accounted for by the fact that the interaction energies are extended to the whole F3CCl molecule and that the ν(C–Cl) stretching vibration is a more localized mode.
4 Conclusions
In this work, the halogen bonded complexes of six carbonyl bases: H2C=O; HFC=O; F2C=O; CH3HC=O; CH3FC=O; (CH3)2C=O with chlorotrifluoromethane have been studied using ab initio MP2 and CCSD(T) as well as density functional theory (B3LYP, B3LYP-D3 and wB97XD) methods. The most important conclusions are the following:
-
1.
In these complexes, the intermolecular O···Cl distances are all smaller than the sum of the van der Waals radii.
-
2.
The interaction energies calculated at the CCSD(T)/aug-cc-pVTZ level vary between −1.22 and −2.62 kcal mol−1. The largest ΔE CCSD(T) is predicted for the (CH3)2CO···ClCF3 system, while the smallest interaction energy has been obtained for the F2CO···ClCF3 complex.
-
3.
The B3LYP-D3 calculated interaction energies are linearly related to the ionization potential and the proton affinity of the carbonyl bases.
-
4.
The interaction results in a contraction of the C–Cl bond and an elongation of the C–F bond. The variation of the bond lengths is accompanied by a blueshift of the C–Cl stretching vibration and a redshift of the C–F stretching vibration.
-
5.
The NBO analysis shows that the increase of the s-character of the C(Cl) atom largely predominates over the decrease of the σ*(C–Cl) occupation in determining the contraction of the C–Cl bond.
-
6.
The elongation of the C–F bonds is explained by a decrease of intramolecular delocalization from one of the lone-pair orbital (LP) of the F atoms to the σ*(C–Cl) orbital.
-
7.
It is suggested that the formation of these complexes originates from a two-step mechanism: the intermolecular charge transfer from the lone-pair orbitals of the O atom (LPsO) to the σ*(C–Cl) antibonding orbital followed by a decrease of the intramolecular charge transfer from the LP on the fluorine atom to the σ*(C–Cl) orbital.
-
8.
The SAPT results for these complexes show that the dispersion and electrostatic contributions cover about 90 % of the total attraction forces. The exchange energy E(exch) is lower than the absolute value of the electrostatic term E(elec). Similar results were found in the case of the carbonyl bases complexed with molecular chlorine [38].
Notes
The correlation computed from the data of Ref. [28] can be written q (Cl) (me) = 0.032 (% s-character C) − 0.0926 (r 2 = 0.993).
References
Pimentel GC, McClellan AL (1960) The hydrogen bond. W.H. Freeman Co., San Francisco, London
Mulliken RS, Person WB (1969) Molecular complexes. A lecture and reprint volume. Wiley-Interscience. Wiley, New York
Metrangolo P, Resnati G (2001) Halogen bonding: a paradigm in supramolecular chemistry. Chem Eur J 7:2511–2519
Chu QL, Wang ZM, Huang QC, Yan CH, Zhu SZ (2001) Fluorine-containing donor-acceptor complexes: infinite chains formed by oxygen···iodine interactions. J Am Chem Soc 123:11069–11070
Nguyen HL, Horton PN, Hursthouse MB, Legon AC, Bruce DW (2004) Halogen bonding: a new interaction for liquid crystal formation. J Am Chem Soc 126:16–17
Wang W, Zhang Y, Ji B, Tian A (2011) On the correlation between bond-length change and vibrational frequency shift in halogen-bonded complexes. J Chem Phys 134:224303
Riley KE, Merz KM (2007) Insights into the strength and origin of halogen bonding: the halobenzene–formaldehyde dimer. J Phys Chem A 111:1688–1694
Amezaga NJM, Pamies SC, Peruchena NN, Sosa GL (2010) Halogen bonding: a study based on the electronic charge density. J Phys Chem A 114:552–562
