
Abstract
This review deals with the drug transporters allowing drugs to enter and leave cells by carrier-mediated pathways. Emphasis is put on liver transporters but systems in gut, kidney, and blood-brain barrier are mentioned as well. Drug-drug interactions on carriers may provoke significant modification in pharmacokinetics as do carrier gene polymorphisms yielding functional carrier protein mutations. An integrated phase concept should reflect the interplay between drug metabolism and drug transport.
Similar content being viewed by others

Historical background
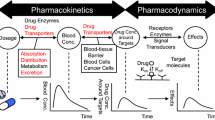
Pharmacokinetics comprises drug liberation, drug absorption, drug distribution, drug metabolism and drug excretion. The issue of xenobiotic elimination, also termed evasion, is defined in pharmacokinetics by two processes, drug metabolism and drug excretion, whereby metabolism is described to comprise sequential biotransformation steps termed phase 1 and phase 2 metabolism (Williams 1959; Gillette 1963; Gerok and Sickinger 1973). The “phase concept” has long been embedded in pharmacokinetics: R.T. Williams divided phenacetin detoxification into two parts, phase I which consists of an oxidative dealkylation reaction to yield 4-acetamidophenol, followed by phase II in which an organic acid is formed, yielding the highly polar 4-acetamidophenyl-β-glucuronide (Fig. 1). Emphasis was put on phase II as an essential detoxification phase because increase of polarity was considered the essential step for drug elimination via bile and urine.

Detoxification of phenacetin taken from Smith and Williams 1949. This description was a hallmark in drug metabolism and the first description of an O-dealkylation reaction of drugs
Since the detoxification reactions occur within cells, it is surprising that the questions of how the drug reaches the endoplasmic reticulum of, for example, a liver parenchymal cell from the blood, and how the drug is released out of the hepatocyte into bile, have not been subject to equally intense studies as those concerning drug metabolism. In analogy to intestinal absorption, which in those days was thought to follow the physical diffusion concept of non-charged molecules (Brodie and Hogben 1957; Hogben et al. 1959; Brodie 1964), the permeation of organic compounds into and out of cells was considered to be governed by non-selective physical diffusion processes (Stein 1967; Diamond and Wright 1969; Elleroy and Lew 1977).
However, it was soon recognized that at least for highly polar organic compounds such as sugars, so-called facilitated passive diffusion, which involves a biological carrier with saturation and competition kinetics (Wilbrandt 1975) or even active, energy-dependent uphill transport by membrane carriers, is mandatory (LeFevre 1948; Crane and Krane 1959). An Na+-dependent intestinal glucose-carrier from a mammalian species was not cloned until 1987 (Hediger et al. 1987; Wright and Turk 2004). In general, biological non-electrolyte transport is a strong pillar of physiological cell functions. Its energetics comprises ion-coupled cotransport or ion-coupled countertransport, allowing secondary uphill transport, or pumping by ABC-carriers (ATP-binding cassette-carriers), allowing primary active transport, or countertransport with endogenous non-electrolytes or even simple facilitated diffusion along the compound´s concentration gradient (Fig. 2).

Classification of solute carriers (SLC), ATP-dependent carriers (ABC-carriers), and channels in the plasma membrane
It has long been established that transporters exist for endogenous compounds such as glucose, amino acids, nucleosides, water soluble hormones and neurotransmitters. However, the perspective that xenobiotics are also substrates of membrane carriers has emerged only in the last two decades. Xenobiotics are by definition compounds which are not essential for the maintenance of a physiological function; they may, however, modulate, ameliorate or damage such functions, depending on whether they are drugs, diagnostics, or toxins. Living organisms, therefore, have also developed processes to eliminate “non-physiological” xenobiotics by carrier mediated transport (LeFevre 1975). This article focuses mainly on transporter-based elimination of drugs by the liver.
Functional indications of drug transporters
Early functional studies on the elimination of drugs by the liver (Klaassen and Watkins 1984), gut (Kramer and Lauterbach 1977; Jackson 1987), and kidney (Ullrich 1997) indicated the existence of carriers for xenobiotics, defined by transport saturation, cell specificity, and mutual transport interferences. Without any molecular knowledge of the carrier proteins themselves, descriptions were made of driving forces for uphill transport such as electrochemical gradients of ions (Heinz 1972; Semenza and Kinne 1985; Alvarado and Van Os 1986), counter- or antiport- transport (Rosenberg and Wilbrandt 1957; Heinz 1978), membrane potential (Ward 1970; Athayde and Ivory 1985) or direct energy transmission due to ATP-cleavage (Caldwell 1956, 1960; Keynes 1961; Carafoli and Scarpa 1982). Although it was suggested very early (Sperber 1959), it was later shown that drug excretion and absorption is strongly dominated by membrane carrier proteins, i.e. in liver (Petzinger et al. 1989; Petzinger 1994), kidney (Greger et al. 1981) and gut (Gilles-Baillieu and Gilles 1983). As with the detoxification enzymes of phase I and phase II, these carriers are subject to postnatal development in neonates (Gao et al. 2004), and regulation and induction by diseases may occur (Trauner and Boyer 2003; Kullak-Ublick et al. 2004). With the availability of cloned carrier genes, pharmacogenomics of drug transporters have recently been the subject of pharmacological and clinical research (Tirona et al. 2001; Tirona and Kim 2002; Ieiri et al. 2004).
Molecular classification of SLC- drug transporters
Drug transporter gene identification started in the 1990s by cloning Oatps (organic anion transporting polypeptides) from the liver (Jacquemin et al. 1994; Hagenbuch and Meier 2003) and Oats/Octs (organic anion transporters/organic cation transporters) from the kidney (Gründemann et al. 1994; Sekine et al. 1997; Sweet et al. 1997; Burckhardt and Burckhardt 2003; Koepsell and Endou 2004). These carriers have now been grouped in the large family of solute carriers SLC (Hediger et al. 2004). Table 1 shows a list of selected families of SLCs, which currently comprise 43 families with an expanding number of subfamilies and individual carriers. Over 300 human genes are assumed to encode SLC carriers. Many of these carriers can be considered to transport physiological endogenous substrates alone; several transport a few foreign compounds in addition, and some even transport xenobiotics predominantly. The SLC-families receive sequential numbers from the HUGO (human genome organisation) nomenclature committee HGNC. Overviews of several families of drug transporters have been published. Since this field is rapidly expanding, this article can only present a momentary snapshot, and several other reviews are recommended (Pritchard and Miller 1993; Burckhardt and Wolff 2000; Kullak-Ublick et al. 2000; Ayrton and Morgan 2001; Dean et al. 2001; Dresser et al. 2001; Borst and Oude Elferink 2002; Russel et al. 2002; Burckhardt and Burckhardt 2003; Daniel and Rubio-Aliaga 2003; Hagenbuch and Meier 2003; Mizuno et al. 2003; Daniel 2004; Koepsell and Endou 2004; Hediger et al. 2004; Pauli-Magnus and Meier 2004, and also reviews in this journal issue, i.e. König et al. 2006 and Geyer et al. 2006).
An extended phase concept
Drug transporter / drug metabolism interplay represents a new challenge in cellular pharmaco- and toxicokinetics (Liu and Pang 2005). Reflecting the meaning of drug transport in cellular pharmacokinetics, the historical two-phase concept, which has only considered the relevance of biotransformation of drugs for drug evasion, needs extension.
Figure 3 shows an extended phase concept encompassing interactions between transporter phases for uptake and excretion, and the metabolic steps. The metabolic phases 1 and 2 are flanked by drug transporter phases 0 and 4, while intracellular cytoplasmic drug traffic is regarded as phase 3. In the following, some implications of this model are presented.