Donald KJ, Wittmaack BK, Crigger C (2010) Tuning σ-holes: charge redistribution in the heavy (group 14) analogues of simple and mixed halomethanes can impose strong propensities for halogen bonding. J Phys Chem A 114:7213–7222
Wang W (2011) Halogen bond involving hypervalent halogen: CSD search and theoretical study. J Phys Chem A 115:9294–9299
Legon AC (2010) The halogen bond: an interim perspective. Phys Chem Chem Phys 12:7736–7747
Zhang X, Zeng Y, Li X, Meng L, Zheng S (2011) A computational study on the nature of the halogen bond between sulfides and dihalogen molecules. Struct Chem 22:567–576
Hill JG, Legon A (2015) On the directionality and non-linearity of halogen and hydrogen bonds. J Phys Chem Chem Phys 17:858–867
Otero-de-la-Roza A, Johnson ER, DiLabio GA (2014) Halogen bonding from dispersion-corrected density-functional theory: the role of delocalization error. J Chem Theor Comput 10:5436–5447
Azofra LM, Scheiner S (2014) Substituent effects in the noncovalent bonding of SO2 to molecules containing a carbonyl group. The dominating role of the chalcogen bond. J Phys Chem A 21:3835–3845
Kozuch S, Martin JML (2013) Halogen bonds: benchmarks and theoretical analysis. J Chem Theory Comput 9:1918–1931
Alkorta I, Elguero J (2014) Characterizing traditional and chlorine-shared halogen bonds in complexes of phosphine derivatives with ClF and Cl2. J Phys Chem A 118(23):4222–4231
Scheiner S (2015) The interplay between charge transfer, rehybridization, and atomic charges in the internal geometry of subunits in noncovalent interactions. Int J Quant Chem 115:28–33
Stone AJ (2013) Are halogen bonded structures electrostatically driven? J Am Chem Soc 135:7005–7009
Sutradhar D, Chandra AK, Zeegers-Huyskens Th (2014) A theoretical investigation of the interaction between fluorinated dimethyl ethers and molecular chlorine. Mol Phys 112:2791–2801
Zou J-W, Jiang Y-J, Guo M, Hu G-X, Zhang B, Liu H-C, Yu Q-S (2005) Ab initio study of the complexes of halogen-containing molecules RX (X=Cl, Br, and I) and NH3: towards understanding the nature of halogen bonding and the electron-accepting propensities of covalently bonded halogen atoms. Chem Eur J 11:740–751
Jahromi WJ, Eskandari K (2013) Halogen bonding: a theoretical study based on atomic multipoles derived from quantum theory of atoms in molecules. Struct Chem 24:1281–1287
Wang W, Wang N-B, Zheng W, Tian A (2004) Theoretical study on the blueshifting halogen bond. J Phys Chem A 108:1799–1805
Wang W, Hobza P (2008) Origin of the X–Hal (Hal=Cl, Br) bond-length change in the halogen-bonded complexes. J Phys Chem A 112:4114–4119
Zhao Q, Feng D, Hao J, Cai Z (2010) Chemical origin of contracted C–Cl bonds in the halogen-bonded complexes. J Mol Struct (THEOCHEM) 958:71–75
Tawfik M, Donald K (2014) Halogen bonding: unifying perspectives on organic and inorganic cases. J Phys Chem A 118:10090–10100
Grabowski SJ (2012) QTAIM characteristics of halogen bond and related interactions. J Phys Chem A 116:1838–1845
Grabowski SJ (2011) Halogen bond and its counterparts: Bent’s rule explains the formation of nonbonding interactions. J Phys Chem A 115:12340–12347
Grabowski SJ (2013) Non-covalent interactions—QTAIM and NBO analysis. J Mol Model 19:4713–4721
Zhang Y, Li A-Y, Cao L-J (2012) Electronic properties of the halogen bonds Z 3CX···Y− between halide anions and methyl halides. Struct Chem 23:627–636
Scheiner S (2013) Detailed comparison of the pnicogen bond with chalcogen, halogen, and hydrogen bonds. Int J Quant Chem 113:1609–1620
Hauchecorne D, Szostak R, Herrebout WA, van der Veken B (2009) C–X···O halogen bonding:interactions of trifluoromethyl halides with dimethyl ether. J Chem Phys Chem 10:2105–2115