Schematic principle of vectorial drug evasion in liver and kidney. Phase 0 = drug uptake out of blood, Phases 1 and 2 = biotransformation exemplified by hydroxylation and glucuronidation, Phase 3 = transport of xenobiotics/metabolites towards excretion, Phase 4 = efflux into excreted fluids and/or backward into blood. ☼ xenobiotic
Phase 0
SLC transporters mediate the earliest phase in drug kinetics on cells, which is here termed phase zero (phase 0). Phase 0 is the first step of drug elimination from blood via uptake across the basolateral membrane into the cells, i.e. of the proximal tubule or into hepatocytes respectively, or the first step of absorption from the gut, i.e. the uptake across the luminal membrane into enterocytes. Thus, SLC transporters are located in both the basolateral and the luminal cell membrane. This type of carrier imposes a selection, although not a really stringent “filter” gate, for certain classes of xenobiotics. SLC-drug carriers are multispecific, which means they allow permeation of a spectrum of compounds with variable chemical structures. An example of this type of multispecificity are members of the organic anion transporting polypeptide carriers Oatps (SLC21 / SLCO) which transport weak organic acids, neutral compounds and even a few cationic compounds (Hagenbuch and Meier 2003). Quantitative structure–activity relationships (QSARs) (Yarim et al. 2005) were elaborated to predict structural criteria of compounds essentially needed to fulfill the requirements for transport by individual carrier proteins. Systems in the liver and kidney have been reviewed in detail (Van Montfoort et al. 2003; Hagenbuch and Meier 2003; Chandra and Brouwer 2004). Similarly, another family of organic anion transporters, the Oat-family, transports negatively and positively charged compounds, and is very closely related to the Oct-family, specialised for organic cation transport (Burckhardt and Wolff 2000; Burckhardt and Burckhardt 2003; Koepsell and Endou 2004). Oats and octs, therefore, are members of the same SLC22 family (Table 1). Phase 0 drug transporters jointly influence compound allocation for drug metabolism.
Phase 4
Phase 4 pathways are well-defined. They comprise final steps of excretion, e.g. in the bile canaliculus of the liver, or secretory steps in the luminal membrane, e.g. in the gut, counteracting absorption. Phase 4 is predominantly maintained by directly driven “primary” uphill transport of xenobiotics across cell membranes, which is achieved by ATP-consuming transport ATPases belonging to the ABC-carrier proteins (Müller and Jansen 1998; Chan et al. 2004) (Table 2). A prominent member of these transport proteins, first detected in drug-resistant tumour cells (Juliano and Ling 1976), is P-glycoprotein. This drug resistance was conferred by a gene named multidrug resistance (MDR) gene, which was characterised in 1986 (Roninson et al. 1986). P-glycoprotein substrates are lipophilic, and are generally non-conjugated compounds, whereas water-soluble drug conjugates (sulfated, glucuronidated and glutathione-conjugated drugs) are transported in the liver by the multidrug resistance-associated protein MRP2 (Homolya et al. 2003; Hoffmann and Kroemer 2004; Fardel et al. 2005). Other members of the ABCC-transporter family (MRPs 1, 3, 4, 5, and 6), in conjunction with the BCRP (breast cancer resistance protein)-carrier belonging to the ABCG-family (Table 2) provide a panel of export pumps in the liver and elsewhere in the body (Haimeur et al. 2004) (Fig. 4). Carrier genes comprise at least 5% of all human genes with 49 genes belonging to human ABC-carrier genes (Dean et al. 2001).

Individual carriers in the hepatocyte which are involved in drug uptake and secretion. OAT/Oat = organic anion transporter belonging to the SLC22 family; OCT/Oct = organic cation transporter belonging to the SLC22 family; NTCP/Ntcp = Na + /taurocholate cotransporting polypeptide belonging to the SLC10 family; OATP/Oatp = organic anion transporting polypeptide belonging to the SLCO family (previously called SLC21). MDR1/Mdr1 = multidrug resistance protein (ABCB1), BCRP/Bcrp = breast cancer resistance protein (ABCG2), MRP2/Mrp2 = multidrug resistance-associated protein (ABCC2), BSEP/Bsep = bile salt export pump (ABCB11) all belonging to ABC-carrier proteins; OA− = organic anion; BS− = bile salt; OC+ = organic cation; DC− = dicarboxylate; GSH = glutathion
ABC-carriers for xenobiotics, i.e. P-glycoprotein, MRP2 and BCRP, are mainly located at the luminal membrane of a cell facing, for example, the bile canaliculus, the lumen of the gut or the tubule lumen of a nephron. The general function which results from this location is drug excretion, providing evidence that these types of ABC-carriers convey protection against xenobiotics (Leslie et al. 2005). In the gut, these ABC-carriers are gatekeepers of drug absorption, limiting drug bioavailability (Dietrich et al. 2003).
Certain ABC-carriers, i.e. MRPs 1, 3, 4, and 6, are mainly found in the basolateral membrane, where they confer secretion of organic compounds into blood. In the liver MRP1 and MRP6 are strongly expressed, whereas MRP3 and MRP4 expression is low in normal liver. MRP1 transports non-metabolized cytotoxic drugs including certain cytostatics (daunomycin, doxorubicin, vincristine, but not cisplatin) known to be also P-glycoprotein transportates. In addition, ATP-dependent efflux of conjugated endogenous compounds, i.e. bilirubin glucuronides, cysteinyl-leukotriene C4, estradiol 17β-glucuronide and dianionic bile salts, is promoted, and efflux of xenobiotic conjugates via MRP1 may also occur (Leslie et al. 2001).
MRP3, normally present in the liver at low expression level, is highly expressed if hepatobiliary drug excretion is impaired due to extrahepatic cholestasis (Ogawa et al. 2000; Soroka et al. 2001; Scheffer et al. 2002). Under cholestasis (Donner and Keppler 2001) and in patients suffering from Dubin-Johnson syndrome (König et al. 1999), this carrier allows reflux of drug metabolites out of the hepatocyte into blood (small arrow of Fig. 3). At certain blood/tissue barriers such as the blood/brain and blood/testis barrier, reflux out of the cell into the blood occurs via P-glycoprotein (MDR1) and other carriers of the ABCC and ABCG family (Fromm 2004). P-glycoprotein could act as a “vacuum cleaner” (Bolhuis et al. 1997), sucking lipophilic drugs out of membrane phospholipids. Such drugs will not have penetrated through the membrane into the cell cytoplasm, and are thus not subjected to phase 1/phase 2 metabolism. This mechanism would impose a strict barrier at the membrane level between blood and tissues.
Perspective: intracellular transport of xenobiotics within a polarised cell
A mandatory step following phase 0 and also the biotransformation phases 1 or 2 is a cytoplasmic shuttle service of xenobiotics between differing cell poles. Pharmacokinetic phases 1 and 2 of drug metabolism preferably, but not exclusively, occur in the endoplasmic reticulum (ER), and are related to drug metabolism by membrane-bound cytochrome P450s (CYPs) and comparable enzymes (phase 1), followed by reactions of phase 2. Here, conjugating enzymes within or outside of the ER, such as glucuronosyltransferases (UGT-GTs), glutathione-S-transferases (GSTs), sulfotransferases (SULTs), cytoplasmic N-acetylases (NATs) and others, generate drug metabolites which are suited for rapid excretion (Fig. 3, main arrow). The metabolism phases afford delivery of drugs from the basolateral cell membrane to the ER and, later, of drug metabolites from the ER to the secretory sites of the cell membrane. This step is referred to as transport phase 3 in this review (Fig. 3). Cytoplasmic phase 3 transport represents a vectorial transport phase which has not yet been described in detail in any of the various pharmacokinetic cell transport models that have to date been published. It appears unlikely that cytoplasmic drug trafficking is a matter of simple physical diffusion, because diffusion would account for neither the velocity nor the efficiency of drug excretion by polarized cells. Thus, phase 3 drug transport needs floating carrier proteins or carrier particles. Previously, a protein named ligandin was considered as an important protein for cytoplasmic drug delivery in the liver (Levi et al. 1969). Later it was recognized that this protein was a glutathione-S-transferase (Habig et al. 1974). Other travelling proteins considered were cytosolic fatty acid-binding proteins (cFABPs), ileal lipid-binding proteins (ILBP) and bile acid-binding proteins (for a review see Petzinger 1994; Kramer et al. 2001). They comprise intracellular steps of the enterohepatic circulation of bile acids and drugs. Such binding proteins are assumed to travel together with transporter ligands along cytoskeletal tracks to their distinctive membrane sites (Bilej and Vetvicka 1989). Modern reflexions consider also endocytic trafficking pathways as important routes of drugs in the cell (Watson et al. 2005). For example, some weakly basic drugs such as doxorubicin may accumulate within acidic lysosomal organelles (Weaver et al. 1991), and thus may follow the traffic via vesicular pathways either in the lysosomal compartment (Lloyd 2000) or after fusion with endosomes in the multivesicular body (MVB) compartment. Vesicular cytoplasmic drug traffic may also require the participation of the cytoskeleton (Murray and Wolkoff 2003), and could explain cholestatic effects of toxins destroying the cytoskeleton (Ohashi et al. 2002). During cholestasis, the redistribution of some membrane carrier proteins would change the hepatobiliary secretion route for drugs which, instead of circulating into bile, reflux back to the blood (small arrow Fig. 3) (Rost et al. 1999). New approaches use fluorescent drugs or drugs coupled to fluorescent dyes for live cell imaging of intracellular transport pathways (Watson et al. 2005). This will better help to elucidate phase 3 drug transport in the future.
Clinical importance of implementing drug transporter phases
Drug transporters of the SLC-families preferably transport hydrophilic and amphiphilic xenobiotics. These carriers could confer some organ selectivity in xenobiotic elimination and toxicity. E.g. the Green Death Cap mushroom toxin phalloidin has long been recognised to be a liver-selective toxin. It was suggested by the author in the early 1980s that the bicyclic heptapeptide phalloidin should be transported by a liver-specific bile acid transporter (Petzinger et al. 1979; Petzinger 1981). In 2003 the liver-specific human OATP1B1, syn. OATP-C was shown to transport phalloidin (Fehrenbach et al. 2003). Only in the hepatocyte can the peptide accumulate to such an extent that the cytoskeletal protein actin is blocked. OATP carriers transport bile acids (Trauner and Boyer 2003) and a plethora of drugs (Hagenbuch and Meier 2003) including HMG-CoA reductase inhibitors such as pravastatin (Ziegler and Stünkel 1992; Ziegler and Hummelsiep 1993; Hsiang et al. 1999). Oatps were addressed by drugs conjugated with bile acids in a bile acid-based drug targeting approach for the liver (Kramer et al. 1992; Meijer 1993; Petzinger et al. 1995; Kramer and Wess 1996).
Drug–drug interferences already occur at the level of phase 0. A known example is inhibition of tubular secretion of β-lactam antibiotics penicillin G and cephalosporine by probenecid (Burckhardt and Burckhardt 2003). In the past this interaction was used clinically to diminish renal excretion of penicillins and to prolong their half-life times in serum. The timely development of β-lactam antibiotics with longer half-lives now makes the co-application of probenecid unnecessary. Dibromosulfophthalein is a substrate of the basolateral Oatp1a1 and of canalicular Mrp2 in rat liver (compare Fig. 4). It inhibited the hepatobiliary excretion of fexofenadine into rat bile by blockage of fexofenadine entry into the hepatocyte and, thereby, markedly reduced fexofenadine liver-tissue concentration (Milne et al. 2000). Decreased absorption of fexofenadine from gut after concomitant intake of grapefruit, orange, or apple juice has been reported (Dresser et al. 2002) and was partly related to transport competition. Further examples of clinically relevant drug interactions versus phase-0 carriers have been noted elsewhere (Ayrton and Morgan 2001).
Better known are drug–drug interactions resulting from transport competition on phase 4 ABC-carriers. For example, hepatobiliary liver excretion of the antihistaminic drug fexofenadine is reduced by 46% if co-administered with erythromycin, due to competition on P-glycoprotein (Milne et al. 2000). In contrast to dibromosulfophthalein (see above), erythromycin did not reduce the intrahepatic concentration of fexofenadine. The β-blocker talinolol is transported by P-glycoprotein (Grammatté et al. 1996). Therefore, the P-glycoprotein inhibitor verapamil inhibited the excretion of talinolol via the gut (Grammatté and Oertel 1999) and enhanced the oral exposure to the drug (Spahn-Langguth et al. 1998). Co-substrates of P-glycoprotein are the commonly used drugs ketoconazole/itraconazole, erythromycin, and quinidine. Previous clinical observations of a significant increase of plasma concentrations of digoxin, a cardiac glycoside, by simultaneous application of the antiarrhythmic drug quinidine (Doering 1979; Dahlquist et al. 1980; Pedersen et al. 1983) are now explained by transport competition on P-glycoprotein (Fromm et al. 1999; Drescher et al. 2003). This clinically important interaction was previously interpreted to be caused predominantly by phase 1 interactions (Dresser et al. 2001). The implementation of drug–carrier interactions (phase 4) into the picture of drug–CYP interactions (phase 1) (Ito et al. 1998) may also resolve certain drug–drug interactions among the modern anti-HIV-protease inhibitors ritonavir and saquinavir. Combinations of ritonavir with saquinavir boosted saquinavir blood levels several-fold (Van Heeswijk et al. 2001). Both compounds are P-glycoprotein substrates (Kim et al. 1998; Washington et al. 2000) but are also metabolized by CYP3A4 enzyme. A recent clinical study indicated that ritonavir exerted both types of interactions, namely inhibition of first-pass metabolism and increased bioavailability due to inhibition of intestinal P-glycoprotein secretion (Kilby et al. 2002).
Conversely, examples of co-stimulation of phase 2 enzyme and MRP-transporter expression under drug therapy are also known (Catania et al. 2004). For example, the herbal medicine St. John’s Wort is a strong inducer of several human drug-metabolizing enzymes (Delgoda and Westlake 2004) but also induces MRP2 transcription in rats and in human HepG2 cells respectively (Shibayama et al. 2004; Krusekopf and Roots 2005). Via the same PXR-mediated transactivation, human CYP3A4 and P-glycoprotein expression is enhanced, too (Synold et al. 2001). It is due to this mode of action that St. John’s Wort had a detrimental effect on therapy with immunosuppressants cyclosporin A and tacrolimus, with antineoplastic agents irinotecan and imatinib mesylate, and with hormonal contraceptives (Mannel 2004). Several drugs, i.e. oltipraz, garlic compound allyl sulphide, rifampicin, tamoxifen, and phenobarbital—already known inducers of phase 1 CYP enzymes—also induce or enhance MRP2- and P-glycoprotein-related drug transport (Fardel et al. 2005). For example, the well-known CYP inducers rifampicin/rifampin and phenobarbital are co-inducers of P-glycoprotein. Such changes complicate the prediction of drug–drug interactions enormously (Ito et al. 1998). Under rifampin, the plasma concentration of digoxin and talinolol decreased due to induction of intestinal digoxin secretion via P-glycoprotein (Greiner et al. 1999; Westphal et al. 2000). It is the extent of either inhibition or stimulation of metabolism, together with effects on transport, which alters the pharmacokinetic balance between the phases and which finally modulates drug–drug interactions in a very subtle manner (Zhang et al. 1998; Wandel et al. 1999).