Evangelisti L, Feng G, Gou Q, Grabov J-U, Caminati W (2014) Halogen bond and free internal rotation: the microwave spectrum of CF3Cl–dimethyl ether. J Phys Chem A 118:579–582
Clark T., Hennemann M., Murray J. S., Politzer P. (2007) Halogen bonding: the sigma-hole. In: Proceedings of modeling interactions in biomolecules II, Prague, 5–9 Sept 2005. J Mol Mod 13:291–296
Politzer P, Murray JS, Clark T (2013) Halogen bonding and other σ-hole interactions: a perspective. Phys Chem Chem Phys 15:11178–11189
Riley KE, Hobza P (2008) Investigations into the nature of halogen bonding including symmetry adapted perturbation theory analyses. J Chem Theory Comput 4:232–242
Gall J-F, Maria P-C (1990) In progress in physical organic chemistry. In: Taft RW (ed) vol 17. Wiley, New York/Chichester/Brisbane, pp 159–238
Møller C, Plesset MS (1934) Note on an approximation treatment for many-electron systems. Phys Rev 46:618–622
Kendall RA, Dunning TH Jr, Harrison RJ (1992) Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J Chem Phys 96:6796–6806
Becke AD (1993) Density-functional thermochemistry. III. The role of exact exchange. J Chem Phys 98:5648–5652
Lee C, Yang W, Parr RG (1988) Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys Rev B 37:785–789
Grimme S, Antony J, Ehrlich S, Krieg H (2010) A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J Chem Phys 132:154104
Chai J-D, Head-Gordon M (2008) Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys Chem Chem Phys 10:6615–6620
Krishnan R, Binkley JS, Seeger R, Pople JA (1980) Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J Chem Phys 72:650–654
Frisch MJ, Pople AJ, Binkley JS (1984) Self-consistent molecular orbital methods 25. Supplementary functions for Gaussian basis sets. J Chem Phys 80:3265–3269
Boys SF, Bernardi F (1970) The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol Phys 19:553–566
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson et al (2009) Gaussian 09. Gaussian Inc, Wallingford
Werner HJ, Knowles PJ, Knizia G, Manby FR, Schütz M, Celani P, Korona T, Lindh R, Mitrushenkov A, Rauhut G et al (2012) MOLPRO, version 2012.1, a package of ab initio programs
Reed AE, Curtiss LA, Weinhold F (1988) Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem Rev 88:899–926
Glendening ED, Badenhoop JK, Reed AE, Carpenter JE, Bohmann JA, Morales CM, Weinhold F (2001) NBO 5.0 Software. Theoretical Chemistry Institute, University of Wisconsin, Madison. http://www.chem.wisc.edu/~nbo5
Todd A, Keith TK (2014) AIMAll (Version 14.11.23). Gristmill Software, Overland Park KS, USA, 2014 (aim.tkgristmill.com)
Zierkiewicz W, Bieńko DC, Michalska D, Zeegers-Huyskens Th (2015) Theoretical investigation of the halogen bonded complexes between carbonyl bases and molecular chlorine. J Comput Chem 36:821–832
Wiberg KB, Marquez M, Castejon H (1994) Lone pairs in carbonyl compounds and ethers. J Org Chem 59:6817–6822
Koch U, Popelier PLA (1995) Characterization of C–H–O hydrogen bonds on the basis of the charge density. J Phys Chem 99:9747
Popelier PLA (1998) Characterization of a dihydrogen bond on the basis of the electron density. J Phys Chem A 102:1873
Chandra AK, Zeegers-Huyskens Th (2012) A theoretical investigation of the interaction between substituted carbonyl derivatives and water: Open or cyclic complexes? J Comput Chem 33:1131–1141
Niu B, Shirley DA, Bai Y (1993) High resolution photoelectron spectroscopy and femtosecond intramolecular dynamics of H2CO+ and D2CO+. J Chem Phys 98:4377–4390
Wittel K (1976) The photoelectron spectrum of formylfluoride. J Electron Spectrosc Relat Phenom 8:245–248