Drug carrier polymorphisms and pathologies
Genetic carrier polymorphisms cause functional alterations of drug kinetics and their effects, and also of hereditary diseases. There are reports addressing pharmacokinetic consequences related to MDR1 polymorphism (Hoffmeyer et al. 2000; Drescher et al. 2002, 2003; Ieiri et al. 2004) and OATP-polymorphisms (Tirona et al. 2001; Nozawa et al. 2002; König et al. 2006). A frequent polymorphism of phase 4 MDR1 carrier occurring in about 12% of patients is the homozygous C3435T exchange. In these patients, (i) less than half of the normal MDR1 gene expression in the intestine, (ii) much lower gene induction by rifampin, and (iii) decreased excretion of digoxin into the gut was reported. Therefore, the homozygous T/T patients had digoxin plasma levels under rifampin stimulation four times higher than those of rifampin-stimulated homozygous C/C MDR1 patients (Hoffmeyer et al. 2000). On the other hand, plasma kinetics of P-glycoprotein substrate fexofenadine was not altered in patients bearing the C3435T or a G2677T mutation, although the activity of P-glycoprotein was markedly reduced (Drescher et al. 2002). Therefore, the known variability of the plasmakinetics of this drug in normal populations may have other reasons, and alterations in the phase 0 uptake transporters were considered (Drescher et al. 2002).
Other authors reported that certain SNPs and haplotypes of MDR1 affecting P-glycoprotein expression are associated with treatment outcome and/or host susceptibility to renal epithelial tumors (Siegsmund et al. 2002), Balkan endemic nephropathie (Atanasova et al. 2004), Parkinson’s disease (Drozdzik et al. 2003), breast cancer (Kafka et al. 2003), inflammatory large-bowel disease (IBD) (Brant et al. 2003) and ulcerative colitis (Schwab et al. 2003).
Many of the known polymorphisms in the human MDR1 gene are either functionally silent, or only marginally alter P-glycoprotein-mediated transport. None of them resulted in a complete loss of transport. This, however, was observed when a deletion mutation was found to occur frequently in the Collie dog MDR1 gene (Mealey et al. 2001; Roulet et al. 2003; Geyer et al. 2005a,b). These dogs suffer from neurotoxic symptoms of the antiparasitic drugs ivermectin and moxidectin that normally do not reach the central nervous system (Geyer et al. 2005a). In these dogs, lethal outcomes following drug application have even occurred (Pulliam et al. 1985; Paul et al. 1987).
Single nucleotide polymorphisms are also frequent in human phase zero OATP carriers. In the OATP-C gene (OATP1B1), which is specifically expressed only in the liver, the cholesterol-lowering effect of pravastatin decreased in patients exhibiting multiple SNPs, due to the decreased drug uptake into hepatocytes where the drug inhibits HMG-Co-A reductase (Kim 2004; Niemi et al. 2004, 2005a). An analogous kinetic change was reported for fexofenadine, which uses this carrier for hepatobiliary clearance. Patients with the T512C-SNP had higher plasma levels of this H1-receptor antihistamine when compared with patients lacking or exhibiting non-functional SNPs (Niemi et al. 2005b). SNPs have been also reported for other human OATP members, namely OATP1A2, 1B3, and 2B1 (Iida et al. 2001).
Pathologies of the liver related to single carrier defects and associated with severe inherited diseases are rare in humans. Of those that have been identified, all are caused by loss of function of phase 4 carriers (Jansen 2001; Kubitz et al. 2005). Examples are Dubin–Johnson syndrome caused by MRP2 (ABCC2) loss (Paulusma and Oude Elferink 1997) and subtypes of progressive familial intrahepatic cholestasis (PFIC), of which PFIC type 2 and PFIC type 3 are the most progressive cholestases caused by functional loss of BSEP (ABCB11) and of MDR3 (ABCB4; syn. Mdr2 in rodents), respectively (DeVree et al. 1998; Maisonnette et al. 2005; Wagner and Trauner 2005). Dubin–Johnson syndrome results in black pigmentation of the liver and high serum levels of glucuronidated bilirubins. Since conjugated bilirubin is not toxic, the MRP2 defect does not elevate serum cytotoxicity markers, and prognosis is good (Jansen 2001). In Dubin–Johnson syndrome, the defect of MRP2 is counteracted by an overexpression of MRP3, enabling the release of glucuronidated bilirubin into blood. PFIC2, also named Byler’s syndrome, is characterised by marginal biliary bile acid secretion (1% of normal), jaundice and progressive cholestasis requiring liver transplantation within the first decade (Wagner and Trauner 2005). PFIC3 is characterised by defective phospholipid, particularly phosphatidylcholine secretion into bile, and high serum γ-glutamyltransferases levels (DeVree et al. 1998). The prognosis of this disease is infaust.
On the other hand, and already mentioned, remarkable reconstruction of carrier expression resulting in synchronous up- and down-regulation of multiple transporters in the basolateral and canalicular membrane is observed in patients with intra- or extrahepatic cholestasis (Trauner et al. 1998, 2005; Shoda et al. 2001). These phenomena reflect adaptive transport modulation aiming to prevent excessive load of liver cells with toxic bile acids and cholephilic xenobiotics. Whilst the understanding of the molecular mechanisms of cholestasis is becoming more and more detailed on this level, this knowledge also offers an intriguing approach for a rational treatment of liver diseases (Wagner and Trauner 2005).
In conclusion
We would like to emphasize an integrated drug metabolism/drug transporter concept consisting of the “old” metabolism phases 1 and 2 but extended for new transport phases: the sequential processes of carrier-mediated drug uptake, intracellular drug transport, and carrier-mediated drug excretion. A carrier-mediated uptake phase 0 precedes the metabolism phases 1 and 2. A transcellular transport of a xenobiotic/metabolite through the cytosol of a polar excretory cell is termed phase 3, and the final carrier-mediated excretory process across the cell membrane is named phase 4. Each phase is prone to drug–drug interactions, both by blockade or induction. Prediction models for drug pharmacokinetics based hitherto on metabolism need to be expanded to include transport phases. Clinical and toxicological relevancies have been reported in the literature.
References
Alvarado F, Van Os CH (1986) (eds) Ion gradient-coupled transport. Elsevier Science Publishers, Amsterdam, pp 442
Atanasova S, von Ahsen N, Dimitrov T, Armstrong V, Oellerich M, Toncheva D (2004) MDR1 haplotypes modify BEN disease risk: a study in Bulgarian patients with Balkan endemic nephropathy compared to healthy controls. Nephron Exp Nephrol 96:7–13
Athayde AL, Ivory CF (1985) Electrical pumping in carrier-mediated membrane transport. J Membr Sci 24:309–323
Ayrton A, Morgan P (2001) Role of transport proteins in drug absorption, distribution and excretion. Xenobiotica 31:469–497
Bilej M, Vetvicka V (1989) The transmembrane and intracellular transport of drugs: interactions with the cytosceletal network. Crit Rev Ther Drug Carrier Syst 6:613–691
Bolhuis H, van Veen HW, Poolman B, Driessen AJ, Konings WN (1997) Mechanisms of multidrug transporters. FEMS Microbiol Rev 21:55–84
Borst P, Oude Elferink R (2002) Mammalian ABC transporters in health and disease. Annu Rev Biochem 71:537–592
Brant SR, Panhuysen CI, Nicolae D, Reddy DM, Bonen DK, Karaliukas R, Zhang L, Swanson E, Datta LW, Moran T, Ravenhill G, Duerr RH, Achkar JP, Karban AS, Cho JH (2003) MDR1 Ala893 polymorphism is associated with inflammatory bowel disease. Am J Hum Genet 73:1282–1292
Brodie BB (1964) Physico-chemical factors in drug absorption. In: Binns TB (ed) Absorption and distribution of drugs. Livingstone, Edinburgh, pp 16–48
Brodie BB, Hogben CAM (1957) Some physico-chemical factors in drug action. J Pharmacy Pharmacol 9:345–380
Burckhardt BC, Burckhardt G (2003) Transport of organic anions across the basolateral membrane of proximal tubule cells. Rev Physiol Biochem Pharmacol 146:95–158
Burckhardt G, Wolff NA (2000) Structure of renal organic anion and cation transporters. Am J Physiol Renal Physiol 278:F853–F866
Caldwell PC (1956) The effects of certain metabolic inhibitors on the phosphate esters of the squid giant axon. J Physiol (London) 132:35
Caldwell PC (1960) The phosphorus metabolism of squid axons and its relationship to the active transport of sodium. J Physiol (London) 152:545–560
Carafoli E, Scarpa A (1982) (eds) Transport-ATPases. Ann N Y Acad Sci, New York (402):604
Catania VA, Sanchez Pozzi EJ, Luquita MG, Ruiz ML, Villanueva SS, Jones B, Mottino AD (2004) Co-regulation of expression of phase II metabolizing enzymes and multidrug resistance-associated protein 2. Ann Hepatol 3:11–17
Chan LMS, Lowes S, Hirst BH (2004) The ABCs of drug transport in intestine and liver: efflux proteins limiting drug absorption and bioavailability. Eur J Pharmaceut Sci 21:25–51
Chandra P, Brouwer KL (2004) The complexities of hepatic drug transport: current knowledge and emerging concepts. Pharm Res 21:719–735
Crane RK, Krane SM (1959) Studies on the mechanism of intestinal active transport of sugars. Biochim Biophys Acta 31:397–401
Dahlquist R, Ejvinsson G, Schenk-Gustaffson K (1980) Effect of quinidine on plasma concentration and renal clearance of digoxin. A clinically important drug interaction. Brit J Clin Pharmacol 9:413–418
Daniel H (2004) Molecular and integrative physiology of intestinal peptide transport. Annu Rev Physiol 66:361–384
Daniel H, Rubio-Aliaga I (2003) An update on renal peptide transporters. Am J Physiol Renal Physiol 284:F885–F892
Dean M, Rzhetsky A, Allikmets R (2001) The human ATP-binding cassette (ABC) transporter superfamily. Genome Res 11:1156–1166
Delgoda R, Westlake AC (2004) Herbal interactions involving cytochrome p450 enzymes: a mini review. Toxicol Rev 23:239–249
DeVree J, Jacquemin E, Sturm E, Creteil D, Bosma PJ, Aten J, DeLeuze JF, Desrochers M, Burdelski M, Bernard O, Oude Elferink RP, Hadchouel M (1998) Mutations in the MDR3 gene cause progessive familial intahepatic cholestasis. Proc Natl Acad Sci USA 95:282–287
Diamond JM,Wright EM (1969) Biological membranes: the physical basis of ion and nonelectrolyte selectivity. Annu Rev Physiol 31:581–646
Dietrich CG, Geier A, Oude Elferink RPJ (2003) ABC or oral bioavailability: transporters as gatekeepers in the gut. Gut 52:1788–1795
Doering W (1979) Quinidine-digoxin interaction: Pharmacokinetics, underlying mechanism and clinical implications. N Engl J Med 301:400–404
Donner MG, Keppler D (2001) Up-regulation of basolateral multidrug resistance protein 3 (Mrp3) in cholestatic rat liver. Hepatology 34:351–359
Drescher S, Schaeffeler E, Hitzl M, Hofmann U, Schwab M, Brinkmann U, Eichelbaum M, Fromm MF (2002) MDR1 gene polymorphisms and disposition of the P-glycoprotein substrate fexofenadine. Br J Clin Pharmacol 53:526–534
Drescher S, Glaser H, Mürdter T, Hitzl M, Eichelbaum M, Fromm MF (2003) P-glycoprotein-mediated intestinal and biliary digoxin transport in humans. Clin Pharmacol Ther 73:223–231
Dresser MJ, Leabman MK, Giacomini KM (2001) Transporters involved in the elimination of drugs in the kidney: organic anion transporters and organic cation transporters. Pharmaceut Sci 90:397–420
Dresser GK, Bailey DG, Leake BF, Schwarz UI, Dawson PA, Freeman DJ, Kim RB (2002) Fruit juices inhibit organic anion transporting polypeptide-mediated drug uptake to decrease the oral availability of fexofenadine. Clin Pharmacol Ther 71:11–20
Drozdzik M, Bialecka M, Mysliwiec K, Honczarenko K, Stankiewicz J, Sych Z (2003) Polymorphism in the P-glycoprotein drug transporter MDR1 gene: a possible link between environmental and genetic factors in Parkinson’s disease. Pharmacogenetics 13:259–263
Elleroy JC, Lew VL (1977) (eds) Membrane transport in red cells. Academic Press, New York, pp 469
Fardel O, Jigorel E, Le Vee M, Payen L (2005) Physiological, pharmacological and clinical features of the multidrug resistance protein 2. Biomed Pharmacother 59:104–114
Fehrenbach T, Cui Y, Faulstich H, Keppler D (2003) Characterization of the transport of the bicyclic peptide phalloidin by human hepatic transport proteins. Naunyn-Schmiedeberg’s Arch Pharmacol 368:415–420
Fromm MF (2004) Importance of P-glycoprotein at blood-tissue barriers. Trends Pharmacol Sci 25:423–429
Fromm MF, Kim RB, Stein CM, Wilkinson GR, Roden DM (1999) Inhibition of P-glycoprotein-mediated drug transport: a unifying mechanism to explain the interaction between digoxin and quinidine. Circulation 99:552–557
Gao B, St Pierre MV, Stieger B, Meier PJ (2004) Differential expression of bile salt and organic anion transporters in developing rat liver. J Hepatol 41:202–208
Gerok W, Sickinger K (1973) (eds) Drugs and the liver. Schattauer Verlag, Stuttgart, pp 441
Geyer J, Döring B, Godoy JR, Moritz A, Petzinger E (2005a) Development of a PCR-based diagnostic test detecting a nt230(del4) MDR1 mutation in dogs: verification in a moxidectin-sensitive Australian Shepherd. J Vet Pharmacol Ther 28:95–99
Geyer J, Döring B, Godoy JR, Moritz A, Petzinger E (2005b) Frequency of the nt230(del4) MDR1 mutation in Collies and related dog breeds from Germany. J Vet Pharmacol Ther 28:545–551
Geyer J, Wilke T, Petzinger E (2006) The solute carrier family SLC10: more than a family of bile acid transporters regarding function and phylogenetic relationships. Naunyn-Schmiedeberg’s Arch Pharmacol, this issue