Buckley TJ, Johnson RD, Huie RE, Zhang Z, Kuo SC, Klemm RB (1995) Ionization Energies, appearance energies, and thermochemistry of CF2O and FCO. J Phys Chem 99:4879–4885
Hernandez R, Mouvier G (1977) Spectroscopie de photoelectrons d’aldehydes et de cetones aliphatiques. J Electron Spectrosc Relat Phenom 10:333–347
Chadwick D, Katrib A (1974) Photoelectron spectra of acetaldehyde and acetyl halides. J Electron Spectrosc Relat Phenom 3:39–52
Trott WM, Blais NC, Walters EA (1978) Molecular beam photoionization study of acetone and acetone-d6. J Chem Phys 69(7):3150–3158
Alkorta I, Blanco F, Solimannejad M, Elguero J (2008) Competition of hydrogen bonds and halogen bonds in complexes of hypohalous acids with nitrogenated bases. J Phys Chem A 12:10856–10863
Wang W, Zhang Y, Ji B (2010) On the difference of the properties between the blue-shifting halogen bond and the blue-shifting hydrogen bond. J Phys Chem A 114:7257–7260
Mohan N, Suresh CH (2014) A molecular electrostatic potential analysis of hydrogen, halogen, and dihydrogen bonds. J Phys Chem A 118:1697–1705
An X, Zhao H, Wang Y, Li Q (2013) Competition between hydrogen bonds and halogen bonds in complexes of formamidine and hypohalous acids. J Mol Mod 19:4529–4535
Wu W, Zeng Y, Li X, Zhang X, Zheng S, Meng L (2013) Interplay between halogen bonds and hydrogen bonds in OH/SH···HOX···HY (X=Cl, Br; Y=F, Cl, Br) complexes. J Mol Mod 19:1069–1077
Scheiner S, Grabowski SJ, Kar T (2001) Influence of hybridization and substitution on the properties of the CH···O hydrogen bond. J Phys Chem A 105:10607–10612
Li X, Schlegel B (2002) On the physical origin of blue-shifted hydrogen bonds. J Am Chem Soc 124:9639–9647
Sosa GL, Peruchena NM, Contreras RH, Castro E (2002) Topological and NBO analysis of hydrogen bonding interactions involving C–H···O bonds. J Mol Struct 577:219–228
Kryachko ES, Zeegers-Huyskens Th (2001) Theoretical Study of the CH···O interaction in fluoromethanes H2O and chloromethanes H2O complexes. J Phys Chem A 105:7118–7125
Alabugin IV, Manoharan M (2003) Electronic basis of improper hydrogen bonding: a subtle balance of hyperconjugation and rehybridization. J Am Chem Soc 125:5973–5987
Nguyen HMT, Peters J, Zeegers-Huyskens Th (2006) Theoretical study of the blue-shifting hydrogen bonds between CH2X2 and CHX3 (X=F, Cl, Br) and hydrogen peroxide. J Mol Struct 792:16–22
Joseph J, Jemmis ED (2007) Red-, blue-, or no-shift in hydrogen bonds: a unified explanation. J Am Chem Soc 129:4620–4632
Li AY (2007) Chemical origin of blue- and redshifted hydrogen bonds: intramolecular hyperconjugation and its coupling with intermolecular hyperconjugation. J Chem Phys 126:154102–154112
Badger RM, Bauer SH (1937) Spectroscopic studies of the hydrogen bond. II. The shift of the O-H vibrational frequency in the formation of the hydrogen bond. J Chem Phys 5:839
Badger RM (1940) The relation between the energy of a hydrogen bond and the frequencies of the O–H bands. J Chem Phys 8:288
Acknowledgments
This work was financed in part by a statutory activity subsidy from the Polish Ministry of Science and Higher Education for the Faculty of Chemistry of Wroclaw University of Technology. A generous computer time from the Wroclaw Supercomputer and Networking Center as well as the Poznan Supercomputer and Networking Center is acknowledged. Th. Z. H thanks the University of Leuven for computer facilities.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Zierkiewicz, W., Bieńko, D.C., Michalska, D. et al. On the nature of halogen bonded complexes between carbonyl bases and chlorotrifluoromethane. Theor Chem Acc 134, 103 (2015). https://doi.org/10.1007/s00214-015-1706-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00214-015-1706-7