Gilles-Baillieu M, Gilles R (1983) (eds) Intestinal transport: fundamental and comparative aspects. Springer-Verlag, Berlin Heidelberg New York, pp 375
Gillette JR (1963) Metabolism of drugs and other foreign compounds by enzymatic mechanisms. Progr Drug Res 6:11–75
Grammatté T, Oertel R (1999) Intestinal secretion of intravenous talinolol is inhibited by luminal R-verapamil. Clin Pharmacol Ther 66:239–245
Grammatté T, Oertel R, Terhaa, B, Kirch W (1996) Direct demonstration of small intestinal secretion and side-dependent absorption of the β-blocker talinolol in human. Clin Pharmacol Ther 59:541–549
Greiner B, Eichelbaum M, Fritz P, Kreichgauer HP, von Richter O, Zundler J, Kroemer HK (1999) The role of intestinal P-glycoprotein in the interaction of digoxin and rifampin. J Clin Invest 104:147–153
Greger R, Lang F, Silbernagl S (1981) (eds) Renal transport of organic substances. Springer-Verlag, Berlin Heidelberg New York, pp 314
Gründemann D, Gorboulev V, Gambaryan S, Veyhl M, Koepsell H (1994) Drug excretion mediated by a new prototype of polyspecific transporter. Nature 372:549–552
Habig WH, Pabst MJ, Fleischner G, Gatmaitan Z, Arias IM, Jacobvy WB (1974) The identity of glutathione S-transferase B with ligandin, a major binding protein of liver. Proc Natl Acad Sci USA 71:3879–3882
Hagenbuch B, Meier PJ (2003) The superfamily of organic anion transporting polypeptides. Biochim Biophys Acta 1609:1–18
Haimeur A, Conseil G, Geeley RG, Cole SP (2004) The MRP-related and BCRP/ABCG2 multidrug resitance proteins: biology, substrate specificity and regulation. Curr Drug Metab 5:21–53
Hediger MA, Coady MJ, Ikeda TS, Wright EM (1987) Expression cloning and cDNA sequencing of the Na+/glucose cotransporter. Nature (London) 330:379–381
Hediger MA, Romero MF, Peng JB, Rolfs A, Takanaga H, Bruford EA (2004) The ABCs of solute carriers: physiological, pathological, and therapeutic implications of human membrane transport proteins. Pfluegers Arch 447:465–468
Heinz E (1972) (eds) Na-linked transport of organic solutes. The coupling between electrolyte and nonelectrolyte transport in cells. Springer-Verlag, Berlin Heidelberg New York, pp 201
Heinz E (1978) (eds) Mechanics and energetics of biological transport. Springer-Verlag, Berlin Heidelberg New York, pp 159
Hoffmann U, Kroemer HK (2004) The ABC transporters MDR1 and MRP2: multiple functions in disposition of xenobiotics and drug resistance. Drug Metab Rev 86:669–701
Hoffmeyer S, Burk O, von Richter O, Arnold H P, Brockmöller J, Johne A, Cascorbi I, Gerloff T, Roots I, Eichelbaum M, Brinkmann U (2000) Functional polymorphisms of the human multidrug-resitance gene: multiple sequence variations and correlation of one allele with P-glycoprotein expression and activity in vivo. Proc Nat Acad Sci U S A 97:3473–3478
Hogben CAM, Tocco DJ, Brodie BB, Schanker LS (1959) On the mechanism of intestinal drug absorption of drugs. J Pharmacol Exp Ther 125:275–282
Homolya L, Varadi A, Sarkadi B (2003) Multidrug resitance-assiciated proteins: export pumps for conjugates with glutathione, glucuronate or sulfate. Biofactors 17:103–114
Hsiang B, Zhu Y, Wang Z, Wu Y, Sasseville V, Yang WP, Kirchgessner TG (1999) A novel human hepatic organic anion transporting polypeptide (OATP2) Identification of a liver-specific human organic anion transporting polypeptide and identification of rat and human hydroxymethylglutaryl-CoA reductase inhibitor transporters. J Biol Chem 274:37161–37168
Ieiri I, Takane H, Otsubo K (2004) The MDR1 (ABCB1) gene polymorphism and its clinical implications. Clin Pharmacokinet 43:553–576
Iida A, Saito S, Sekine A, Mishima C, Kondo K, Kitamura Y, Harigae S, Osawa S, Nakamura Y (2001) Catalog of 258 single-nucleotide polymorphisms (SNPs) in genes encoding three organic anion transporters, three organic anion transporting polypeptides, and three NADH : ubichinone oxidoreductase flavoproteins. J Hum Genet 46:668–683
Ito K, Iwatsuro T, Kanamitsu S, Ueda K, Suzuki H, Sugiyama Y (1998) Prediction of pharmacokinetic alterations caused by drug-drug interactions: metabolic interaction in the liver. Pharmacol Rev 50:387–411
Jackson MJ (1987) Drug transport across gastrointestinal epithelia. In: LR Johnson (ed) Physiology of the gastrointestinal tract. Raven Press, New York, pp 1597–1621
Jacquemin E, Hagenbuch B, Stieger B, Wolkoff AW, Meier PJ (1994) Expression cloning of a rat liver Na(+)-independent organic anion transporter. Proc Natl Acad Sci USA 91:133–137
Juliano RL, Ling V (1976) A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim Biophys Acta 455:152–162
Jansen PLM (2001) Hereditary defects of hepatobiliary transport. In: Matern S, Boyer JL, Keppler D, Meier-Abt PJ (eds) Hepatobiliary transport: from bench to bedside. Kluwer Acad. Publ., Dordrecht, pp 91–97
Kafka A, Sauer G, Jaeger C, Grundmann R, Kreienberg R, Zeillinger R, Deissler H (2003) Polymorphism C3435T of the MDR-1 gene predicts response to preoperative chemotherapy in locally advanced breast cancer. Int J Oncol 22:1117–1121
Keynes RD (1961) The energy source for active transport in nerve and muscle. In: A Kleinzeller, A Kotyk (eds) Membrane transport and metabolism. Academic Press, London, pp 131–139
Kilby JM, Hill A, Buss N (2002) The effect of ritonavir on saquinavir plasma concentration is independent of ritonavir dosage: combined analysis of pharmacokinetic data from 97 subjects. HIV Med 3:97–104
Kim RB (2004) 3-hydroxy-3-methylglutaryl-coenzyme A reductase inhibitors (statins) and genetic variability (single nucleotide polymorphisms) in a hepatic drug uptake transporter: what’s it all about? Clin Pharmacol Ther 75:381–385
Kim RB, Fromm MF, Wandel C, Leake B, Wood AJJ, Roden DM, Wilkinson GR (1998) The drug transporter p-glycoprotein limits oral absorption and brain entry of HIV-1 protease inhibitors. J Clin Invest 101:289–294
Klaassen CD, Watkins JB (1984) Mechanisms of bile formation, hepatic uptake, and biliary excretion. Pharmacol Rev 36:1–67
Koepsell H, Endou H (2004) The SLC22 drug transporter family. Pflügers Arch 447:666–676
König J, Rost D, Cui Y, Keppler D (1999) Characterisation of the human multidrug resistance protein isoform MRP3 localized to the basolateral hepatocyte membrane. Hepatology 29:1156–1163
König J, Seithel A, Gradhand U, Fromm MF (2006) Pharmacogenomics of human OATP transporters. Naunyn-Schmiedeberg’s Arch Pharmacol, this issue
Kramer M, Lauterbach F (1977) (eds) Intestinal permeation. Excerpta Medica, Amsterdam, pp 449
Kramer W, Wess G (1996) Bile acid transport systems as pharmaceutical targets. Eur J Clin Invest 26:715–732
Kramer W, Wess G, Schubert G, Bickel M, Girbig F, Gutjahr U, Kowalewski S, Baringshaus K-H, Ehnsen A, Glombik H, Müllner S, Neckermann G, Schulz S, Petzinger E (1992) Liver-specific drug trageting by coupling with bile acids. J Biol Chem 267:18598–18604
Kramer W, Sauber K, Baringhaus K-H, Kurz M, Stengelin S, Lange G, Corsiero D, Girbig F, König W, Weyland C (2001) Identification of the bile acid-binding site of the ileal lipid-binding protein by photoaffinty labelling, matrix-assisted laser desorption ionization-mass spectrometry, and NMR structure. J Biol Chem 276:7291–7301
Krusekopf S, Roots I (2005) St. John’s wort and its constituent hyperforin concordantly regulate expression of genes encoding enzymes involved in basic cellular pathways. Pharmacogenetics 15:817–829, Genomics
Kubitz R, Keitel V, Häussinger D (2005) Inborn errors of biliary canalicular transport systems. Meth Enzymol 400:558–569
Kullak-Ublick GA, Beuers U, Paumgartner G (2000) Hepatobiliary transport. J Hepatol 32:3–18
Kullak-Ublick GA, Stieger B, Meier PJ (2004) Enterohepatic bile salt transporters in normal physiology and liver disease. Gastroenterology 126:322–342
LeFevre PG (1948) Evidence of active transfer of certain non-electrolytes across the human red cell membrane. J Gen Physiol 31:505–507
LeFevre PG (1975) The present state of the carrier hypothesis. In: Bronner F, Kleinzeller A (eds) Current topics in membranes and transport. Academic Press, London, pp 109–215
Leslie EM, Deeley RG, Cole SP (2001) Toxicological relevance of the multidrug resistance protein 1, MRP1 (ABCC1) and related transportes. Toxicology 167:3–23
Leslie EM, Deeley RG, Cole SP (2005) Multidrug resistance proteins: role of P-glycoprotein, MRP1, MRP2 and BCRP (ABCG2) in tissue defense. Toxicol Appl Pharmacol 204:216–237
Levi AJ, Gatmaitan Z, Arias IM (1969) Two hepatic cytoplasmic protein fractions, Y and Z, and their possible role in the hepatic uptake of bilirubin, sulfobromophthalein, and other anions. J Clin Invest 48:2156–2167
Liu L, Pang KS (2005) The roles of transporters and enzymes in hepatic drug processing. Drug Metab Dispos 33:1–9
Lloyd JB (2000) Lysosome membrane permeability: implications for drug delivery. Adv Drug Deliv Rev 41:189–200
Maisonnette F, Abita T, Barriere E, Pichon N, Vincensini JF, Descottes B (2005) The MDR3 gene mutation: a rare cause of progressive familial intrahepatic cholestasis (PFIC). Ann Chir 130:581–583
Mannel M (2004) Drug interactions with St John’s wort: mechanisms and clinical implications. Drug Saf 27:773–797
Mealey KL, Bentjen SA, Gay JM, Cantor GH (2001) Ivermectin sensitivity in collies is associated with a deletion mutation in the mdr1 gene. Pharmacogenetics 11:727–733
Meijer DK (1993) Drug targeting to the liver with bile acids: the “Trojan horse” resurrected? Hepatology 17:945–948
Milne RW, Larsen LA, Jorgensen KL, Bastlund JF, Stretch GL, Evans AM (2000) Hepatic disposition of fexofenadine: influence of the transport inhibitors erythromycin and dibromosulphothalein. Pharmaceutical Res 17:1511–1515
Mizuno N, Niwa T, Yotsumoto Y, Sugiyama Y (2003) Impact of drug transporter studies on drug discovery and development. Pharmacol Rev 55:425–461
Müller M, Jansen PLM (1998) The secretory step of the liver: new aspects of hepatobiliary transport. J Hepatol 28:344–354
Murray JW, Wolkoff AW (2003) Roles of the cytoskeleton and motor proteins in endocytic sorting. Adv Drug Deliv Rev 55:1385–1403
Niemi M, Schaeffeler E, Lang T, Fromm MF, Neuvonen M, Kyrklund C, Backman J T, Kerb R, Schwab M, Neuvonen PJ, Eichelbaum M, Kivistö KJ (2004) High plasma pravastatin concentrations are associated with single nucleotide polymorphisms and haplotypes of organic anion transporting polypeptide-C (OATP-C, SCLO1B1). Pharmacogenetics 14:429–440
Niemi M, Neuvonen PJ, Hofmann U, Backman JT, Schwab M, Lütjohann D, von Bergmann K, Eichelbaum M, Kivistö KT (2005a) Acute effects of pravastatin on cholesterol synthesis are associated with SCLO1B1 (encoding OATP1B1) haplotype *17. Pharmacogen Genom 15:303–309
Niemi M, Kivistö KT, Hofmann U, Schwab M, Eichelbaum M, Fromm M (2005b) Fexofenadine pharmacokinetics are associated with a polymorphism of the SCLO1B1 gene (encoding OATP1B1). Br J Clin Pharmacol 59:602–604
Nozawa T, Nakajima M, Tamai I, Noda K, Nezu JI, Sai Y, Tsuji A, Yokoi T (2002) Genetic polymorphisms of human organic anion transporters OATP-C (SLC21A6) and OATP-B (SLC21A9): allele frequencies in the Japanese population and functional studies. J Pharmacol Exp Ther 302:804–813
Ogawa K, Suzuki H, Hirohashi T, Ishikawa T, Meier PJ, Hirose K, Akizawa T, Yoshioka M, Sugiyama Y (2000) Characterization of inducible nature of MRP3 in rat liver. Am J Physiol Gastrointest Liver Physiol 278:G438–G446
Ohashi M, Sano N, Takikawa H (2002) Effects of phalloidin on the biliary excretion of cholephilic compounds in rats. Pharmacology 66:31–35
Paul AJ, Tranquilli WJ, Seward RL, Todd KS, DiPietro JA (1987) Clinical observations in collies given ivermectin orally. Am J Vet Res 48:684–685
Pauli-Magnus C, Meier PJ (2004) Pharmacogenetics of hepatocellular transporters. Pharmacogenetics 13:189–198
Paulusma LC, Oude Elferink RP (1997) The canalicular multispecific anion transporter and conjugated hyperbilirubinemia in rat and man. J Mol Med 75:420–428
Pedersen KE, Christansen BD, Klittgaard NA, Nielsen-Kudsk F (1983) Effect of quinidine of digoxin bioavailability. Eur J Clin Pharmacol 24:41–47
Petzinger E (1981) Competitive inhibition of the uptake of demethylphalloin by cholic acid in isolated hepatocytes. Evidence for a transport competition rather than a binding competition. Naunyn-Schmiedeberg’s Arch Pharmacol 316:345–349
Petzinger E (1994) Transport of organic anions in the liver. An update on bile acid, fatty acid, monocarboxylate, anionic amino acid, cholephilic organic anion, and anionic drug transport. Rev Physiol Biochem Pharmacol 123:47–211
Petzinger E, Ziegler K, Frimmer M (1979) Inhibition of 3H-demethylphalloin uptake in isolated rat hepatocytes under various experimental conditions. Naunyn-Schmiedeberg’s Arch Pharmacol 307:275–281
Petzinger E, Kinne RKH, Sies H (1989) (eds) Hepatic transport of organic substances. Springer-Verlag, Berlin Heidelberg New York, pp 435
Petzinger E, Nickau L, Horz JA, Schulz S, Wess G, Ehnsen A, Falk E, Baringhaus K-H, Glombik H, Hoffmann A, Müllner S, Nekermann G, Kramer W (1995) Hepatobiliary transport of hepatic 3-hydroxy-3-methylglutaryl Coenzyme A reductase inhibitors conjugated with bile acids. Hepatology 22:1801–1811
Pritchard JB, Miller DS (1993) Mechanisms mediating renal secretion of organic anions and cations. Physiol Rev 73:765–796
Pulliam JD, Seward RL, Henry RT, Steinberg SA (1985) Investigating ivermectin toxicity in Collies. Vet Med 80:33–40
Roninson IB, Chin JE, Choi KG, Gros P, Housman DE, Fojo A, Shen DW, Gottesman MM, Pastan I (1986) Isolation of human mdr DNA sequences amplified in multidrug resistant KB carcinoma cells. Proc Natl Acad Sci U S A 83:4538–4542
Rosenberg T, Wilbrandt W (1957) Uphill transport induced by counterflow. J Gen Physiol 41:289–296
Rost D, Kartenbeck J, Keppler D (1999) Changes in the localization of the rat canalicular conjugate export pump Mrp2 in phalloidin-induced cholestasis. Hepatology 29:814–821
Roulet A, Puel O, Gesta S, Lepage JF, Drag M, Soll M, Alvinerie M, Pineau T (2003) MDR1-deficient genotype in Collie dogs hypersensitive to the P-glycoprotein substrate ivermectin. Eur J Pharmacol 460:85–91
Russel FG, Masereeuw R, Van Aubel RA (2002) Molecular aspects of renal anionic drug transport. Annu Rev Physiol 64:563–594
Scheffer GL, Kool M, de Haas M, de Vree JM, Pijnenborg AC, Bosman DK, Oude Elferink R, Van Borst P, Scheper RJ (2002) Tissue distribution and induction of human multidrug resistant protein 3. Lab Invest 82:193–201
Schwab M, Schaeffeler E, Marx C, Fromm MF, Kaskas B, Metzler J, Stange E, Herfarth H, Schoelmerich J, Gregor M, Walker S, Cascorbi I, Roots I, Brinkmann U, Zanger UM, Eichelbaum M (2003) Association between the C3435T MDR1 gene polymorphism and susceptibility for ulcerative colitis. Gastroenterology 124:26–33
Sekine T, Watanabe N, Hosoyamada M, Kanai Y, Endou H (1997) Expression cloning and characterization of a novel multispecific organic anion transporter. J Biol Chem 272:18526–18529
Semenza G, Kinne R (1985) (eds) Membrane transport driven by ion gradients. Ann N Y Acad Sci 456:459
Shibayama Y, Ikeda R, Motoya T, Yamada K (2004) St John’s Wort (Hypericum perforatum) induces overexpression of multidrug resistance protein2 (MRP2) in rats: a 30-day ingestion study. Food Chem Toxicol 42:995–1002
Shoda J, Kano M, Oda K, Kamiya J, Nimura Y, Suzuki H, Sugiyama Y, Miyazaki H, Todoroki T, Stengelin S, Kramer W, Matsuzaki Y, Tanaka N (2001) The expression levels of plasma membrane transporters in the cholestatic liver of patients undergoing biliary drainage and their association with the impairment of biliary secretory function. Am J Gastroenterol 96:3368–3378
Siegsmund M, Brinkmann U, Schaffeler E, Weirich G, Schwab M, Eichelbaum M, Fritz P, Burk O, Decker J, Alken P, Rothenpieler U, Kerb R, Hoffmeyer S, Brauch H (2002) Association of the P-glycoprotein transporter MDR1(C3435T) polymorphism with the susceptibility to renal epithelial tumors. J Am Soc Nephrol 13:1847–1854
Smith JN, Williams RT (1949) The metabolism of phenacetin (p-ethoxyacetanilide) in the rabbit and a further observation on acetanilide metabolism. Biochem J 44:239–242
Soroka CJ, Lee JM, Azzaroli F, Boyer JL (2001) Cellular localization and up-regulation of multidrug resistance-associated protein 3 in hepatocytes and cholangiocytes during obstructive cholestasis in rat liver. Hepatology 33:783–791
Spahn-Langguth H, Baktir G, Radschuweit A, Okyar A, Terhaag B, Ader P, Hanafy A, Langguth P (1998) P-glycoprotein transporters and the gastrointestinal tract: evaluation of the potential in vivo relevance of in vitro data employing talinolol as model compound. Int J Clin Pharmacol Ther 36:16–24
Sperber I (1959) Secretion of organic anions in formation of urine and bile. Pharmacol Rev 11:109–134
Stein W (1967) (ed) The movement of molecules across membranes. Academic Press, New York, pp 367
Sweet DH, Wolff NA, Pritchard JB (1997) Expression cloning and characterization of ROAT1 The basolateral organic anion transporter in rat kidney. J Biol Chem 272:30088–30095
Synold TW, Dussault I, Forman BM (2001) The orphan nuclear receptor SXR coordinately regulated drug metabolism and efflux. Nature Med 7:584–590
Tirona RG, Kim RB (2002) Pharmacogenomics of drug transporters. In: J Licinio, M-L Wing (eds) Pharmacogenomics: the search for individualized therapies. Wiley-VCH, pp 179–213
Tirona RG, Leake BF, Merino G, Kim RB (2001) Polymorphisms in OATP-C: Identification of multispecific allelic variants associated with altered transport activity among European- and African-Americans. J Biol Chem 276:35669–35675
Trauner M, Boyer JL (2003) Bile salt transporters: molecular characterization, function, and regulation. Physiol Rev 83:633–671
Trauner M, Meier PJ, Boyer JL (1998) Molecular pathogenesis of cholestasis. N Engl J Med 339:1217–1227
Trauner M, Wagner M, Fickert P, Zollner G (2005) Molecular regulation of hepatobiliary transport systems: clinical implications for understanding and treating cholestasis. J Clin Gastroenterol 39:S111–S124
Ullrich KJ (1997) Renal transporters for organic anions and organic cations. Structural requirements for substrates. J Membr Biol 158:95–107
Van Heeswijk RP, Veldkamp A, Mulder JW, Meenhorst PL, Lange JM, Beijnen JH, Hoetelmans RM (2001) Combination of protease inhibitors for the treatment of HIV-1-infected patients: a review of pharmacokinetics and clinical experience. J Antivir Ther 6:201–229
Van Montfoort JE, Hagenbuch B, Groothuis GMM, Koepsell H, Meier PJ, Meijer DKF (2003) Drug uptake systems in liver and kidney. Curr Drug Metab 4:185–211
Wagner M, Trauner M (2005) Transcriptional regulation of hepatobiliary transport systems in health and disease: implications for a rationale approach to the treatment of intrahepatic cholestasis. Ann Hepatol 4:77–99
Wandel C, Kim RB, Kajiji S, Guengerich FP, Wilkinson GR, Wood AJJ (1999) P-glycoprotein and cytochrome P-450 3 A inhibition: dissociation of inhibitory potencies. Cancer Res 59:3944–3948
Ward WJ III (1970) Electrically induced carrier transport. Nature 227:162–163
Washington CB, Wiltshire HR, Man M, Moy T, Harris SR, Worth E, Weigl P, Liand Z, Hall D, Marriot L, Blaschke TF (2000) The disposition of saquinavir, in normal and P-glycoprotein deficient mice, rats, and cultured cells. Drug Metab Disp 28:1058–1062
Watson P, Jones AT, Stephens DJ (2005) Intracellular trafficking pathways and drug delivery: fluorescence imaging of living and fixed cells. Adv Drug Deliv Rev 57:43–61
Weaver JL, Pine PS, Aszalos A, Schoenlein PV, Currier SJ, Padmanabhan R, Gottesman MM (1991) Laser scanning and confocal microscopy of daunorubicin, doxorubicin, and rhodamine 123 in multi-drug resistant cells. Exp Cell Res 196:323–329
Westphal K, Weinbrenner A, Zschiesche M, Franke G, Knoke M, Oertel R, Fritz P, von Richter O, Warzok R, Hachenberg T, Kaumann HM, Schrenk D, Terhaag B, Kroemer HK, Siegmund W (2000) Induction of P-glycoprotein by rifampin increases intestinal secretion of talinolol in human beings: a new type of drug/drug interaction. Clinical Pharmacol Ther 68:345–355
Wilbrandt W (1975) Criteria in carrier transport. In: H Eisenberg, E Katchalski-Kazir, LA Manson (eds) Biomembranes vol 7. Plenum Press New York, London, pp 11–31
Williams RT (1959) (ed) Detoxification mechanisms the metabolism and detoxification of drugs, toxic substances and other organic compounds. Chapman & Hall, London, pp 796
Wright EM, Turk E (2004) The sodium/glucose cotransport family SLC5. Pflügers Arch-Eur J Physiol 447:510–518
Yarim M, Moro S, Huber R, Meier PJ, Kaseda C, Kashima T, Hagenbuch B, Folkers G (2005) Application of QSAR analysis to organic anion polypeptide 1a5 (Oatp 1a5) substrates. Bioorg Med Chem 13:463–471
Zhang Y, Guo X, Tin ET, Benet LZ (1998) Overlapping substrate specificities of cytochrome P450 3A and P-glycoprotein for a novel cysteine protease inhibitor. Drug Metab Disp 26:360–366
Ziegler K, Hummelsiep S (1993) Hepatoselective carrier-mediated sodium-independent uptake of pravastatin and pravastatin-lactone. Biochim Biophys Acta 1153:23–33
Ziegler K, Stünkel W (1992) Tissue-selective action of pravastatin due to hepatocellular uptake via a sodium-independent bile acid transporter. Biochim Biophys Acta 1139:203–209
Acknowledgement
We wish to thank Dr. Bruce Boschek for critically reading the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Petzinger, E., Geyer, J. Drug transporters in pharmacokinetics. Naunyn Schmied Arch Pharmacol 372, 465–475 (2006). https://doi.org/10.1007/s00210-006-0042-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00210-006-0042-9